

Abstract
Fibroblast growth factor-23 (FGF23), a hormone central to renal phosphate handling, is elevated in multiple hypophosphatemic disorders. Initial FGF23-dependent Erk1/2 activity in the kidney localizes to the distal convoluted tubule (DCT) with the co-receptor α-Klotho (KL), distinct from Npt2a in proximal tubules (PT). The Hyp mouse model of XLH is characterized by hypophosphatemia with increased Fgf23, and patients with XLH elevate FGF23 following combination therapy of phosphate and calcitriol. The molecular signaling underlying renal FGF23 activity, and whether these pathways are altered in hypophosphatemic disorders, is unknown. To examine Npt2a in vivo, mice were injected with FGF23. Initial p-Erk1/2 activity in the DCT occurred within 10 min, however Npt2a protein was latently reduced in the PT at 30–60 min, and was independent of Npt2a mRNA changes. KL-null mice had no DCT p-Erk1/2 staining following FGF23 delivery. Under basal conditions in Hyp mice, c-Fos and Egr1, markers of renal Fgf23 activity, were increased, however KL mRNA was reduced 60% (P<0.05). Despite the prevailing hypophosphatemia and elevated Fgf23, FGF23 injections into Hyp mice activated p-Erk1/2 in the DCT. FGF23 injection also resulted in phospho-β-catenin (p-β-cat) co-localization with KL in WT mice, and Hyp demonstrated strong p-β-cat staining under basal conditions, indicating potential cross-talk between Mapk and Wnt signaling. Collectively, these studies refine the mechanisms for FGF23 bioactivity, and demonstrate novel suppression of Wnt signaling in a KL-dependent DCT-PT axis, which is likely altered in XLH. Finally, the current treatment of phosphate and calcitriol for hypophosphatemic disorders may increase FGF23 activity.
Keywords: FGF-23, Klotho, Hyp, phospho-ERK1/2, hypophosphatemia, XLH, MAPK
Introduction
Fibroblast growth factor-23 (FGF23) has been identified as the key regulator of phosphate metabolism. The importance of FGF23 in phosphate metabolism is underscored by disorders of hypophosphatemia associated with increased circulating FGF23 including autosomal dominant hypophosphatemic rickets (ADHR), X-linked hypophosphatemic rickets (XLH), autosomal recessive hypophosphatemic rickets (ARHR), tumor induced osteomalaica (TIO), as well as through syndromes of reduced FGF23, primarily hyperphosphatemic tumoral calcinosis (TC). In situations of elevated FGF23, renal phosphate wasting occurs through down regulation of the proximal tubule membrane Type-IIa (Npt2a) and -IIc (Npt2c) sodium-phosphate co-transporters (Larsson, et al. 2004; Shimada, et al. 2004c). In parallel, FGF23 decreases expression of the proximal tubule (PT) vitamin D 1-α-hydroxylase (CYP27b) and increases the 24-hydroxylase (CYP24), thereby reducing the serum concentrations of 1,25(OH)2 vitamin D (Shimada, et al. 2001). These changes in gene expression account for the paradoxical low circulating 1,25(OH)2 vitamin D in the face of prevailing hypophosphatemia in syndromes associated with increased FGF23.
The co-receptor α-Klotho (KL) has been identified as necessary for FGF23 activity (Kuro-o 2006; Urakawa, et al. 2006). The hypothesized mechanism for FGF23 bioactivity with KL is the recruitment of canonical FGF receptors (FGFRs) to form heteromeric complexes, that then signal through the mitogen activated protein kinase (MAPK) cascade and phospho-ERK1/2 (p-ERK1/2) (Kuro-o 2006; Urakawa et al. 2006). In support of FGF23-KL interactions, the Fgf23- and KL-null animals have highly similar biochemical and skeletal phenotypes (Shimada, et al. 2004b; Sitara, et al. 2004), and a recessive, inactivating mutation in the KL gene resulted in impaired KL expression and a severe human TC phenotype, most likely due to end-organ resistance to FGF23 (Ichikawa, et al. 2007). Although it has been shown that interactions between KL and a canonical FGF receptor (FGFR) are necessary to mediate FGF23 signaling in vitro, the mechanisms underlying FGF23 bioactivity in vivo are unclear, as KL localizes to the renal distal convoluted tubule (DCT) and FGF23 mediates its effects on NPT2a and vitamin D metabolizing enzymes within the PT (Larsson et al. 2004; Shimada et al. 2001). We previously demonstrated that initial renal FGF23-dependent activity, as measured by p-ERK1/2 expression, occurs in the DCT localizing with Klotho expression (Farrow, et al. 2009). These findings indicate the presence of an intra-renal signaling axis controlling the biological effects of FGF23 on renal phosphate and vitamin D metabolism. Previous studies demonstrated a maximal decrease in serum phosphate concentrations at 8 h following injection of mice with FGF23(Shimada, et al. 2004a). Additionally, acute administration of parathyroid hormone (PTH) results in a decrease in Npt2a expression in the apical membrane 30 minutes after injection (Bacic, et al. 2003). Although initial FGF23 activity occurs in the renal distal tubule within 10 minutes (Farrow et al. 2009), the temporal control of Npt2a and the downstream mediators of FGF23-dependent effects between the DCT and PT remain unclear.
