

Abstract
Ynamides react with ketenes in [2+2] cycloadditions leading to a variety of substituted 3-aminocyclobut-2-en-1-ones. The ynamides employed in these reactions are readily available via the copper-promoted N-alkynylation of carbamates and sulfonamides with alkynyl bromides and iodides. The scope of the [2+2] cycloaddition with regard to both the ketene and ynamide component is described.
Keywords: Ynamides, Ketenes, Cyclobutenones, [2+2] Cycloadditions
1. Introduction
Cyclobutenones are valuable synthetic intermediates that participate in a variety of novel and useful synthetic transformations.1 The most direct and convenient approach to the synthesis of cyclobutenones employs the [2+2] cycloaddition of ketenes with alkynes.2 Unfortunately, unactivated alkynes only engage in efficient cycloadditions with highly electrophilic ketenes such as dichloroketene.3 Electron-rich alkynes (e.g., alkoxy-4 and silyloxy-substituted acetylenes5) combine with a wider range of ketenes and provide access to 3-alkoxycyclobutenones, generally in good yield. Ynamines6 also combine readily with ketenes; however, these reactions often lead to mixtures of the desired cyclobutenones accompanied by allenyl amides.7 The formation of these allene byproducts is believed to result from initial addition of the ynamine across the ketene carbonyl group via a stepwise pathway to form an alkylideneoxete. Electrocyclic ring opening then transforms this strained intermediate to the allenyl carboxamide.
In connection with our interest in benzannulation strategies based on the reaction of alkynes with aryl- and vinyl-ketenes,8 we undertook a study of the reactions of various ynamine derivatives with ketenes. In order to suppress the `abnormal' reaction leading to oxetes and allenes, we have focused our attention on reactions of ynamides, in which the nucleophilicity of the aminoalkyne is attenuated by the electron-withdrawing substituent on the nitrogen atom. Herein, we report the results of our systematic investigation of the [2+2] cycloaddition of several classes of ketenes with ynamides.
2. Results and discussion
2.1. Synthesis of ynamides
Recent advances in copper-promoted amide coupling reactions have provided the basis for the efficient and convenient synthesis of a variety of ynamides. The ynamides employed in the present study were prepared as described in Table 1 using the N-alkynylation method recently developed in our laboratory.9 The terminal ynamide 9 was obtained in 70% yield by desilylation of 7 as shown in Scheme 1. The conditions shown in Table 1 and detailed in Section 4 represent a minor modification of our original procedure. In this revised protocol, we now employ THF as the reaction solvent with 25 equiv of pyridine, use only a small excess of the alkynyl halide, and utilize an improved workup procedure. Although this protocol requires the use of 1 equiv of CuI, coupling proceeds smoothly at rt and this method thus accommodates the synthesis of a wide range of alkyne derivatives including thermally unstable systems. For many of these ynamides, similar results can be obtained by using the method of Hsung, which employs catalytic CuCN or CuSO4 in conjunction with diamine ligands and requires reaction at elevated temperatures.10 We have found both methods to be reliable and reproducible for reactions on both small and large (i.e., multigram) scale.
Table 1.
Synthesis of ynamides by alkynylation of carbamates
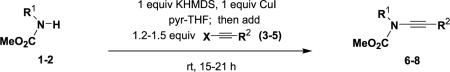
| Entry | Carbamate | Halo alkyne | Ynamide | Yield (%)a |
|---|---|---|---|---|
| 1 | 1 R1=CH3 | 3 X=Br, R2=Hex |
|
61 |
| 2 | 1 R1=CH3 | 4 X=I, R2=SiMe3 |
|
64 |
| 3 | 2 R1=(CH2)2CH=CH2 | 5 X=Br, R2=C(CH3)=CH2 |
|
68 |
Isolated yields of products purified by column chromatography.
Scheme 1.
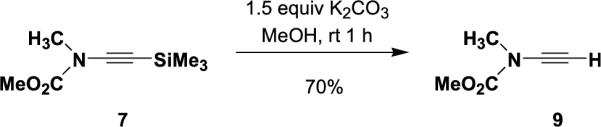
Synthesis of ynamide 9.
Recently, Tam and co-workers have reported an alternative N-alkynylation protocol that involves a melding of the procedures previously developed in our laboratory and that of Hsung.11 Tam was motivated to introduce this variant following difficulties he encountered in reproducing results previously reported by both our group and that of Hsung. For example, Tam reported obtaining none of the desired ynamide from the reaction of 1-bromo-2-phenylacetylene with BnNHCO2Me using either our method9 or the method of Hsung.10b Hsung had previously reported obtaining this ynamide in 73% yield, and we had reported the synthesis of the corresponding Boc derivative in 61% yield.
We have investigated the coupling of BnNHCO2Me with 1-bromo-2-phenylacetylene in some detail in an attempt to identify the experimental variables that might be responsible for Tam's unsuccessful results. We obtained the expected ynamide (N-benzyl-N-methoxycarbonyl-2-phenylethynylamine) in 63% yield using our method, and in 79% yield by employing the method of Hsung.10a In our experience, both procedures have proved to be highly reproducible. We have noted, however, that results employing our method are affected by the quality of the pyridine used in the reaction. Thus, the yields reported here were obtained using pyridine freshly distilled from CaH2 or KOH as recommended in our original report.9 When pyridine (Alfa Aesar, 99%, 0.1% water content) from a freshly opened bottle (without distillation) was used instead, the yield of ynamide product declined to 52%. Most significantly, when distilled pyridine that had then been exposed to the atmosphere for several days was used for the N-alkynylation, none of the desired ynamide was formed.
