

Abstract
The process of amyloid-β (Aβ) fibril formation is genetically and pathologically linked to Alzheimer's disease (AD). Thus, a selective and sensitive method for the quantification of Aβ amyloid fibrils in complex biological samples enables a variety of hypotheses to be tested. Herein we report the basis for a quantitative in vitro kinetic aggregation assay that detects seeding-competent Aβ aggregates in mammalian cell culture media, in Caenorhabditis elegans lysate and in mouse brain homogenate. Sonicated, proteinase K treated Aβ-fibril-containing tissue homogenates or cell culture media were added to an initially monomeric Aβ1–40 reporter peptide to seed an in vitro nucleated aggregation reaction. The reduction in the half time (t50) of the amyloid growth phase is proportional to the quantity of seeding-competent Aβ aggregates present in the biological sample. An ion exchange resin amyloid isolation strategy from complex biological samples is demonstrated as an alternative to improve the sensitivity and linearity of the kinetic aggregation assay.
In Alzheimer's disease (AD), the most common human neurodegenerative disorder, enzyme-mediated endoproteolysis of the amyloid precursor protein (APP) in the secretory and endocytic pathways produces a family of closely related peptides collectively referred to as the amyloid β peptide (Aβ). Compelling genetic and pathological evidence indicates that AD is mechanistically linked to the production and aggregation of Aβ, particularly Aβ1–42 (1–3). Another major risk factor for the development of AD is aging (4–6).
No drugs that alter disease progression are approved for AD. When such treatments become available, it is likely that their efficiency will be critically dependent upon early diagnosis, as substantial neuronal cell loss and brain volume decreases occur at the intermediate stages of AD. Intracellular Tau tangles are also observed in AD (7, 8), suggesting there may be a more general loss of proteostasis (5, 6, 9–13). At present, routine methods are not available to specifically detect Aβ aggregates and tau deposits before autopsy, and while some progress is being made towards the detection of extracellular Aβ amyloid (14–17), these compounds usually bind to amyloid fibrils composed of several different proteins, making a definitive diagnosis more challenging (18). Thus, highly sensitive and selective methodology for the quantitative detection of Aβ aggregates in cells, animal models, and in living patient tissue and biological fluids would be welcomed.
Aβ amyloidogenesis appears to proceed by a nucleated polymerization mechanism in vitro (19–23). The rate limiting step of this process is the formation of an oligomeric nucleus, the highest energy species on the amyloidogenesis pathway, whose formation is required before fibril formation becomes thermodynamically favorable (Figure 1a). Once a nucleus is formed, additional monomeric Aβ peptides or oligomers can be added to the fibril in a step that is thermodynamically favorable, leading to a fast growth phase (Figure 1a, inset) (21). Once the monomer concentration is depleted below the critical concentration, Kc, the amyloidogenesis reaction enters a stationary phase (Figure 1a, inset). The duration of the lag phase associated with nucleation is reduced proportional to the quantity of preformed amyloid fibrils, or seeds, added to an in vitro aggregation reaction (Figure 1a, inset) (23), and this is the basis of the kinetic aggregation assay of focus within.
Figure 1.

The effect of adding preformed Aβ1–40 or Aβ1–42 fibrils on an Ab1–40 amyloidogenesis reaction—the basis for the Ab1–40 kinetic aggregation assay. (a) Free energy amyloidogenesis reaction coordinate diagram for the nucleation-dependent aggregation of Aβ1–40 (solid line). Addition of a sufficient quantity of preformed Ab1–40 fibrils or seeds (dashed line) eliminates the requirement for nucleation, which is the rate-limiting step for Aβ1–40 amyloidogenesis. Inset: A typical nucleated polymerization time course. The addition of amyloid fibril seeds reduces the t50 proportional to the quantity of seed termini added. (b) Aβ1–40 (10 μM) aggregation kinetics (pH 7.4, 37 °C) with agitation (5s of shaking every 10 min) in the absence or presence of the indicated amount of preformed Aβ1–40 amyloid seeds (sonicated for 40 min before addition). The amyloidogenesis reaction is followed by the binding of ThT to the fibril which dramatically increases its fluorescence quantum yield. Fluorescence is normalized to the value of the plateau phase in a given reaction. (c) The observed amyloidogenesis t50 as a function of the amount of Aβ1–40 amyloid seed added. Data are reported as mean ± SD of triplicate results.