X-linked hypophosphatemic rickets is the most common form of heritable hypophosphatemia (HYP-Consortium 1995), and most patients have chronically elevated serum FGF23 (Weber, et al. 2003). The current therapy for XLH includes an oral regimen of phosphate and calcitriol, however the treatment itself can have significant side effects including hyperparathyroidism (Lyles, et al. 1982). Of significance, phosphate and 1,25(OH)2 vitamin D stimulate the production of Fgf23 in animals (Azam, et al. 2003; Shimada et al. 2004c), and this therapeutic regimen has recently been shown to increase circulating FGF23 concentrations in XLH patients (Imel EA 2010). The Hyp mouse model of XLH, in parallel with the human condition, has significantly elevated Fgf23 serum concentrations and Fgf23 bone mRNA expression (Liu, et al. 2006b). These animals also increase FGF23 in response to dietary phosphate supplementation and vitamin D injections (Liu, et al. 2006a). However, the molecular effects of chronically elevated FGF23 on the renal mechanisms of phosphate handling are unknown and whether FGF23 is capable of further signaling effects in the face of elevated Fgf23 in Hyp is also unclear. Therefore, the goals of the present studies were to identify novel pathways involved in renal FGF23 bioactivity as a portion of a multi-nephron segment process, and to test whether FGF23-dependent signaling is altered in syndromes associated with FGF23 excess.
Materials and Methods
Animal studies
All animal studies were performed according to the Indiana University Institutional Animal Care and Use Committee guidelines. Wild type (WT) C57/Bl6, Hyp (Jackson Laboratories; Bar Harbor, ME) or α-Klotho (KL)-null (Lexicon Genetics, Inc.; The Woodlands, Texas) mice were injected with either vehicle (saline) or 10 μg of recombinant FGF23 (gift from Genzyme Corp., Framingham, MA) either intravenously (i.v.) or intraperitoneally (i.p.). The KL-null mice carry the β-galactosidase gene cassette in place of mouse KL exons 2 and 3. Human FGF23 with the R176Q ADHR mutation was produced in insect cells (d-Mel-2), and is fully glycosylated. Animals were sacrificed at 10, 30, and 60 minutes following injection. Kidneys were harvested for preparation of RNA and immunofluorescence and serum was collected for serum FGF23 analysis.
Cell culture
HEK293 cells (American Type Culture Collection, Manassas, VA) were cultured in D-MEM/F-12 (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (Hyclone, Hudson, NH), 1 mM sodium pyruvate, 2 mM L-glutamine, and 25 mM penicillin-streptomycin at 37°C and 5% CO2. Cells stably expressing mKL were generated as previously described (Farrow et al. 2009). For bioactivity assays, mKL cells (1.5×105) were seeded in 12 well plates. Cells were serum starved a minimum of 2 hours before treatment with vehicle or 100 ng/mL of FGF23 for 0, 30, or 60 minutes.
Immunofluorescence
Kidneys were fixed with 4% PFA, mounted in OCT (Sakura Finetek, Torrance, CA), and sliced into 10 μm sections. Sections were treated with pepsin (Biocare Medical, Concord, CA) for antigen retrieval and probed with anti-mouse KL (Santa Cruz Biotechnology, Santa Cruz, CA), phospho-ERK1/2 (p-ERK1/2, Cell Signaling, Danvers, MA), phospho-Beta-catenin (p-B-cat, Cell signaling, Danvers, MA), or a highly specific antibody to mouse Npt2a (Custer, et al. 1994) followed by incubation with the appropriate fluorescent secondary antibodies (Alexa Fluor, Invitrogen, Carlsbad, CA). Where indicated, immunofluorescent signal amplification was performed using the Tyramide signal amplification (TSA) systems kit according to manufacturer’s directions (Perkin Elmer, Waltham, MA). Slides were mounted using DAPI (Vector Laboratories, Burlingame, CA). Imaging was performed using a Leica DM5000B fluorescent microscope (Leica Microsystems, Inc, Bannockburn, IL), and SPOT camera and computer program (RTKE Diagnostic Instruments, Inc, Sterling Heights, MI). A minimum of n=3–4 animals per time point, and 8–12 sections per animal were examined for each primary antibody set.
Quantitative RT-PCR (qPCR)
Kidney RNA was harvested using Trizol (Invitrogen, Carlsbad, CA) according to manufacturer’s protocol. RNA was harvested from the cellular lysates using the RNeasy kit (Qiagen, Inc., Valencia, CA). MAP kinase signal (PAMM-061) and signal transduction pathway finder (PAMM-014) array plates were obtained from SABiosciences (Frederick, MD) and used according to manufacturer’s protocol. To confirm array results, RNA samples were tested with intron-spanning primers specific for mouse Egr1 mRNA (forward 5′-agccgagcgaacaaccctat-3′, reverse 5′-cgccttctcattattcagagcg-3′, probe 5′-agcacctgaccacagagtccttttctgaca-3′), mouse c-Fos (Applied Biosystems, Carlsbad, CA), mouse Klotho (forward 5′-cagctccaggctcgggta-3′, reverse 5′-aggtgttgtagagatgccagacttt-3′, probe 5′-ttgcccacaacctacttttggctcatg-3′), and mouse Actin (forward 5′-ggctcctagcaccatgaag-3′, reverse 5′-cgccttctcattattcagagcg-3′, probe 5′-agcacctgaccacagagtccttttctgaca-3′) was used as an internal control. Intron-spanning primers specific for human EGR1 mRNA (forward 5′-ggacacgggcgagcag-3′, reverse 5′-cgttgttcagagagatgtcagga-3′, probe 5′-cctacgagcacctgaccgcagagtct-3′), human c-FOS (Applied Biosystems), and human actin (forward 5′-ggcacccagcacaatgaag-3′, reverse 5′-gccgatccacacggagtact-3′, probe 5′-tcaagatcattgctcctcctgagcgc-3′ were used for in vitro experiments with HEK293 cells. The TaqMan One-Step RT-PCR kit (Applied Biosystems, Carlsbad, CA) was used to perform qPCR. PCR conditions for all experiments were: 30 min 48°C, 10 min 95°C, followed by 40 cycles of 15 sec 95°C and 1 min 60°C. The data was collected and analyzed by the 7500 Real Time PCR system and software (Applied Biosystems, Carlsbad, CA). The expression levels of Egr1, c-Fos, Klotho, and β-actin mRNAs were calculated relative to wild type littermates or vehicle-treated controls. All primer sets were tested for specific amplification of mRNA by parallel analyses of controls that included omitting RT or template, and resulted in no fluorescent signal detection. Each RNA sample was analyzed in at least triplicate, and each experiment was performed independently at least three times. The 2−ΔΔCT method described by Livak was used to analyze the data (Livak and Schmittgen 2001).