2.2. Cycloaddition studies
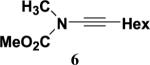
N-Methoxycarbonyl-N-methyl-1-octynylamine (6) was employed as an initial test substrate to investigate the reactivity of ynamides with different classes of ketenes. As shown in Scheme 2, we found that this ynamide combines readily with ketene itself to afford the expected cyclobute-none 10 in high yield after purification by column chromatography. For this reaction, ketene was generated by pyrolysis of acetone in a Hurd `ketene lamp' as described previously12 and bubbled into a 0.5 M solution of the ynamide in acetonitrile. Although reactions of ketene with alkoxyacetylenes are well known,4a,c,d to our knowledge only a few examples of reactions with ynamines,7c,13 and none with ynamides, have previously been reported. In contrast to the reactions of ynamines, no evidence for the formation of oxete or allene byproducts was detected in the reaction of ynamide 6 with ketene.
Scheme 2.

Dichloroketene is considerably more reactive in [2+2] cycloadditions than ketene itself,14 and its reaction with a variety of alkynes has previously been described.3 As shown in Scheme 2, generation of dichloroketene via reductive dechlorination of trichloroacetyl chloride with zinc–copper couple3c in the presence of ynamide 6 provides the 3-amino-4,4-dichlorocyclobutenone 11 in 88% yield. As noted previously, the 1,2-dechlorination protocol constitutes the superior method for the generation of dichloroketene for cycloaddition with alkynes. In the present case, when dichloroketene was generated via the dehydrohalogenation of dichloroacetyl chloride with Et3N, cyclobutenone 11 was obtained in only 35% yield. To our knowledge, no previous examples of the addition of dichloroketene to ynamines or ynamides have previously been reported.
Reaction of ynamide 6 with (phenylthio)ketene also proceeded smoothly to furnish 12 in 76% yield when this ketene was generated in situ by our Rh2(OAc)4-catalyzed `thia-Wolff rearrangement' beginning with PhSCOCHN2.15 Finally, addition of dimethylketene to ynamide 6 also proceeded in excellent yield when the ketene was prepared in situ via the triethylamine-promoted dehydrohalogenation of isobutyryl chloride. Best results were obtained using CH2Cl2 as solvent; in Et2O the desired cyclobutenone was obtained in only 11% yield. Previously, the cycloaddition of dimethylketene (generated by pyrolysis of tetramethyl-cyclobutane-1,3-dione) with ynamines has been reported to occur in only low to moderate yield.7a As expected, allene byproducts were not detected in any of the above reactions. It should also be noted that the yields in the above cycloadditions are all based on the ynamide component and these reactions were all conducted employing the ketenophile as the limiting reactant. In many ketene cycloadditions, satisfactory yields are only obtained when the ketenophile reaction partner is used in significant excess.
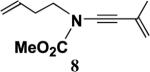
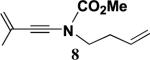
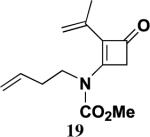
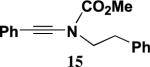
Table 2 presents the results of our investigation of the reaction of ketene with several types of ynamides. In the case of most ynamides, these cycloadditions proceed smoothly in acetonitrile (0.5 M), and CH2Cl2, THF, and toluene can also be used with similar results. The [2+2] cycloaddition of ynamides 8 and 15 with ketene proved sluggish under these standard conditions; we believe that the reactivity of these ynamides is attenuated by the inductive effect of the sp2 unsaturated substituents attached to the alkyne. These cycloadditions did proceed at a reasonable rate when conducted in the absence of solvent, and under these conditions the desired cyclobutenones 19 and 20 could be obtained in good yield. It is noteworthy that the addition of ketene to ynamide 8 occurs exclusively at the triple bond; no products could be detected resulting from addition of ketene to either the conjugated double bond or the terminal olefin of the butenyl substituent.
Table 2.
[2+2] Cycloadditions of ynamides with ketene
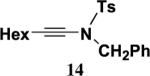
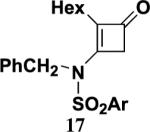
Isolated yields of products purified by column chromatography.
Yield based on recovered starting material.
As discussed earlier, alkoxyacetylenes function as excellent ketenophiles in reactions with ketene itself. For example, addition of ketene to 1-ethoxyoctyne (22) proceeds in 80% yield to afford cyclobutenone 23 after purification by column chromatography on triethylamine-deactivated silica gel (Scheme 3). In order to compare the reactivity of ynamides and alkoxyalkynes in [2+2] cycloadditions, we carried out a competition experiment in which a solution of equal amounts of ynamide 6 and 1-ethoxyoctyne in benzene-d6 were reacted with excess ketene in the presence of 1,4-dibromobenzene as an internal standard. As shown in Figure 1, analysis of aliquots by 1H NMR indicated that the ynamide reacts with ketene at a similar but slightly slower rate as compared to the alkoxyacetylene.
Scheme 3.
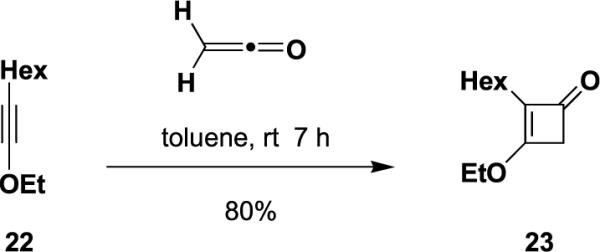
Cycloaddition of ketene with 1-ethoxyoctyne.
Figure 1.