We have previously used the half time (t50) of the growth phase of the in vitro Aβ amyloidogenesis reaction to semi-quantitatively detect the seeding-competent Aβ aggregates, including amyloid in tissue homogenates from Caenorhabditis elegans and murine AD models (5, 6). Herein, we report a substantially optimized kinetic aggregation assay featuring new procedures that make the assay more reliable, and importantly allows for the quantification of the seeding competent Aβ aggregate load in tissues and in cells. The kinetic aggregation assay is at least three orders of magnitude more sensitive than a Western blot without antigen retrieval at quantifying Aβ aggregate load in mouse and worm tissue. Unlike the small molecules that semi-quantify amyloid load by PET imaging (14), the kinetic aggregation assay does not sensitively detect amyloid composed of other proteins.
Experimental Procedures
Preparation of seed-free Aβ peptide
Aβ1–40 was synthesized using standard Fmoc-chemistry for solid-phase peptide synthesis (24). The resulting peptide was purified by reverse phase C18 HPLC and characterized by MALDI-MS. The Aβ1–40 peptide utilized in the kinetic aggregation assay was monomerized as described previously (25). Briefly, lyophilized Aβ powder was dissolved in aqueous NaOH (2 mM) and the pH was adjusted to 10.5 with aqueous NaOH (100 mM). The solution was sonicated (20 min, 25°C), then filtered sequentially through 0.2 μm and 10-kDa cutoff filters. The concentration of protein was determined by UV absorption at 280 nm (1,280 M−1cm−1). The final peptide solution was seed free by atomic force microscopy (AFM) and dynamic light scattering analysis.
Mice strains and preparation of brain homogenate
An AD mouse model expressing both a mutant chimeric mouse/human APPswe and a mutant human presenilin 1 (Delta E9), both driven by the prion protein promoter, was purchased from Jackson Laboratory (Strain B6C3-Tg, (APPswe PSEN1dE9) 85Dbo/J, Stock number 004462). Long-lived male mice harboring only one Igf1r copy (26) were obtained from Dr. Jeffery Friedman (TSRI, La Jolla, CA). Details of crosses and genotyping are provided in Supporting Information. Mouse brain was homogenized in cold PBS using a glass tissue grinder (885482, Kontes, Vineland, NJ) to a final concentration of 10% w/v. The total protein concentration in mouse brain homogenate was measured with a BCA kit (Pierce, Rockford, IL).
Worm growth and preparation of PDS
Wild type (N2) and CL2006 Aβ worm strains were obtained from the Caenorhabditis Genetics Center (Minneapolis, MN). Worms were grown on bacterial strains at 20 °C. At day 1 of adulthood, the worms were washed twice with M9 media and once more with phosphate buffered saline (PBS) (RT), resuspended in 300 μL ice cold PBS, transferred to a 2 mL tissue grinder (885482, Kontes, Vineland, NJ) and homogenized. Crude homogenates were spun in a desktop microfuge (3,000 rpm, 3 min) and the resulting supernatant, the post debris supernatant (PDS), was transferred to new tubes and the total protein concentration was measured with a BCA kit (Pierce, Rockford, IL).
APP-overexpressing cell lines
Genes encoding human wild type APP (APPwt) and APP carrying the Swedish mutations (APPswe) were cloned into the tetracycline-inducible mammalian expression vector pcDNA4/TO (Invitrogen). T-REx-293 cells (Invitrogen) were stably transfected with empty pcDNA4/TO (EV), pcDNA4/TO/APPwt, or pcDNA4/TO/APPswe vectors. Following transfection, clones were selected and subcloned to ensure genetic homogeneity. For expression, 3×106 cells were adhered to 10 cm plates overnight, after which 100 ng/mL tetracycline (Sigma-Aldrich) was added to the culture media to induce overexpression. Conditioned culture media were aspirated from individual culture plates over the course of 4 days following tetracycline induction and stored at −20 °C. The total protein concentration in cell culture media was measured with a BCA kit (Pierce, Rockford, IL). See Supporting Information for details of cloning and cell culture.