FGF23 serum measurements
Serum intact FGF23 concentrations were assessed using an ELISA according to the manufacturer’s protocol (Kainos Laboratories Int’l; Tokyo, Japan). This two-site, monoclonal antibody ELISA has previously been shown to recognize rodent Fgf23 (Yamazaki, et al. 2002).
Statistical analysis
Statistical analysis of the data was performed by Student’s t-test and significance for all tests was set at P<0.05. Data are presented as means ± standard error of the mean (SEM).
Results
Temporal Control of Npt2a by FGF23
We previously demonstrated that initial FGF23 bioactivity, as assessed by ERK1/2 phosphorylation (p-ERK1/2), co-localizes with Klotho in the renal DCT within 10 minutes after injection (Farrow et al. 2009). However, it has been shown that FGF23 delivery results in a latent reduction in serum phosphate concentrations, with a maximum decrease apparent at 8 h (Shimada et al. 2004a). Thus the temporal control of Npt2a in the renal PT by FGF23 is not clear. To determine the effect of FGF23 on Npt2a expression, wild type mice were injected with vehicle or 10 μg of recombinant human FGF23 for 10, 30, and 60 minutes and the kidneys were harvested for RNA and for immunofluroscence (IF) analyses. Npt2a expression was diminished in the proximal tubule apical membrane within 30 min following FGF23 injection, and remained decreased 60 min after injection (Figure 1a). No change in Npt2a protein expression was observed in vehicle injected animals at any time point (Figure 1a). Kidney sections were co-stained with actin to delineate the apical membrane, which confirmed that the majority of Npt2a was removed from the apical membrane by 60 minutes (Figure 1a). Robust p-ERK1/2 staining was confirmed to localize to the renal DCT at 10 min (Figure 1b). Immunofluorescent signal amplification was used to test for p-ERK1/2 expression in the PT at 30 and 60 min. However, we were unable to identify a p-ERK1/2 signal co-localized with Npt2a at these time points (Figure 1b).
Figure 1. Control of Npt2a by FGF23.




a) Mice were injected with FGF23 or vehicle for 10, 30, or 60 minutes. Under standard staining conditions Npt2a expression was tested. Npt2a was reduced 30–60 minutes following injection with FGF23 (top row). Sections were co-stained with actin (red), to mark the apical membrane, and co-localization of Npt2a in the apical membrane was detected (yellow). By 60 minutes Npt2a was not detected in the apical membrane (middle row). Higher magnification of the proximal tubule shows the progressive removal of Npt2a from the apical membrane (bottom row); b) Sections were co-stained for Npt2a and p ERK1/2. p-ERK1/2 activity was absent from the vehicle injected animals. p-ERK1/2 was confirmed in the 10 min animals and localization was distinct from Npt2a expression in the PT; no p-ERK1/2 activity was detected in the PT at any time point tested. At the 30 and 60 min time points, Npt2a staining was not detected as reduced (as in Figure 1a) due to the effects of signal amplification for Npt2a and pERK1/2. c) The reduction of Npt2a at 60 min was independent of transcription, as no change in Npt2a mRNA was detected (P=0.7). d) WT or KL-null mice were injected with vehicle or 10 μg of FGF23 (WT vehicle, left panel; WT FGF23, middle panel) to test for renal MAPK activity. In the KL-null animals, the LacZ gene is inserted into the Klotho locus, which enabledβ-galactosidase (βgal) immunofluorescence to be used as a surrogate marker for KL (green, right panel). Following FGF23 delivery to KL-null mice, p-ERK1/2 activity was absent from the DCT labeled with β-gal (KL) (right panel). As confirmed, strong p-ERK1/2 activity co-localized with KL in WT animals injected with FGF23 (middle).
The change in Npt2a protein localization within the apical membrane was independent of a transcriptional change, as a reduction in Npt2a mRNA was not detected between vehicle and FGF23-injected animals (P=0.7; Figure 1c). In contrast, the positive control for FGF23 activity, Early growth response gene-1 (Egr1) mRNA was dramatically increased (~50-fold; P<0.05; not shown). Thus, initial FGF23 activity in the DCT through p-ERK1/2 occurs within 10 min, whereas Npt2a protein expression in the PT is a latent effect (30–60 min) that does not require changes Npt2a mRNA levels.
To test whether KL was required for initial FGF23 activity through MAPK in the DCT, KL null mice were injected with 10 μg FGF23 and sacrificed at 10 min. These analyses indicated that ERK1/2 was not activated in the tubule segments of vehicle-injected WT mice (Figure 1d, left), or in KL-null mice that express a surrogate marker for KL expression, β-galactosidase (Figure 1d, right). The positive control WT mice showed strong p-ERK1/2 activity (Figure 1d, middle).