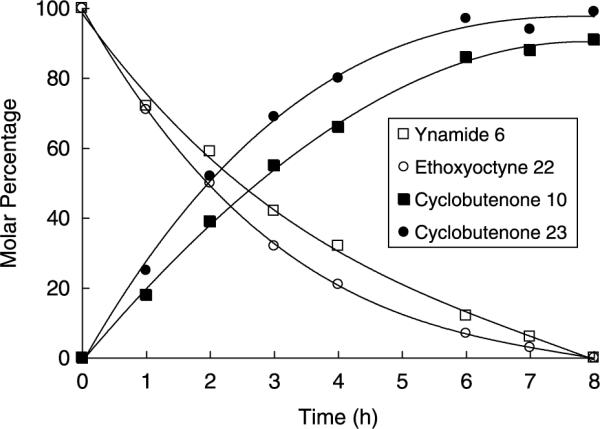
Comparison of rate of [2+2] cycloaddition of ketene with 1-ethoxyoctyne (22) and ynamide 6 in benzene-d6 at 25 °C.
3. Conclusions
3-Aminocyclobutenones have previously been prepared via addition–elimination reactions of amines with 3-alkoxyand 3-acyloxycyclobutenones,4d,16 and by the reaction of cyclobutane-1,3-diones with primary amines.17 Although a few examples of [2C2] cycloadditions of ketenes with ynamines have been reported previously,7,13 these reactions are often complicated by the formation of allene byproducts arising from stepwise addition pathways. We have shown here that the cycloaddition of several classes of ketenes with ynamide derivatives proceeds in good yield to provide access to a variety of substituted 3-aminocyclobutenones.
4. Experimental
4.1. General
All reactions were performed in flame-dried or oven-dried glassware under a positive pressure of argon. Reaction mixtures were stirred magnetically unless otherwise indicated. Air- and moisture-sensitive liquids and solutions were transferred by syringe or cannula and introduced into reaction vessels through rubber septa. Reaction product solutions and chromatography fractions were concentrated by rotary evaporation at ca. 20 mmHg and then at ca. 0.1 mmHg (vacuum pump) unless otherwise indicated. Thin-layer chromatography was performed on Merck precoated glass-backed silica gel 60 F-254 0.25 mm plates. Column chromatography was performed on EM Science silica gel 60 or Silicycle silica gel 60 (230–400 mesh).
4.2. Materials
Commercial grade reagents and solvents were used without further purification except as indicated below. CH2Cl2,Et2O and THF were purified by pressure filtration through activated alumina. Toluene was purified by pressure filtration through activated alumina and Cu(II) oxide. Benzene-d6 was degassed by purging with argon for 10 min prior to use. Et3N, CH3CN, and pyridine were distilled under argon from CaH2 prior to use. Copper(I) iodide was extracted with THF for 24 h in a Soxhlet extractor and then dried under vacuum (0.1 mmHg). DME was predried over sodium and then distilled from sodium–benzophenone ketyl prior to use. Trichloroacetyl chloride and isobutyryl chloride were distilled at atmospheric pressure under argon. Zinc–copper couple was prepared from Zn and CuSO4·5H2O.3c PhSCOCHN2 was prepared from phenyl thioacetate by diazo transfer as described previously.15
4.3. Instrumentation
Melting points were determined with a Fisher-Johns melting point apparatus and are uncorrected. Infrared spectra were obtained using a Perkin Elmer 2000 FT-IR spectrophotometer. 1H NMR spectra were recorded on Bruker Avance-400 (400 MHz) and Varian Inova-500 (500 MHz) spectrometers. 1H NMR chemical shifts are expressed in parts per million (δ) downfield from tetramethylsilane (with the CHCl3 peak at 7.27 ppm used as a standard). 13C NMR spectra were recorded on Bruker Avance-400 (100 MHz) and Varian Inova-500 (125 MHz) spectrometers. 13C NMR chemical shifts are expressed in parts per million (δ) downfield from tetramethylsilane (with the central peak of CHCl3 at 77.23 ppm used as a standard). High-resolution mass spectra (HRMS) were measured on a Bruker Daltonics APEXII 3 T Fourier transform mass spectrometer.
4.4. General procedure for the synthesis of ynamides
4.4.1. N-Methoxycarbonyl-N-methyl-1-octynylamine (6)
A 250 mL, three-necked, round-bottomed flask equipped with an argon inlet adapter, rubber septum, and an addition funnel fitted with a rubber septum was charged with carbamate 118 (1.20 g, 13.5 mmol), 60 mL of THF, and 27.3 mL of pyridine. The colorless solution was cooled at 0 °C and a solution of KHMDS (0.91 M in THF, 14.8 mL, 13.5 mmol) was added dropwise via syringe over 4 min. After 15 min, CuI (2.57 g, 13.5 mmol) was added and the resulting green reaction mixture was allowed to warm to rt (ca. 40 min) and stirred for a total of 2 h. A solution of bromo alkyne 39 (3.06 g, 16.2 mmol) in 16 mL of THF was added via the addition funnel over 1 h and the reaction mixture was stirred for 20 h. The resulting red mixture was diluted with 50 mL of Et2O and washed with three 100-mL portions of a 2:1 mixture of brine and concentrated aqueous NH4OH solution. The combined aqueous phases were extracted with two 100-mL portions of Et2O, and the combined organic phases were washed with two 100-mL portions of 3 M HCl solution and 100 mL of brine, dried over MgSO4, filtered, and concentrated to afford 5.87 g of dark red oil. Column chromatography on 100 g of silica gel (gradient elution with 0–20% EtOAc–hexanes) provided 1.62 g (61%) of ynamide 6 as an orange liquid: IR (neat) 3584, 2956, 2930, 2858, 2265, 1729, 1446, and 1377 cm−1; 1H NMR (500 MHz, CDCl3) δ 3.80 (s, 3H), 3.14 (s, 3H), 2.29 (t, J=7.2 Hz, 2H), 1.49–1.55 (m, 2H), 1.36–1.42 (m, 2H), 1.26–1.35 (m, 4H), 0.90 (t, J=7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 156.6, 54.1, 38.2, 31.8, 31.6, 29.2, 28.7, 22.8, 18.7, 14.4, and 14.3; HRMS-ESI m/z [M+Na]+ calcd for C11H19NO2, 220.1308; found 220.1310.