Preparation of Aβ1–40 seeds
Aβ1–40 fibrillar seeds were prepared by incubating a monomerized Aβ1–40 peptide solution (50 μM) in PBS (50 mM Na-phosphate, 150 mM NaCl, pH 7.4) at 37 °C in a 1.5 mL reaction tube on a rotating shaker (20 rpm) for 5–6 days. Fibril morphology was confirmed by AFM analysis (see Supporting Information for details on AFM).
In vitro Aβ1–40 kinetic aggregation assay with added preformed Aβ1–40 seeds
The preformed Aβ1–40 fibrils were sonicated for 40 min in a water bath sonicator (FS60, Fisher Scientific, Pittsburg, PA). A known quantity of these seeds was added to a monomerized Aβ1–40 peptide solution diluted to a final concentration of 10 μM in phosphate buffer (50 mM Na-phosphate, 150 mM NaCl, pH 7.4) containing ThT (20 μM). Three aliquots (100 μL) were transferred into wells of a 96-well microplate (Costar #3631, black, clear bottom), the plate was sealed with a microplate cover and loaded into a Gemini SpectraMax EM fluorescence plate reader (Molecular Devices, Sunnyvale, CA), where it was incubated at 37 °C. The fluorescence (excitation at 440 nm, emission at 485 nm) was measured from the bottom of the plate at 10 min intervals, with 5s of agitation before each reading. Half maximal fluorescence time points (t50) were defined as the time point at which ThT fluorescence reached the midpoint between pre- and post-aggregation baselines. t50 values represent means of triplicate results.
In vitro Aβ1–40 kinetic aggregation assay with added Aβ-fibril-doped biological samples
Preformed Aβ1–40 fibrils were added to mouse brain homogenates or worm PDS in known amounts and the mixture was sonicated for 40 min in an ice water bath before being added to an Aβ1–40 monomer solution (10 μM) for the kinetic aggregation assay. To treat with proteinase K (PK), samples spiked with known amounts of preformed Aβ1–40 amyloid were sonicated for 40 min in an ice water bath, and then incubated with PK (w/w ratio of PK to total protein in the sample is 1:500) for 2 h at 25 °C. The solution was then supplemented with Roche complete protease inhibitor cocktail (PI) (cat#1836170) at four times the recommended concentration to inhibit PK activity. The mixed solution was then sonicated for 20 min, and added to the kinetic aggregation assay. The concentration of total protein from the mouse brain homogenate or worm PDS before PK treatment was 10 μg/mL in the kinetic aggregation assay.
In vitro Aβ1–40 kinetic aggregation assay with other biological samples
For assays with PK treatment, samples were sonicated for 20 min (N2 and Aβ worm PDS, and cell culture media) or 40 min (WT and AD mouse brain homogenates) in an ice water bath, treated for 2 h with PK at 25 °C (w/w ratio of PK to total protein in the sample is 1:500) and then PI was added at four times the recommended concentration to inhibit PK activity. The mixed solution was sonicated for an additional 20 min, and then added to the kinetic aggregation assay. The concentration of total protein in the final kinetic aggregation assay was 10 μg/mL (mouse brain homogenate and worm PDS) and 25 μg/mL (cell culture media) before PK treatment. For the assays without PK and PI treatment, samples were sonicated for 20 min (worm PDS and cell culture media) or 40 min (mouse brain homogenate) before being added to the kinetic aggregation assay.
Aβ amyloid ion exchange isolation
Preformed Aβ1–40 amyloid (13 ng) was suspended in 200 μL H2O, sonicated for 40 min in an ice water bath, and then incubated with 8 μL weak cation CM beads (CM sepharose fast flow, GE) in a 0.6 mL microcentrifuge tube on a rotating shaker (10 rpm) overnight at 4 °C. The CM beads were washed 3 times with H2O, and then added to the kinetic aggregation assay. To isolate the Aβ1–40 amyloid doped into mouse brain homogenate, preformed Aβ1–40 fibrils were added to WT mouse brain homogenate samples in known amounts (total protein concentration from mouse brain homogenate was 15 μg/mL). The mixture was sonicated for 40 min in an ice water bath, and then incubated with 8 μL CM beads in 200 μL H2O in a 0.6 mL microcentrifuge tube on a rotating shaker (10 rpm) overnight at 4 °C. The CM beads were washed 3 times with H2O, and then added to the kinetic aggregation assay. To isolate endogenous Aβ1–40 amyloid in WT and AD mouse brain homogenates, homogenates prepared as described above (total protein concentration from mouse brain homogenate 50 μg/mL) were sonicated for 40 min in an ice water bath, then incubated with 8 μL CM beads in 200 μL H2O in a 0.6 mL microcentrifuge tube on a rotating shaker (10 rpm) overnight at 4 °C. The beads were washed three times with H2O and added to the kinetic aggregation assay.