FGF23-mediated signaling under normal conditions and in disease
To identify genes responsive to FGF23, we tested for changes in transcription factors other than Egr1 that are known to be downstream of canonical FGF signaling, including c-Fos (Kang, et al. 2005). In this regard, WT mice were injected with either vehicle or 10 μg of recombinant human FGF23 i.p. (n=3–4/group) and sacrificed at 60 min. qPCR was performed for c-Fos and Egr1 on the kidney RNA isolated from the injected animals. c-Fos showed a 13-fold increase in mRNA expression. The positive control, Egr1 increased over 70-fold in FGF23-injected mice when compared to the vehicle controls (P<0.02; Figure 2a). To confirm these results, HEK293 cells stably expressing mKL were treated with 100 ng/mL of recombinant human FGF23 for 0, 30, and 60 minutes. A time-dependent increase was observed for both c-Fos and EGR1 mRNA with a maximum increase of 30- and 200-fold, respectively (P<0.05; Figure 2b). c-Fos is known to be downstream of ERK1/2 in canonical FGF signaling, however it has not been reported previously as responsive to FGF23. Taken together, these in vivo and in vitro results confirm the utilization of c-Fos as a marker of FGF23 bioactivity.
Figure 2. In vivo and in vitro MAPK signaling.

a) To test for novel markers of renal activity, mice were injected with 10 μg of FGF23 i.p. for 1 hour and the kidneys harvested for RNA preparation and qPCR. Egr1, measured as a positive control, was increased 70-fold over vehicle injected mice. c-Fos, was increased 13-fold (P<0.05). b) HEK293 cells stably expressing mKL were treated with 100 ng/mL of FGF23 for 30 and 60 min. EGR1 mRNA increased 80- and 200-fold at 30 and 60 min, respectively in the FGF23-treated mKL cells compared to vehicle control (P<0.05). c-Fos increased 20- and 30-fold at 30 and 60 min, respectively (P<0.05).
Patients with XLH have increased circulating FGF23, however the molecular consequences in renal phosphate handling due to this elevation are unclear. Building upon the results directly above, we next tested basal levels of genes downstream of FGF23 activity in the Hyp model of XLH. These animals are characterized by hypophosphatemia and significantly increased serum Fgf23 levels (~12-fold over wild type littermates P<0.05; Figure 3a), however whether this leads to alterations in markers of FGF23 activity are unknown. Using c-Fos and Egr1 to test FGF-dependent activity in Hyp kidneys, we observed a 200% increase in expression of these genes in Hyp in the basal state compared to sex- and age-matched wild type littermates (P<0.05; Figure 3b), suggesting that FGF23-dependent activity is enhanced in Hyp mice. Further, KL mRNA was reduced by 60% in these mice compared to wild type littermates (P<0.05; Figure 3b), indicating a potential compensatory mechanism is induced in an attempt to down-regulate FGF23-dependent signaling.
Figure 3. Mapk signaling in Hyp mice.

a) Hyp mice had intact serum FGF23 levels approximately 12-fold higher than WT controls; b) Hyp mice had a 200% increase in Egr1 and c-Fos mRNA, and Klotho expression was reduced by 60% compared to wild type controls (P<0.05).
MAPK activity in the Hyp mouse model
The standard treatment for patients with XLH is a combination therapy of phosphate and calcitriol, which is used to raise serum phosphate. This regimen has recently been associated with increasing circulating FGF23 concentrations in patients with XLH (Imel EA 2010), however the molecular effects of elevated FGF23 on the renal mechanisms of phosphate handling in this disorder are unknown. To test whether Hyp mice are able to respond to increased levels of FGF23, Hyp were injected with vehicle or FGF23 (10 μg/animal). Despite the decreased mRNA levels of KL, KL was detectable by immunofluorescence (Figure 4), however no p-ERK1/2 activity was detected in the basal state in Hyp mice. In contrast, Hyp mice injected with FGF23 demonstrated strong p-ERK1/2 staining (Figure 4), and this activity co-localized with KL in the renal DCT. These findings demonstrate that Hyp mice are capable of responding to increased serum FGF23 levels, consistent with the idea that the current treatment for XLH could be associated with elevated renal FGF23 bioactivity.
Figure 4. Response of Hyp to FGF23 delivery.

Results from vehicle-injected Hyp mice are shown in the upper panels, and FGF23-injected Hyp mice in the lower panels at 10 min post-injection. Phospho-ERK1/2 staining was only observed in the FGF23-injected animals; Klotho (KL) was positive in both vehicle- and FGF23-injected animals. p-ERK1/2 staining (red) localized to the nucleus in the same nephron segment as KL (green) in FGF23-injected mice as shown by p-ERK1/2 and KL co-staining (‘Merge’ column; arrows show positive nuclear p-ERK1/2 co-localized with KL). Nuclei were stained blue using DAPI in the Merge column.
FGF23-dependent effects on renal Wnt signaling
To screen for novel pathways involved in renal FGF23-dependent signaling, PCR-based array technology was utilized in initial experiments. This Pathway-Finder array contains primers recognizing 84 genes from 18 signaling pathways, including the positive control for Fgf23 activity, Egr1. Wild type mice were injected with 10 μg of recombinant human FGF23 and sacrificed at 60 min, and the kidneys were harvested for RNA preparation. Consistent with our earlier results, Egr1 mRNA was elevated approximately 20-fold in the array following injection. Unexpectedly, these initial analyses also identified Engrailed-1 (En1) and Wnt1 as elevated following injection, with 3.5- and 5-fold increases in mRNA compared to vehicle-injected mice (not shown). En1 is a well-characterized repressor of Wnt signaling (Bachar-Dahan, et al. 2006). The role of Wnts, as well as the localization of this signaling cascade in FGF23-dependent renal activity has not been previously defined.