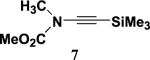
4.4.2. N-Methoxycarbonyl-N-methyl-2-(trimethylsilyl)-ethynylamine (7)
Reaction of a solution of carbamate 118 (1.48 g, 16.6 mmol) in 66 mL of THF with KHMDS (18 mL, 16.6 mmol), pyridine (33 mL, 415 mmol), CuI (3.20 g, 16.6 mmol), and alkynyl iodide 419 (5.20 g, 23.3 mmol) in 20 mL of THF according to the general procedure gave 4.5 g of black oil. Column chromatography on 60 g of silica gel (gradient elution with 0–5% EtOAc–hexanes) afforded 1.96 g (64%) of ynamide 7 as a dark yellow oil: IR (CH2Cl2) 2959, 2179, 1733, 1447, and 1344 cm−1; 1H NMR (400 MHz, CDCl3) δ 3.76 (s, 3H), 3.12 (s, 3H), and 0.14 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 156.0, 96.8, 71.2, 54.2, 37.9, and 0.29; HRMS-ESI m/z [M+Na]+ calcd for C8H15NO2Si, 208.0674; found 208.0674.
4.4.3. N-Methoxycarbonyl-N-(3-butenyl)-3-methyl-3-buten-1-ynylamine (8)
Reaction of a solution of carbamate 220 (0.994 g, 7.70 mmol) in 30 mL of THF with KHMDS (8.5 mL, 7.70 mmol), pyridine (15 mL, 193 mmol), CuI (1.47 g, 7.70 mmol), and alkynyl bromide 59 (1.84 g, 12.6 mmol) in 12 mL of THF according to the general procedure gave 4.2 g of dark brown oil. Column chromatography on 55 g of silica gel (gradient elution with 0–5% EtOAc–hexanes) afforded 1.02 g (68%) of ynamide 8 as a yellow oil: IR (CH2Cl2) 2955, 2235, 1732, 1615, 1445 and 1308 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.76–5.83 (m, 1H), 5.07–5.20 (m, 4H), 3.81 (s, 3H), 3.57 (t, J=7.0 Hz, 2H), 2.43 (app q, J=7.1 Hz, 2H), and 1.92 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 155.8, 134.3, 126.5, 119.6, 117.6, 82.2, 72.4, 54.2, 49.4, 32.3, and 23.9; HRMS-ESI m/z [M+Na]+ calcd for C11H13NO2, 216.0995; found 216.0999.
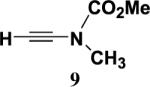
4.4.4. N-Methoxycarbonyl-N-methylethynylamine (9)
A 100 mL, one-necked, round-bottomed flask fitted with an argon inlet adapter was charged with ynamide 7 (1.08 g, 5.84 mmol) and 30 mL of methanol. K2CO3 (1.21 g, 8.76 mmol) was added in one portion and the reaction mixture was stirred at rt for 1 h. The resulting cloudy mixture was diluted with 30 mL of H2O and 30 mL of Et2O. The aqueous layer was separated and extracted with three 30-mL portions of Et2O and the combined organic phases were washed with 50 mL of brine, dried over MgSO4, filtered, and concentrated to provide 0.456 g (69%) of 9 as a flaky yellow solid: IR (CH2Cl2) 2959, 2145, 1729, 1448, and 1341 cm−1; 1H NMR (400 MHz, CDCl3) δ 3.76 (s, 3H), 3.11 (s, 3H), and 2.78 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 156.3, 105.2, 58.0, 54.4, and 37.7; HRMS-ESI m/z [M+Na]+ calcd for C5H7NO2, 136.0369; found 136.0372. A sample recrystallized from hexane had mp 43–45 °C.
4.5. General procedure for the [2+2] cycloaddition of ketene with ynamides21
4.5.1. 2-Hexyl-3-[N-(methoxycarbonyl)-N-methylamino]-2-cyclobuten-1-one (10)
Ketene was generated by pyrolysis of acetone over an electrically heated metal filament using the apparatus described by Williams and Hurd.12 A two-necked, 25 mL, pear flask fitted with a rubber septum and an argon inlet adapter was charged with ynamide 6 (0.152 g, 0.77 mmol) in 1.5 mL of CH3CN. The argon inlet adapter was replaced with an adapter fitted with a glass pipette connected via Tygon tubing to the ketene generator. The septum was fitted with an outlet needle connected via tubing to a column of CaSO4 leading to a trap of H2O. Ketene was bubbled into the reaction mixture at rt over a period of 5 h. The reaction mixture was then concentrated to afford 0.279 g of brown oil. Purification by column chromatography on 16 g of silica gel (elution with 25% EtOAc–hexanes) gave 0.184 g (94%) of 10 as a yellow oil: IR (CH2Cl2) 2957, 2930, 2858, 1738, 1611, 1382, 1326, and 1202 cm−1; 1H NMR (400 MHz, CDCl3) δ 3.83 (s, 3H), 3.44 (s, 2H), 3.39 (s, 3H), 2.15 (t, J=7.8 Hz, 2H), 1.45–1.49 (m, 2H), 1.25 (app s, 6H), and 0.85 (t, J=7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 188.3, 160.4, 153.9, 127.3, 54.5, 51.0, 35.4, 32.0, 29.6, 29.4, 24.1, 23.0, and 14.5; HRMS-EI m/z [M]+ calcd for C13H21NO3, 239.1516; found 239.1524.