Western blot
Worm PDS or mouse brain homogenate samples spiked with added Aβ1–40 amyloid were boiled with SDS loading buffer for 10 min, and then were loaded onto SDS-PAGE gels and blotted onto nitrocellulose paper. The blots were incubated with primary antibody 6E10 (Covance, San Diego, CA) at a 1:10,000 dilution overnight, and then incubated with secondary goat anti-mouse HRP-conjugated antibody (Pierce, Rockford, IL) at a 1:10,000 dilution. Blots were visualized by enhanced chemiluminescence using SuperSignal West Pico Substrate (Pierce).
Results
Sonicated Aβ1–40 amyloid fibrils dose dependently accelerate Aβ1–40 amyloidogenesis in vitro
To evaluate the influence of known quantities of sonicated Aβ1–40 amyloid fibrils or seeds on the in vitro amyloidogenesis kinetics of an initially monomeric Aβ1–40 peptide (10 μM), we compared the Aβ1–40 lag phase duration as a function of the amount of added Aβ1–40 fibrillar seeds (prepared as described in Experimental Procedures) in phosphate buffer (50 mM Na phosphate, 150 mM NaCl, pH 7.4) containing thioflavin (ThT, 20 μM). These experiments were carried out in a 96-well microtiter plate (Costar #3631, black, clear bottom) housed in a fluorescent plate reader over the time course of the experiment (37 °C). The fluorescence at 485 nm was measured from the bottom of the plate at 10 min intervals, with 5s of agitation before each reading. The Aβ1–40 fibrils (Figure S1a) were sonicated in a water bath sonicator for 40 min to generate a more uniform (70–150 nm) length distribution (Figure S1b), based on AFM imaging. Since seeding has been demonstrated to be mediated by the fibril ends (27, 28), sonication to afford a uniform length distribution is critical to be able to quantify the amount of fibrils of different lengths in tissues or cells. The time course of Aβ1–40 fibril formation in vitro was monitored using ThT fluorescence. ThT is an environmentally sensitive fluorophore, whose selective binding to amyloid fibrils dramatically increases its fluorescence quantum yield (29).
Addition of sonicated Aβ-fibrils or nuclei shortens the lag phase of Aβ amyloidogenesis (Figure 1b). This is most conveniently measured as the half time, or t50, of the growth phase, which is defined as the time at which the fluorescence intensity reaches the midpoint between the pre- and post-aggregation baselines (Figure 1a, inset). We sonicated Aβ1–40 amyloid for 40 min to prepare seeds, as longer sonication does not further reduce the t50 appreciably. The kinetic aggregation assay is based on the hypothesis that the aggregation half time of the growth phase is proportional to the amount of sonicated Aβ amyloid fibrils added (Figure 1b). As shown in Figure 1c, the growth phase t50 quantitatively correlates with the quantity of sonicated Aβ1–40 amyloid fibril seeds added to the in vitro kinetic aggregation assay, confirming the predicted quantitative effect of seeding on the lag phase of Aβ1–40 fibrillization in vitro (19–23). This quantitative relationship is the first innovation we report, which makes this assay more valuable, as previously it was employed to simply demonstrate that there were aggregates present in the sonicated tissue.