To build upon the mRNA results at the protein level, FGF23-dependent suppression of renal Wnt signaling was tested. In this regard, kidney sections from vehicle and FGF23-injected mice (10 μg/animal for 10 min) were co-stained for phospho-β-catenin (p-β-catenin), a well-characterized marker of suppressed of Wnt signaling, as well as KL. A strong p-β-catenin signal was only detected in the renal glomeruli of the vehicle-injected animals (Figure 5a). In contrast, p-β-catenin signaling was observed in the glomeruli and also co-localized with KL in the renal DCT in the FGF23-injected animals (Figure 5b). Thus, in a similar manner to p-ERK1/2, p-β-catenin activity was spatially separate from PT Npt2a expression.
Figure 5. Wnt pathway in renal FGF23 signaling.
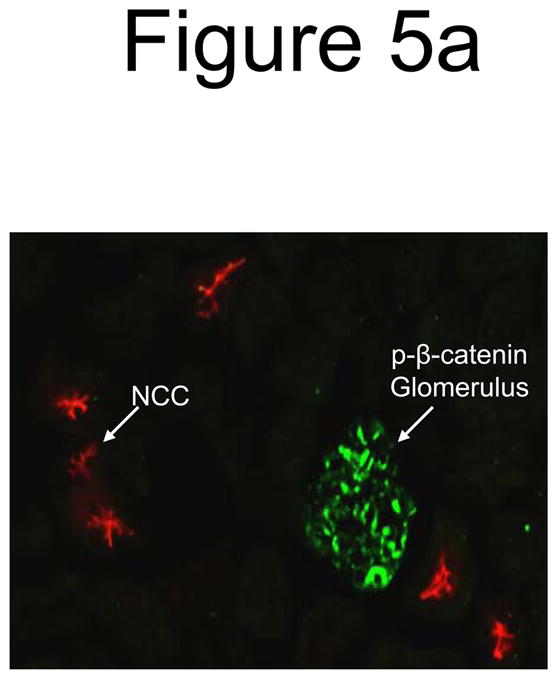

a) Strong p-β-catenin signaling was detected in the glomeruli of all animals (green), separate from the DCT, which was demarcated using anti-NCC IF (red). b) p-β-catenin signaling was tested by IF in vehicle and FGF23-injected mice at 10 min following injection. p-β-catenin signaling was not detected in the vehicle injected animals. In the FGF23-injected animals, p-β-catenin (red) was detected in the distal tubule, co-localized with Klotho (green). This activity did not co-localize with Npt2a in the renal PT (right column).
Due to the strong Wnt signaling response in WT mice, we next tested for the presence of p-β-catenin signaling in Hyp mice, which would be consistent with the increased Egr1 and c-Fos mRNA indicating elevated MAPK signaling in the basal state (see Figure 3b). Indeed, uninjected Hyp mice expressed robust p-β-catenin in the DCT co-localized with KL (Figure 6). Further, pβ-catenin was specifically co-localized with the sodium chloride cotransporter (NCC), a reliable DCT marker (Bostanjoglo, et al. 1998). Importantly, the DCT-specific staining pattern for p-β-catenin paralleled that of WT mice injected with FGF23, i.e., being distinct from Npt2a in the PT (Figure 6). Thus, these findings revealed that FGF23 acts to suppress Wnt signaling in the renal DCT, further supporting an intra-renal axis for FGF23 activity.
Figure 6. Renal p-β-catenin expression in Hyp mice.

a) p-β-catenin (red) was readily detectable in Hyp kidney in the basal state, localized to the renal DCT with KL (green staining; left panel); b) p-β-catenin (red) was spatially separate from Npt2a (green; middle panel). c) In the Hyp basal state, strong p-β-catenin signal (green) was co-localized with NCC, a marker for the renal DCT (red; right panel).
Discussion
FGF23 controls renal phosphate handling through directing the membrane localization of Npt2a and Npt2c (Larsson et al. 2004; Shimada et al. 2004c) in the PT. Previously, we demonstrated that initial FGF23-dependent MAPK activity occurs within minutes in the renal DCT (Farrow et al. 2009). It has also been shown that serum phosphate levels are maximally decreased in mice approximately 9 hours after FGF23 injection (Shimada et al. 2004a). Herein, we show that following FGF23 delivery, in a manner similar to PTH (Bacic et al. 2003), Npt2a protein expression is decreased in the PT by 30 min and fully at 60 min (Figure 1a), and is associated with p-ERK1/2 activity in the DCT (Figure 1b). This relatively rapid reduction in Npt2a is likely due to the endocytic retrieval of the cotransporter from the apical membrane, as no transcriptional changes in Npt2a were detected at 60 min (Figure 1c). Although FGF23 rapidly down regulates Npt2a; changes in serum phosphate concentrations and 1,25(OH)2 vitamin D are known to be latent (Shimada et al. 2004a). Our findings support the idea that elevations in FGF23 may also be associated with indirect endocrine changes that manifest over longer time intervals, as opposed to balancing minute to minute serum phosphate changes. Importantly, FGF23 activity through MAPK in the DCT was dependent upon the presence of KL. In this regard, p-ERK1/2 activity was not detected that co-localized with DCT segments expressing a surrogate marker for KL, β-galactosidase (Figure 1d) in KL-null animals injected with FGF23. These results are consistent with the fact that KL and a canonical FGF receptor (FGFR) are required for FGF23-dependent MAPK signaling (Urakawa et al. 2006). Further, the lack of FGF23-dependent p-ERK1/2 activity in the DCT in the absence of KL provides mechanistic support for a report demonstrating a likely end-organ deficiency in FGF23-dependent signaling in a patient with severe tumoral calcinosis harboring a homozygous KL loss of function missense mutation (His193Arg) (Ichikawa et al. 2007).