4.5.2. 2-Hexyl-3-{N-benzyl-N-[4-(methylphenyl)sulfonyl]-amino}-2-cyclobuten-1-one (17)
Reaction of ynamide 149 (0.124 g, 0.34 mmol) in 0.7 mL of CH3CN with ketene over 6 h according to the general procedure21 afforded 0.138 g of reddish-brown oil, which was dissolved in 1 mL of CH2Cl2 and concentrated onto 1 g of silica gel. The free-flowing powder was added to the top of a column of 20 g of silica gel and eluted with 15% EtOAc–hexanes to provide 0.109 g (79%) of 17 as a pale yellow oil: IR (neat) 2928, 2856, 1753, 1602, 1380, and 1168 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.61 (d, J=7.9 Hz, 2H), 7.27–7.33 (m, 5H), 7.20 (d, J=7.3 Hz, 2H), 5.05 (s, 2H), 3.50 (s, 2H), 2.42 (s, 3H), 1.80 (t, J=7.6 Hz, 2H), 1.15 (app q, J=7.1 Hz, 4H), 0.98–1.08 (m, 4H), and 0.79 (t, J=7.3 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 187.0, 157.7, 145.3, 136.0, 135.4, 130.3, 129.0, 128.0, 127.5, 126.8, 126.2, 52.6, 50.3, 31.4, 29.2, 28.3, 23.7, 22.5, 21.7, and 14.1; HRMS-ESI m/z [M+Na]+ calcd for C24H29NO3S, 434.1760; found 434.1770.
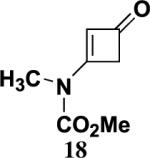
4.5.3. 3-(N-Methoxycarbonyl-N-methylamino)-2-cyclobuten-1-one (18)
Reaction of ynamide 9 (0.456 g, 4.03 mmol) in 8 mL of CH3CN with ketene for 10 h according to the general procedure afforded 1.49 g of dark brown liquid. Column chromatography on 20 g of silica gel (elution with 50% EtOAc–hexanes) yielded 0.496 g (80%) of cyclobutenone 18 as a reddish-brown solid: mp 52–53 °C; IR (CH2Cl2) 2960, 1741, 1570, 1445, 1365, and 1226 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.16 (s, 1H), 3.71 (s, 3H), 3.42 (s, 2H), and 3.25 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 184.3, 166.4, 153.1, 112.2, 49.9, and 34.7; HRMS-ESI m/z [M+Na]+ calcd for C7H9NO3, 178.0475; found 178.0472.
4.5.4. 3-[N-(3-Butenyl)-N-(methoxycarbonyl)amino]-2-isopropenyl-2-cyclobuten-1-one (19)
Reaction of ynamide 8 (0.109 g, 0.564 mmol) with ketene in the absence of solvent for 44 h according to the general procedure21 provided 0.166 g of dark red oil, which was purified by column chromatography on 10 g of silica gel (gradient elution with 0–20% EtOAc–hexanes) to furnish 0.087 g (65%) of 19 as a pale yellow oil: IR (neat) 2958, 1737, 1642, 1595, 1416, 1390, 1368, and 1220 cm−1; 1H NMR (500 MHz, CDCl3) δ 5.66–5.75 (m, 1H), 5.17 (qn, J=1.6 Hz, 1H), 5.08–5.10 (m, 1H), 5.06 (app t, J=1.4 Hz, 1H), 4.89–4.91 (m, 1H), 3.87–3.91 (m, 2H), 3.86 (s, 3H), 3.51 (s, 2H), 2.31–2.36 (m, 2H), and 1.94 (dd, J=1.5, 1.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 185.8, 157.9, 153.3, 134.6, 133.6, 127.9, 117.9, 117.8, 54.2, 51.1, 46.6, 32.9, and 22.3; HRMS-EI m/z [M]+ calcd for C13H17NO3, 236.1281; found 236.1291.
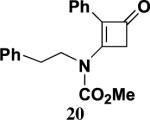
4.5.5. 3-[N-Methoxycarbonyl-N-(2-phenylethyl)amino]-2-phenyl-2-cyclobuten-1-one (20)
Reaction of ynamide 15 (0.100 g, 0.377 mmol) with ketene in the absence of solvent for 42 h according to the general procedure21 provided 0.125 g of dark red oil, which was purified by column chromatography on 6 g of silica gel (gradient elution with 0–20% EtOAc–hexanes) to furnish 0.078 g (67%) of 20 as a pale yellow solid: mp 99–101 °C; IR (CH2Cl2) 3028, 2956, 2361, 1749, 1735, 1619, 1590, and 1399 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.35–7.41 (m, 3H), 7.31 (app d, J=0.9 Hz, 2H), 7.16 (t, J=2.6 Hz, 3H), 6.67 (dd, J=5.8, 2.4 Hz, 2H), 3.94 (t, J=7.8 Hz, 2H), 3.83 (s, 3H), 3.59 (s, 2H), and 2.74 (t, J=7.8 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 185.7, 158.8, 153.4, 137.3, 129.6, 129.3, 128.9, 128.7, 128.6, 128.3, 127.0, 126.4, 54.4, 51.8, 49.4, and 34.9; HRMS-EI m/z [M]+ calcd for C20H19NO3, 322.1438; found 322.1446.
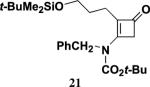
4.5.6. 3-[N-Benzyl-N-(tert-butoxycarbonyl)amino]-2-(3-tert-butyldimethylsiloxybutyl)-2-cyclobuten-1-one (21)
Reaction of ynamide 16 (0.100 g, 0.248 mmol) in 0.5 mL of CH3CN with ketene for 10 h according to the general procedure21 provided 0.114 g of dark red oil, which was purified by column chromatography on 6 g of silica gel (gradient elution with 0–10% EtOAc–hexanes) to furnish 0.095 g (86%) of 21 as an orange oil: IR (neat) 2955, 2930, 2857, 1756, 1732, 1606, 1370, 1239, and 1153 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.35 (app t, J=7.4 Hz, 2H), 7.29 (app d, J=7.3 Hz, 1H), 7.18 (d, J=7.2 Hz, 2H), 4.96 (s, 2H), 3.56 (t, J=1.6 Hz, 2H), 3.51 (t, J=6.1 Hz, 2H), 2.07 (t, J=7.6 Hz, 2H), 1.60–1.66 (m, 2H), 1.46 (s, 9H), 0.84 (s, 9H), and −0.01 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 188.4, 160.7, 151.5, 137.0, 129.0, 127.7, 126.0, 125.7, 84.3, 62.6, 51.4, 50.9, 31.4, 28.1, 26.1, 20.5, 18.5, and −5.1; HRMS-EI m/z [M]+ calcd for C25H39NO4Si, 446.2721; found 446.2737.