A quantitative kinetic aggregation assay should be highly selective; i.e., Aβ amyloidogenesis should not be seeded by amyloids of distinct sequence (30, 31). Sonicated α-synuclein fibrils (14.5 μg/mL) formed from the α-synuclein monomer (50 μM) have less seeding capacity than Aβ1–40 fibrils (4.3 ng/mL), making them < 1/1000 as seeding competent as Aβ fibrils (Figure S2a). Sonicated 8 kDa gelsolin fibrils (7.9 μg/mL) formed from the 8 kDa gelsolin monomer (50 μM) have a seeding capacity equivalent to that of 43 ng/mL Aβ1–40 fibrils, making them 1/100 as seeding competent as Aβ fibrils (Figure S2b). Therefore, the Aβ1–40 kinetic aggregation assay exhibits the selectivity expected of an assay that will be used to quantify Aβ fibril load in tissues that could possibly have other amyloids present. In contrast to α-synuclein and 8 kDa gelsolin, the sequence of the islet amyloid polypeptide (IAPP) exhibits substantial homology with Aβ1–40. However, it is established that IAPP fibrils exhibit weight-normalized efficiencies of only 1–2% relative to Aβ1–40 seeding of Aβ1–40 amyloidogenesis (30). Moreover, IAPP is not expressed in the brain of mice. For these reasons, it is very unlikely that cross-seeding of Aβ1–40 amyloidogenesis by IAPP fibrils would be problematic in the work presented below.
To confirm that ThT fluorescence accurately reflects the amount of aggregates formed, and to verify that there are no non- or weakly-fluorescent aggregates present, we chose to quantify the amount of Aβ aggregates formed following seeding by RP HPLC. A 150,000 × g ultracentrifugation for 1 h successfully separates soluble Aβ monomers from pelleted Aβ fibrils and aggregates (Figure S2c). When the kinetic aggregation assay reached the stationary phase, samples were spun at 150,000 × g, and total Aβ1–40 present in pellets was quantified by RP HPLC. Seeding with 14.5 μg/mL of a-synuclein fibrils yields the same amount of pelletable Aβ1–40 as seeding with 4.3 ng/mL of Aβ1–40 fibrils (Figure S2d). Likewise, seeding with 7.9 μg/mL of 8 kDa gelsolin fibrils yields the same amount of pelletable Aβ1–40 as seeding with 43 ng/mL of Aβ1–40 fibrils (Figure S2e). These data support the hypothesis that non-homologous seeds are not facilitating the formation of non-fibrillar Aβ aggregates.
While the Aβ1–40 and Aβ1–42 amyloid structures are subtly different (32–34), it is well-established that Aβ1–40 takes on the structure of the Aβ1–42 fibril when seeded with sonicated Aβ1–42 fibrils (35, 36). Since the aggregation of Aβ1–42 from a monomeric solution is dramatically faster than Aβ1–40, without an obvious lag phase (35), we utilized the Aβ1–40 peptide as the readout peptide in the kinetic aggregation assay to detect aggregates of Aβ1–40 and/or Aβ1–42 that are competent to seed Aβ1–40 fibrillization. In principle, shorter Aβ peptides could also be used as the readout peptide in the kinetic aggregation assay, although this was not explored. To minimize the influence of the other components in cell or tissue homogenates on the observed t50 in the Aβ1–40 kinetic aggregation assay, two methods were employed—proteinase K treatment to degrade the soluble proteome, and ion exchange capture of amyloid to beads so that the soluble proteome, membrane components, and other components can be washed away.
Proteinase K treatment of tissue homogenates improves quantitative linearity
To assess whether the Aβ1–40 kinetic aggregation assay can be used to quantify the amount of Aβ fibrils in a given tissue or cell, we first probed whether Aβ-fibril-doped biological samples can be accurately quantified using this assay. Known amounts of Aβ1–40 amyloid fibrils were added to wild type (WT) mouse brain homogenate samples (non-transgenic), then the mixture was sonicated in a water bath sonicator and added to an Aβ1–40 monomer solution for the kinetic aggregation assay (Figure 2a). The t50 was shortened proportional to the amount of fibrillar seeds added above ~40 ng/mL , but the kinetic difference becomes small when < 40 ng/mL of sonicated Aβ1–40 fibrils were added (Figure 2b, open triangles). This limited sensitivity is likely a consequence of the high protein and lipid content of mouse brain. In an attempt to improve the sensitivity and linearity of the assay, proteinase K (PK) was used to digest the soluble proteome within the brain homogenate. There is ample precedent that amyloid fibrils are resistant to PK digestion (37–40). We verified that when mouse brain homogenate was incubated with PK, the amount of soluble protein remaining was decreased dramatically (Figure S3). We then confirmed that Aβ amyloid is PK resistant under the conditions used herein by quantifying total Aβ1–40 following PK digestion by RP-HPLC; 84% of Aβ1–40 remains undigested following incubation with PK for 2 h. (Figure S4a, b).