We have now demonstrated the presence of early FGF23 signaling in the DCT with both p-ERK1/2 and Wnt involvement (see below). Nonetheless, the mechanisms underlying a potential transfer of FGF23 bioactivity across a spatial separation to the PT remain unclear. Although the second step or process following initial FGF23 signaling is unknown, our results support the fact that it likely occurs relatively quickly (under 30 min) after initial signaling (5 minutes), although physiological changes in serum phosphate are known to occur over at least 8–9 hours (Shimada et al. 2004a). Collectively, these findings provide a narrowed time frame for the identification of candidate pathways guiding intra-renal FGF23-dependent activity. At this time, however, the possibility that FGF23 has a direct effect in the PT that does not involve p-ERK1/2 (or Wnts) cannot be ruled out.
To test for additional markers of FGF23 bioactivity, the transcription factor c-Fos was identified as being up-regulated following FGF23 delivery in vivo and in vitro (Figure 2b). Although c-Fos is known to be downstream of the MAPK pathway initiated by canonical FGF signaling (autocrine and paracrine FGFs), it had not previously been associated with renal FGF23 bioactivity. Hyp mice have significantly elevated serum FGF23 and hypophosphatemia, and are a well established model of the human syndrome XLH. The standard treatment for XLH currently includes supplemental oral phosphate and high dose calcitriol (Tenenhouse 2001). This therapeutic regimen has now been realized to result in further increases in circulating FGF23 in this patient group (Imel EA 2010). Similarly, administration of phosphate or vitamin D potently increases FGF23 production in wild type and in Hyp mice (Azam et al. 2003; Shimada et al. 2004c). Although the Hyp mouse has been well studied, the levels of relative renal FGF23 bioactivity under a state of chronic hypophosphatemia in this model remained unclear. We therefore examined FGF23 signaling in Hyp in the basal state and found that the markers of MAPK signaling, Egr1 and c-Fos mRNAs, were elevated in Hyp kidney compared to wild type littermates (Figure 3b). These findings are consistent with the idea that FGF23-dependent signaling is increased in Hyp mice under ‘normal’ circumstances, consistent with their significantly increased circulating Fgf23 ((Figure 3a and (Liu et al. 2006b)). Although renal KL mRNA concentrations were reduced by 60% in Hyp compared to wild type littermates, when Hyp were injected with FGF23, robust p-ERK1/2 signal was detected in the DCT with KL, similar to the response observed in WT mice (Figure 1d). Consistent with our findings in Hyp, KL mRNA was reported to be the most significant down-regulated kidney gene in a markedly hypophosphatemic transgenic mouse over expressing human FGF23 in osteoblasts and osteocytes (Marsell, et al. 2008). Conversely, KL transcripts were up-regulated in a model of hyperphosphatemic tumoral calcinosis, the GALNT3-null mouse (Ichikawa, et al. 2009), which suggests that FGF23 may potentially be regulated at the level of its receptor complex in kidney depending upon phosphate status. Taken together, these results indicate that in the basal state, Hyp mice have altered renal MAPK activity, but overall, FGF23-dependent signaling remains intact. These findings have implications for XLH, as increased FGF23 following treatment could suppress renal 1,25(OH)2 vitamin D production through decreasing 25-vitamin D 1-α-hydroxylase and increasing 24-hydroxylase expression, or have direct actions on the parathyroid, further lending to a scenario for secondary hyperparathyroidism, a common and often severe complication of XLH (Tenenhouse 2001).
To screen for novel pathways associated with FGF23 renal activity, we used an array approach and identified markers of suppressed Wnt signaling. Wnt signaling is critical for proper kidney development (Herzlinger, et al. 1994; Stark, et al. 1994), and altered Wnt activity has been implicated in the development of several renal diseases including renal cell carcinoma and cystic kidney disease (Kim, et al. 1999; Kim, et al. 2000). Importantly, following FGF23 delivery, p-β-catenin signaling co-localized to the DCT with KL and with NCC (Figures 5b and 6) within the same time course for DCT p-ERK1/2 expression (10 min). In Hyp mice in the basal state, p-ERK1/2 was not detected by immunofluorescence (Figure 4), consistent with the modest increases in Egr1 and c-Fos mRNAs (Figure 3b). However, in the basal state, strong p-β-catenin signaling was detected in the Hyp DCT, indicating that p-β-catenin may be a sensitive marker in XLH and in disease models of altered FGF23 activity.