4.5.7. 4,4-Dichloro-2-hexyl-3-[N-(methoxycarbonyl)-N-methylamino]-2-cyclobuten-1-one (11)
A one-necked, 10 mL, pear flask fitted with a rubber septum and an argon inlet needle was charged with ynamide 6 (0.100 g, 0.507 mmol) in 1.7 mL of Et2O and cooled to 0 °C. Zinc–copper couple (0.149 g, 2.28 mmol) was then added in one portion followed by a solution of trichloroacetyl chloride (0.17 mL, 0.276 g, 1.52 mmol) in 0.5 mL of DME dropwise via syringe over 15 min. The reaction mixture was allowed to warm to rt over 3.5 h, diluted with 10 mL of Et2O, and then extracted with 4 mL of ice-cold 0.5 M HCl solution followed by 4 mL of ice-cold 5% NaOH solution. The combined aqueous layers were extracted with two 5-mL portions of Et2O, and the combined organic phases were washed with 10 mL of brine, dried over MgSO4, filtered, and concentrated to give 0.165 g of dark yellow oil. Column chromatography on 10 g of silica gel (elution with 10% EtOAc–hexanes) afforded 0.138 g (88%) of cyclobutenone 11 as a yellow oil: IR (CDCl3) 2958, 2931, 1785, 1754, 1601, 1448, 1384, and 1295 cm−1; 1H NMR (500 MHz, CDCl3) δ 3.94 (s, 3H), 3.61 (s, 3H), 2.35 (t, J=6.7 Hz, 2H), 1.55–1.56 (m, 2H), 1.26–1.32 (m, 6H), 0.88 (t, J=6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 180.1, 163.0, 152.9, 133.1, 88.6, 54.7, 35.1, 31.5, 29.7, 29.2, 24.9, 22.6, and 14.1; HRMS-EI m/z [M]+ calcd for C13H19Cl2NO3, 307.0737; found 307.0749.
4.5.8. 2-Hexyl-3-[N-(methoxycarbonyl)-N-methylamino]-4-phenylsulfanyl-2-cyclobuten-1-one (12)
A 25 mL, two-necked, pear flask equipped with a rubber septum and a reflux condenser fitted with an argon inlet adapter was charged with ynamide 6 (0.100 g, 0.507 mmol), 6 mL of CH2Cl2, and Rh2(OAc)4 (0.002 g, 0.005 mmol). The rubber septum was replaced with a 5 mL addition funnel, which was then charged with a solution of PhSCOCHN2 (0.145 g, 0.811 mmol) in 1.5 mL of CH2Cl2. The green reaction mixture was heated at reflux and the diazo thiol ester solution was added dropwise over 1 h (the funnel was rinsed with 0.5 mL of CH2Cl2). The resulting mixture was heated at reflux for an additional 20 min and then allowed to cool to rt. The reaction mixture was concentrated and the resulting brown oil was filtered through a column of 2 g of silica gel with the aid of 40 mL of CH2Cl2. The filtrate was concentrated to give 0.244 g of orange oil, which was purified by column chromatography on 10 g of silica gel (elution with 10% EtOAc–hexanes) to give 0.133 g (76%) of 12 as an orange oil: IR (neat) 2956, 2929, 1758, 1738, 1612, and 1379 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.40 (d, J=7.0 Hz, 2H), 7.27–7.33 (m, 3H), 4.85 (s, 1H), 3.88 (s, 3H), 3.20 (s, 3H), 1.93 (t, 2H), 1.05–1.28 (m, 8H), and 0.86 (t, 3H); 13C NMR (125 MHz, CDCl3) δ 186.0, 161.2, 153.3, 136.4, 132.3, 129.1, 129.0, 128.7, 66.0, 54.4, 35.1, 31.5, 29.2, 28.2, 23.7, 22.5, and 14.1; HRMS-ESI m/z [M+Na]+ calcd for C19H25NO3S, 370.1447; found 370.1440.
4.5.9. 4,4-Dimethyl-2-hexyl-3-[N-(methoxycarbonyl)-N-methylamino]-2-cyclobuten-1-one (13)
A 10 mL, one-necked, pear flask equipped with a rubber septum and an argon inlet needle was charged with ynamide 6 (0.100 g, 0.507 mmol), 2 mL of CH2Cl2, and isobutyryl chloride (0.108 g, 0.106 mL, 1.01 mmol). A solution of Et3N (0.113 g, 0.156 mL, 1.12 mmol) in 0.3 mL of CH2Cl2 was transferred into the reaction mixture via cannula over 3 min (the flask was rinsed with 0.2 mL of CH2Cl2). The septum was replaced with a cold finger condenser and the pink solution was heated at reflux for 24 h. The resulting heterogeneous orange mixture was allowed to cool to rt, diluted with 20 mL of CH2Cl2, and washed with 10 mL of 1 M HCl solution and 15 mL of H2O. The combined aqueous phases were extracted with two 10-mL portions of CH2Cl2 and the combined organic phases were washed with 20 mL of 10% K2CO3 solution and 20 mL of brine, dried over MgSO4, filtered, and concentrated to afford 0.161 g of orange oil. Column chromatography on 10 g of silica gel (elution with 20% EtOAc–hexanes) provided 0.118 g (87%) of cyclobutenone 13 as a yellow liquid: IR (neat) 2957, 2928, 2361, 1751, 1602, 1449, 1379, and 1198 cm−1; 1H NMR (500 MHz, CDCl3) δ 3.87 (s, 3H), 3.40 (s, 3H), 2.17 (t, J=7.6 Hz, 2H), 1.46–1.52 (m, 2H), 1.31 (s, 6H), 1.24– 1.34 (m, 6H), and 0.87 (t, J=3.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 197.1, 169.5, 152.8, 124.6, 62.3, 53.9, 35.5, 31.7, 29.3, 29.1, 23.8, 22.7, 21.5, and 14.2; HRMS-ESI m/z [M+Na]+ calcd for C15H25NO3, 290.1727; found, 290.1725.