Figure 2.

Aβ1–40 kinetic aggregation assay to which wild type mouse brain homogenate (spiked with a known amount of preformed and sonicated Aβ1–40 amyloid fibrils) was added. (a) Aβ1–40 (10 μM) kinetic aggregation traces detected by ThT fluorescence (pH 7.4, 37 °C) with agitation (5s of shaking every 10 min) without and in the presence of known quantities of preformed Aβ1–40 amyloid seeds (0.043–43 ng/mL) added to mouse brain tissue. Fluorescence is normalized to the value of the plateau phase in a given reaction. The mouse brain homogenate without or with added Aβ fibrils was sonicated for 40 min before being added to the kinetic aggregation assay. (b) Comparison of Aβ1–40 amyloidogenesis t50 without (open triangles) or with PK and PI pretreatment (filled diamonds). Data are reported as the mean ± SD of triplicate experiments. A P value of < 0.005 is realized when comparing the mean t50 values in the absence and presence (0.043 ng/mL) of Aβ seeds (indicated with an * in the figure) utilizing the PK and PI treatment protocol (filled diamonds). In contrast, a P value of > 0.8 is observed when the t50 mean values obtained in the absence and in the presence (0.043 ng/mL) of Aβ seeds are compared without PK and PI treatment (unfilled triangles). (c) Aβ1–40 kinetic aggregation traces of proteinase K (PK) treated mouse brain tissue without and in the presence of known quantities of preformed Aβ1–40 amyloid seeds. The mouse homogenate without or with added Aβ fibrils was sonicated for 40 min, treated with PK for 2 h (w/w ratio of PK to total protein in the mouse brain sample is 1:500), inhibited by Roche protease inhibitor cocktail (PI) at four times the recommended concentration, then sonicated for another 20 min before being added to the kinetic aggregation assay. (d) Dilution series of Aβ1–40 amyloid spiked into wild type mouse brain tissue as detected by Western blot analysis using the 6E10 antibody (antigen retrieval procedures not employed).
Wild type mouse brain homogenate spiked with Aβ1–40 amyloid prepared in vitro was first sonicated in a water bath sonicator for 40 min, and then incubated with PK for 2 h at 25 °C. Roche complete protease inhibitor cocktail (PI) was then added to block PK activity (to avoid degradation of the initially monomeric Aβ1–40 readout peptide used in the kinetic aggregation assay). The PK-treated mouse brain homogenate was then sonicated again in a water bath sonicator for 20 min before being added to the kinetic aggregation assay (Figure 2c). PK treatment substantially improved the sensitivity at < 40 ng/mL of added sonicated Aβ1–40 fibrils and the linearity of the decrease in t50 values with increasing amount of added seeds (Figure 2b, filled diamonds). It is notable that the t50 difference as a function of added amyloid seed is greater in the PK treated samples (cf. Figure 2c to 2a), also reflected by the slope increase in Figure 2b. A P value of < 0.005 is realized when comparing the mean t50 values in the absence and presence of Aβ seeds (0.043 ng/mL, indicated with an * in the Figure 2b) utilizing the PK treatment protocol (filled diamonds). In contrast, a P value of > 0.8 is observed when the mean t50 values obtained in the absence and in the presence (0.043 ng/mL) of Aβ seeds are compared without PK treatment (unfilled triangles). This increased sensitivity and improved linearity of the kinetic aggregation assay utilizing the PK treatment protocol allowed quantification of added amyloid in the range of 0.04–40 ng/mL. This assay is at least three orders of magnitude more sensitive than the SDS-PAGE/Western blot analysis using the 6E10 antibody (cf. ability to detect fibrils in Figure 2d to Figure 2b, filled diamonds). No antigen retrieval protocol was employed, which should increase the sensitivity of the Western blot approach.