The localization of Wnt signaling has not been defined in terms of renal phosphate homeostasis in vivo, and its role in the DCT for normal phosphate handling is unclear. It was reported that GSK3-β phosphorylation was increased in vitro in cells treated with FGF23 and a recombinant KL (Medici, et al. 2008) in parallel with p-ERK1/2 (Farrow et al. 2009; Medici et al. 2008). In canonical Wnt signaling, GSK3-β is upstream of p-β-catenin, consistent with our results that p-β-catenin is localized to the DCT with p-ERK following FGF23 delivery, spatially distinct from Npt2a in the PT. Complementary signaling between the FGF-dependent MAPK and Wnt pathways has been well documented during development (ten Berge, et al. 2008), however the molecules at the origin of the branching point between these two pathways is less clear. Recent reports indicated that GSK3β may serve as a mediator between FGF and non-canonical Wnt signaling during carcinogenesis (Katoh 2006), and during osteoblast proliferation AKT may signal between MAPK and Wnts (Raucci, et al. 2008). Thus, our identification of FGF23-dependent control of Wnt activity in the kidney may also be useful for the further understanding of MAPK-Wnt interactions. Genes known to be involved in Wnt and FGF signaling are also altered in Hyp mouse bone (Liu, et al. 2009), thus in addition to renal pathogenesis, these systems may also be associated with increased skeletal Fgf23 (Liu, et al. 2003).
In summary, we have identified novel markers of renal FGF23 bioactivity, defined the temporal control of Npt2a by FGF23, and have localized alterations in MAPK and Wnt signaling to the renal DCT in the Hyp mouse. These studies have important implications for understanding altered FGF23-dependent signaling in XLH, as well as in other disorders associated with elevated FGF23.
Acknowledgments
Funding
The authors would like to acknowledge support from NIH grants DK051496 (DHE), and DK063934 and an ARRA Supplement to DK063934 (KEW), the Showalter Foundation, Genzyme Corporation, the Indiana Genomics Initiative (INGEN) of Indiana University supported in part by the Lilly Endowment, Inc. EGF is supported in part by a National Kidney Foundation (NKF) Research Fellowship.
We would like to thank Drs. Heini Murer and Jurg Biber at the Institute of Physiology and Zurich Center for Integrative Human Physiology, Zurich, Switzerland, for generously providing the anti-Npt2a antibody.
Footnotes
Declaration of Interest
EF, LS, SD, JM, and DE, have nothing to declare. SS is employed by Genzyme, Corp. KW receives research funding from Genzyme, and royalties for licensing the FGF23 gene to Kyowa Hakko Kirin, Ltd.
References Cited
- Azam N, Zhang MY, Wang X, Tenenhouse HS, Portale AA. Disordered regulation of renal 25-hydroxyvitamin D-1alpha-hydroxylase gene expression by phosphorus in X-linked hypophosphatemic (hyp) mice. Endocrinology. 2003;144:3463–3468. doi: 10.1210/en.2003-0255. [DOI] [PubMed] [Google Scholar]
- Bachar-Dahan L, Goltzmann J, Yaniv A, Gazit A. Engrailed-1 negatively regulates beta-catenin transcriptional activity by destabilizing beta-catenin via a glycogen synthase kinase-3beta-independent pathway. Mol Biol Cell. 2006;17:2572–2580. doi: 10.1091/mbc.E06-01-0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacic D, Schulz N, Biber J, Kaissling B, Murer H, Wagner CA. Involvement of the MAPK-kinase pathway in the PTH-mediated regulation of the proximal tubule type IIa Na+/Pi cotransporter in mouse kidney. Pflugers Arch. 2003;446:52–60. doi: 10.1007/s00424-002-0969-8. [DOI] [PubMed] [Google Scholar]
- Bostanjoglo M, Reeves WB, Reilly RF, Velazquez H, Robertson N, Litwack G, Morsing P, Dorup J, Bachmann S, Ellison DH. 11Beta-hydroxysteroid dehydrogenase, mineralocorticoid receptor, and thiazide-sensitive Na-Cl cotransporter expression by distal tubules. J Am Soc Nephrol. 1998;9:1347–1358. doi: 10.1681/ASN.V981347. [DOI] [PubMed] [Google Scholar]
- Custer M, Lotscher M, Biber J, Murer H, Kaissling B. Expression of Na-P(i) cotransport in rat kidney: localization by RT-PCR and immunohistochemistry. Am J Physiol. 1994;266:F767–774. doi: 10.1152/ajprenal.1994.266.5.F767. [DOI] [PubMed] [Google Scholar]
- Farrow EG, Davis SI, Summers LJ, White KE. Initial FGF23-mediated signaling occurs in the distal convoluted tubule. J Am Soc Nephrol. 2009;20:955–960. doi: 10.1681/ASN.2008070783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzlinger D, Qiao J, Cohen D, Ramakrishna N, Brown AM. Induction of kidney epithelial morphogenesis by cells expressing Wnt-1. Dev Biol. 1994;166:815–818. doi: 10.1006/dbio.1994.1360. [DOI] [PubMed] [Google Scholar]
- HYP-Consortium. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nat Genet. 1995;11:130–136. doi: 10.1038/ng1095-130. [DOI] [PubMed] [Google Scholar]
- Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH, Goetz R, Mohammadi M, White KE, Econs MJ. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. 2007;117:2684–2691. doi: 10.1172/JCI31330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa S, Sorenson AH, Austin AM, Mackenzie DS, Fritz TA, Moh A, Hui SL, Econs MJ. Ablation of the Galnt3 gene leads to low-circulating intact fibroblast growth factor 23 (Fgf23) concentrations and hyperphosphatemia despite increased Fgf23 expression. Endocrinology. 2009;150:2543–2550. doi: 10.1210/en.2008-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imel EADL, Hui SL, Carpenter TO, Econs MJ. Treatment of XLH with Calcitriol and Phosphate Increases Circulating FGF23 Concentrations. J Clin Endo Metab. 2010 doi: 10.1210/jc.2009-1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HB, Kim JS, Kwon HJ, Nam KH, Youn HS, Sok DE, Lee Y. Basic fibroblast growth factor activates ERK and induces c-fos in human embryonic stem cell line MizhES1. Stem Cells Dev. 2005;14:395–401. doi: 10.1089/scd.2005.14.395. [DOI] [PubMed] [Google Scholar]