4.5.10. 3-Ethoxy-2-hexyl-2-cyclobuten-1-one (23)
Reaction of 1-ethoxyoctyne (22)22 (0.078 g, 0.51 mmol) in 2 mL of toluene with ketene for 7 h according to the general procedure21 afforded 0.157 g of brown oil, which was dissolved in 2 mL of CH2Cl2 and several drops of Et3N and concentrated onto 0.6 g of silica gel. This material was added to the top of a column of 8 g of silica gel and eluted with 0–30% EtOAc–hexanes containing 1% Et3N to give 0.080 g (80%) of 23 as a pale yellow oil: IR (neat) 2956, 2928, 2857, 1758, 1635, 1379, and 1340 cm−1; 1H NMR (400 MHz, CDCl3) δ 4.31 (q, J=7.1 Hz, 2H), 3.16 (t, J=1.8 Hz, 2H), 2.02 (app tt, J=7.6, 1.8 Hz, 2H), 1.44–1.52 (m, 2H), 1.44 (t, J=7.0 Hz, 3H), 1.23–1.34 (m, 6H), and 0.88 (t, J=6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 185.4, 175.8, 122.3, 69.0, 46.8, 31.6, 29.3, 27.9, 22.7, 22.4, 15.5, and 14.2; HRMS-ESI m/z [M+Na]+ calcd for C12H20O2, 219.1356; found 219.1362.
4.6. Competition experiment
A solution of ynamide 6 (0.081 g, 0.41 mmol) and 1-ethoxyoctyne (22) (0.063 g, 0.41 mmol) in 1.6 mL of benzene-d6 containing 1,4-dibromobenzene (0.096 g, 0.41 mmol) as an internal standard was treated with ketene according to the general procedure. Aliquots (ca. 0.1 mL) of the reaction mixture were taken at intervals, diluted with benzene-d6, and examined by 1H NMR (500 MHz) with relaxation time d1=20 s to ensure accurate integration and auto phasing or manual phasing to ensure a level baseline. In this experiment, chemical shifts are expressed in parts per million downfield from tetramethylsilane with the C6H6 peak at 7.16 ppm used as the standard. For cyclobutenone 10, the average of the resonances at 3.21, 3.17, and 2.68 ppm were used; for cyclobutenone 23, the resonance at 2.80 ppm was integrated. For alkoxyacetylene 22, the methylene at 3.63 ppm was used, and for ynamide 6, the relative amount was determined by integration of the overlapping methylenes for 6 and 22 at 2.18 ppm (after correcting for the amount of 22 determined by integration of the resonance at 3.63 ppm).
Acknowledgements
We thank the National Institutes of Health (GM 28273) and Merck Research Laboratories for generous financial support.
References and notes
- 1.For reviews, see: Wong HNC, Lau K-L, Tam K-F. Top. Curr. Chem. 1986;133:84.. Bellus D, Ernst B. Angew. Chem., Int. Ed. Engl. 1988;100:820.. de Meijere A, editor. Methoden der Organischen Chemie (Houben Weyl) Vol. E17f. Thieme; Stuttgart: 1997. . Moore HW, Yerxa BR. In: Advances in Strain in Organic Chemistry. Halton B, editor. Vol. 4. Jai; London: 1995. pp. 81–162.. Namyslo JC, Kaufmann DE. Chem. Rev. 2003;103:1485. doi: 10.1021/cr010010y..
- 2.For reviews, see: Hyatt JA, Raynolds PW. In: Organic Reactions. Paquette LA, editor. Vol. 45. Wiley; New York: 1994. pp. 159–646.. Tidwell TT. Ketenes. Wiley; New York: 1995. Danheiser RL, editor. Science of Synthesis: Houben Weyl Methods of Molecular Transformations. Vol. 23. Thieme; Stuttgart: 2006. .
- 3.(a) Hassner A, Dillon JL., Jr. J. Org. Chem. 1983;48:3382. [Google Scholar]; (b) Danheiser RL, Sard H. Tetrahedron Lett. 1983;24:23. [Google Scholar]; (c) Danheiser RL, Savariar S, Cha DD. Organic Syntheses. Vol. VIII. Wiley; New York: 1993. pp. 82–86. Collect. [Google Scholar]
- 4.For representative examples, see: Rosebeck B, Arens JF. Recl. Trav. Chim. Pays-Bas. 1962;81:549. Hasek RH, Gott GP, Martin JC. J. Org. Chem. 1964;29:1239.. Ficini J, Genêt JP. Tetrahedron Lett. 1975;16:2633.. Wasserman HH, Piper JU, Dehmlow EV. J. Org. Chem. 1973;38:1451.. Danheiser RL, Gee SK. J. Org. Chem. 1984;49:1672..