Similarly, PK treatment of wild type C. elegans post debris supernatant (PDS), spiked with known amounts of Aβ1–40 amyloid, (Figure 3a, cf. filled diamonds (PK treated) to open triangles (untreated)) markedly enhanced assay sensitivity and linearity of the kinetic aggregation assay. A P value of < 0.004 is realized when comparing the mean t50 values in the absence and presence (0.43 ng/mL) of Aβ seeds (indicated with an * in Figure 3a) utilizing the PK treatment protocol (filled triangles). In contrast, a P value > 0.07 between the mean t50 values in the absence and the presence (0.43 ng/mL) of Aβ seeds is observed without PK treatment (unfilled triangles). The C. elegans kinetic aggregation assay is at least three orders of magnitude more sensitive than employing a SDS/PAGE-6E10 antibody-based Western blot approach for Aβ fibril detection, cf. Figure 3b to Figure 3a, filled diamonds. No antigen retrieval protocol was employed, which should increase the sensitivity of the Western blot approach.
Figure 3.

Aβ1–40 kinetic aggregation assay utilizing wild type C. elegans worm PDS with known quantities of preformed Aβ1–40 amyloid fibrils added as seeds. (a) Amyloidogenesis t50 of an Aβ1–40 kinetic aggregation assay without or with the addition of known quantities of Aβ fibrils added to worm PDS. The assay was performed as described in Figure 2. Triplicate data are reported as mean ± SD. A P value of < 0.004 is observed when comparing the mean t50 values in the absence and presence (0.43 ng/mL) of Aβ seeds (indicated with an * in the figure) utilizing the PK and PI treatment protocol (filled triangles). In contrast, a P value > 0.07 between the mean t50 values in the absence and the presence (0.43 ng/mL) of Aβ seeds is observed without PK and PI treatment (unfilled triangles). (b) Dilution series of Aβ1–40 amyloid spiked into worm PDS as detected by Western blot analysis using the 6E10 antibody (antigen retrieval procedures not employed).
Detection of Aβ amyloid in an Alzheimer's transgenic (AD) mouse brain homogenate
We next examined whether the kinetic aggregation assay can be employed to semiquantify Aβ amyloid fibrils formed in vivo, that differ from fibrils formed in vitro owing to the bound amyloid P component and bound extracellular matrix components (such as glycosaminoglycans). We compared brain homogenates from wild type mice to those from a well-characterized human transgenic Alzheimer's Aβ mouse model that shows slow progressive age-onset Aβ fibril formation in the brain (6, 41).
Without PK treatment, the aggregation t50 of the WT and Borchelt Alzheimer's mouse brain homogenates were not distinguishable (Figure 4a), suggesting that the proteins and lipids mask the seeding capabilities of the Aβ fibrils present in the AD mouse brains. After PK treatment to digest the soluble mouse brain proteome, differences in the kinetic aggregation assay t50 between the AD and WT murine samples were evident (Figure 4b). Addition of AD mouse brain homogenate resulted in a faster aggregation time course relative to WT mouse brain homogenate (Figure 4b), consistent with seeding by Aβ amyloid known to be present in the murine AD transgenic tissue.
Figure 4.
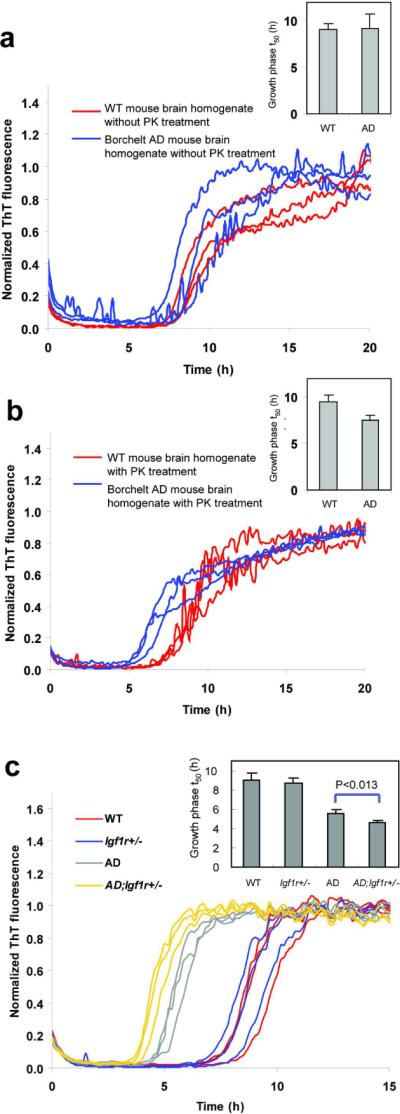
Aβ1–40 kinetic aggregation traces with added mouse brain homogenate. Triplicate results are shown in each figure. (a) Wild type (WT) or AD mouse brain homogenate without PK treatment. Inset: t50 of the kinetic aggregation traces. Data are reported as mean ± SD of triplicate results. (b) WT or AD mouse brain homogenate with PK and PI treatment. Inset: t50 of the kinetic aggregation traces with data reported as mean ± SD of triplicate results. (c) WT, Igf1r+/−, AD and AD;Igf1r+/− mouse brain homogenates with PK and thermal denaturation treatment. The mouse brain homogenates were sonicated for 40 min, treated with PK for 2 h, then boiled for 10 min, sonicated for an additional 20 min, and added to the Aβ1–40 kinetic aggregation assay. Inset: t50 of the kinetic aggregation traces with data reported as mean ± SD. p < 0.013 between AD and AD;Igf1r+/− (analyzed using a two-tailed student's t-test).