- Katoh M. Cross-talk of WNT and FGF signaling pathways at GSK3beta to regulate beta-catenin and SNAIL signaling cascades. Cancer Biol Ther. 2006;5:1059–1064. doi: 10.4161/cbt.5.9.3151. [DOI] [PubMed] [Google Scholar]
- Kim E, Arnould T, Sellin LK, Benzing T, Fan MJ, Gruning W, Sokol SY, Drummond I, Walz G. The polycystic kidney disease 1 gene product modulates Wnt signaling. J Biol Chem. 1999;274:4947–4953. doi: 10.1074/jbc.274.8.4947. [DOI] [PubMed] [Google Scholar]
- Kim YS, Kang YK, Kim JB, Han SA, Kim KI, Paik SR. beta-catenin expression and mutational analysis in renal cell carcinomas. Pathol Int. 2000;50:725–730. doi: 10.1046/j.1440-1827.2000.01111.x. [DOI] [PubMed] [Google Scholar]
- Kuro-o M. Klotho as a regulator of fibroblast growth factor signaling and phosphate/calcium metabolism. Curr Opin Nephrol Hypertens. 2006;15:437–441. doi: 10.1097/01.mnh.0000232885.81142.83. [DOI] [PubMed] [Google Scholar]
- Larsson T, Marsell R, Schipani E, Ohlsson C, Ljunggren O, Tenenhouse HS, Juppner H, Jonsson KB. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology. 2004;145:3087–3094. doi: 10.1210/en.2003-1768. [DOI] [PubMed] [Google Scholar]
- Liu S, Guo R, Simpson LG, Xiao ZS, Burnham CE, Quarles LD. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem. 2003;278:37419–37426. doi: 10.1074/jbc.M304544200. [DOI] [PubMed] [Google Scholar]
- Liu S, Tang W, Fang J, Ren J, Li H, Xiao Z, Quarles LD. Novel regulators of Fgf23 expression and mineralization in Hyp bone. Mol Endocrinol. 2009;23:1505–1518. doi: 10.1210/me.2009-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, Quarles LD. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol. 2006a;17:1305–1315. doi: 10.1681/ASN.2005111185. [DOI] [PubMed] [Google Scholar]
- Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab. 2006b;291:E38–49. doi: 10.1152/ajpendo.00008.2006. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lyles KW, Harrelson JM, Drezner MK. The efficacy of vitamin D2 and oral phosphorus therapy in X-linked hypophosphatemic rickets and osteomalacia. J Clin Endocrinol Metab. 1982;54:307–315. doi: 10.1210/jcem-54-2-307. [DOI] [PubMed] [Google Scholar]
- Marsell R, Krajisnik T, Goransson H, Ohlsson C, Ljunggren O, Larsson TE, Jonsson KB. Gene expression analysis of kidneys from transgenic mice expressing fibroblast growth factor-23. Nephrol Dial Transplant. 2008;23:827–833. doi: 10.1093/ndt/gfm672. [DOI] [PubMed] [Google Scholar]
- Medici D, Razzaque MS, Deluca S, Rector TL, Hou B, Kang K, Goetz R, Mohammadi M, Kuro OM, Olsen BR, et al. FGF-23-Klotho signaling stimulates proliferation and prevents vitamin D-induced apoptosis. J Cell Biol. 2008;182:459–465. doi: 10.1083/jcb.200803024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raucci A, Bellosta P, Grassi R, Basilico C, Mansukhani A. Osteoblast proliferation or differentiation is regulated by relative strengths of opposing signaling pathways. J Cell Physiol. 2008;215:442–451. doi: 10.1002/jcp.21323. [DOI] [PubMed] [Google Scholar]
- Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004a;19:429–435. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004b;113:561–568. doi: 10.1172/JCI19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98:6500–6505. doi: 10.1073/pnas.101545198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Urakawa I, Yamazaki Y, Hasegawa H, Hino R, Yoneya T, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun. 2004c;314:409–414. doi: 10.1016/j.bbrc.2003.12.102. [DOI] [PubMed] [Google Scholar]
- Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, Erben RG, Juppner H, Lanske B. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. 2004;23:421–432. doi: 10.1016/j.matbio.2004.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark K, Vainio S, Vassileva G, McMahon AP. Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature. 1994;372:679–683. doi: 10.1038/372679a0. [DOI] [PubMed] [Google Scholar]
- ten Berge D, Brugmann SA, Helms JA, Nusse R. Wnt and FGF signals interact to coordinate growth with cell fate specification during limb development. Development. 2008;135:3247–3257. doi: 10.1242/dev.023176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenenhouse HS, Econs MJ. The Metabolic Bases of Inherited Disease. Chapter 197. Mendelian Hypophosphatemias; 2001. [Google Scholar]
- Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–774. [Google Scholar]
- Weber TJ, Liu S, Indridason OS, Quarles LD. Serum FGF23 levels in normal and disordered phosphorus homeostasis. J Bone Miner Res. 2003;18:1227–1234. doi: 10.1359/jbmr.2003.18.7.1227. [DOI] [PubMed] [Google Scholar]
- Yamazaki Y, Okazaki R, Shibata M, Hasegawa Y, Satoh K, Tajima T, Takeuchi Y, Fujita T, Nakahara K, Yamashita T, et al. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab. 2002;87:4957–4960. doi: 10.1210/jc.2002-021105. [DOI] [PubMed] [Google Scholar]