- 5.(a) Danheiser RL, Nishida A, Savariar S, Trova MP. Tetrahedron Lett. 1988;29:4917. [Google Scholar]; (b) Kowalski CJ, Lal GS. J. Am. Chem. Soc. 1988;110:3693. [Google Scholar]
- 6.For representative examples, see: Ficini J. Tetrahedron. 1976;32:1449.. Himbert G. In: Methoden der Organischen Chemie (Houben Weyl) Kropf E, Schaumann E, editors. Vol. E15e. Stuttgart: Germany: 1993. pp. 3267–3443.. Zificsak CA, Mulder JA, Hsung RP, Rameshkumar C, Wei L-L. Tetrahedron. 2001;57:7575..
- 7.For examples, see: Kuehne ME, Sheeran PJ. J. Org. Chem. 1968;33:4406.. Truce WE, Bavry RH, Bailey PS., Jr. Tetrahedron Lett. 1968:5651.. Delaunois M, Ghosez L. Angew. Chem., Int. Ed. Engl. 1969;8:72.. Ficini J, Pouliquen J. Tetrahedron Lett. 1972:1135.. Himbert G. Liebigs Ann. Chem. 1979:829.. Barbaro G, Battaglia A, Giorgianni P. J. Org. Chem. 1987;52:3289.. Schulte N, Möller MH, Rodewald U, Würthwein E-U. Chem. Ber. 1994;127:1287..
- 8.(a) Danheiser RL, Gee SK. J. Org. Chem. 1984;49:1672. [Google Scholar]; (b) Danheiser RL, Brisbois RG, Kowalczyk JJ, Miller RF. J. Am. Chem. Soc. 1990;112:3093. [Google Scholar]; (c) Dudley GB, Takaki KS, Cha DD, Danheiser RL. Org. Lett. 2000;20:3407. doi: 10.1021/ol006561c. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 9.Dunetz JR, Danheiser RL. Org. Lett. 2003;5:4011. doi: 10.1021/ol035647d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Zhang Y, Hsung RP, Tracey MR, Kurtz KCM, Vera EL. Org. Lett. 2004;6:1151. doi: 10.1021/ol049827e. [DOI] [PubMed] [Google Scholar]; (b) Frederick MO, Mulder JA, Tracey MR, Hsung RP, Huang J, Kurtz KCM, Shen L, Douglas CJ. J. Am. Chem. Soc. 2003;125:2368. doi: 10.1021/ja021304j. [DOI] [PubMed] [Google Scholar]
- 11.Riddell N, Villeneuve K, Tam W. Org. Lett. 2005;7:3681. doi: 10.1021/ol0512841. [DOI] [PubMed] [Google Scholar]
- 12.(a) Williams JW, Hurd CD. J. Org. Chem. 1940;5:122. [Google Scholar]; (b) Hanford WE, Sauer JC. In: Organic Reactions. Adams R, editor. Vol 3. Wiley; New York: 1946. pp. 108–140. [Google Scholar]
- 13.(a) Henn L, Himbert G. Chem. Ber. 1981;114:1015. [Google Scholar]; (b) Henn L, Himbert G, Diehl K, Kaftory M. Chem. Ber. 1986;119:1953. [Google Scholar]
- 14.Reviewed in: Brady WT. Synthesis. 1971:415.. Brady WT. Tetrahedron. 1981;37:2949.. Schaumann E, Scheiblich S. In: Methoden der Organischen Chemie (Houben Weyl) Kropf E, Schaumann E, editors. Vol. E15c. Thieme; Stuttgart, Germany: 1993. pp. 2933–2958..
- 15.Lawlor MD, Lee TW, Danheiser RL. J. Org. Chem. 2000;65:4375. doi: 10.1021/jo000227c. [DOI] [PubMed] [Google Scholar]
- 16.(a) Hasek RH, Gott PG, Martin JC. J. Org. Chem. 1964;29:2510. [Google Scholar]; (b) von Leander T, Weilenmann M, Dallwigk E. Helv. Chim. Acta. 1977;60:975. [Google Scholar]; (c) Turnbull P, Moore HW. J. Org. Chem. 1995;60:644. [Google Scholar]
- 17.Brand S, de Candole BC, Brown JA. Org. Lett. 2003;5:2343. doi: 10.1021/ol034701n. [DOI] [PubMed] [Google Scholar]
- 18.Carbamate 1 was prepared as described byPirkle WH, Simmons KA, Boeder CW. J. Org. Chem. 1979;44:4891..
- 19.Alkynyl iodide 4 was prepared according to Amatore C, Blart E, Genêt JP, Jutand A, Lemaire-Audoire S, Savignac M. J. Org. Chem. 1995;60:6829..
- 20.The carbamate 2 used in these experiments was prepared in 66% yield via Curtius rearrangement of 4-pentenoic acid with subsequent trapping by methanol (1.0 equiv Et3N, 1.0 equiv DPPA, toluene, 80 °C, 2 h then 10 equiv MeOH, 50 °C, 16 h). For alternative methods for the preparation of 2, see: Esch PM, de Boer RF, Hiemstra H, Boska IM, Speckamp WN. Tetrahedron. 1991;47:4063.. Jego JM, Carboni B, Vaultier M. Bull. Chim. Soc. Fr. 1992;6:554.. Moriarty RM, Chany CJ, Vaid RV, Prakash O, Tuladhar SM. J. Org. Chem. 1993;58:2478.. Kise N, Yamazaki H, Mabuchi T, Shono T. Tetrahedron Lett. 1994;35:1561..
- 21.For reactions employing ca. 0.2–0.6 mmol of ynamide, a 5 mL, thick-walled, conical vial was used as the reaction vessel and ketene was introduced via a 10 cm long, 20-gauge stainless steel needle.
- 22.1-Ethoxyoctyne was prepared as described by Pons J-M, Kocieński P. Tetrahedron Lett. 1989;30:1833..