Thermal denaturation of PK to optimize detection of Aβ amyloid in AD mouse brain
We hypothesized that heating might more fully inactivate PK, and likely other proteases present in the murine brain homogenate relative to protease inhibitor cocktail treatment, improving the kinetic aggregation assay. To determine the effect of the thermal pretreatment alone on Aβ fibril seeding capacity, we sonicated preformed seeds for 40 min, diluted them to concentrations ranging from 0.043 ng/mL to 4,300 ng/mL, heated the seeds in a boiling water bath for 10 min, and sonicated them for an additional 20 min before adding the resulting seeds to the Aβ1–40 kinetic aggregation assay. Heating various quantities of preformed Aβ fibrils in buffer leads to a decrease in their seeding capacity (Figure S5), but this effect is nominal compared to the sensitivity gained by PK pretreatment and subsequent thermal inactivation (Figure 4c). We next sonicated mouse brain homogenates for 40 min, treated with PK, and thermally denatured the PK by heating as described above. Samples were sonicated for an additional 20 min and added to the kinetic aggregation assay. There is a greater kinetic distinction between the boiled AD and WT murine brain homogenates relative to the PI-treated samples (cf. Figure 4c red vs. gray to Figure 4b red vs. blue). We surmise that heating improves the kinetic aggregation assay because it more completely prevents PK, and perhaps other endogenous enzymes, from degrading the monomeric Aβ1–40 readout peptide. This improved protocol, relative to that published (6), demonstrates that the AD mice with half the amount of insulin growth factor receptor signaling (AD;Igf1r+/−) exhibit a statistically significant difference in their t50 values relative to AD mice (p<0.013) (Figure 4c, cf. gray vs. yellow traces). These results demonstrate that AD;Igf1r+/− mouse brain, exhibiting protection from proteotoxicity, harbors more seeding-competent Aβ amyloid than the AD mouse brain, strictly analogous to the results in our Igf1r RNAi experiments in C. elegans (5, 6).
To estimate the quantity of amyloid in the Alzheimer's transgenic mouse samples, we chose to express the results in terms of “synthetic fibril equivalents of amyloid”, rather than absolute amounts, because Aβ amyloid in the Alzheimer's transgenic sample would have glycosaminoglycans, amyloid P component, and potentially other accessory molecules bound to it. These accessory molecules are less likely to accompany the amyloid in the non-transgenic mouse brain sample doped with Aβ fibrils-wherein the standard curve for quantification was generated. To control for day-to-day assay variation (t50 variation), we normalized the t50 values of WT mouse homogenates spiked with known quantities of synthetic Aβ amyloid seeds to the t50 of the control sample–the wild type mouse brain homogenate sample without Aβ amyloid added. The normalized t50 values of the transgenic mouse samples were compared to the normalized t50 values of WT mouse homogenates spiked with known quantities of synthetic Aβ amyloid seeds (the source of the standard curve for quantification), yielding the “synthetic fibril equivalents of amyloid” in the transgenic sample. The normalized t50 from the Alzheimer's transgenic mouse sample corresponds to 0.5 ng/mL “synthetic fibril equivalents of amyloid”. The estimated amount of “synthetic fibril equivalents of amyloid” in the original mouse brain homogenate is ~250 ng/mL, as the mouse brain homogenate is diluted 500 fold in the kinetic aggregation assay.
Optimization of Aβ amyloid detection in the Alzheimer's Aβ worm model
Wild type worms (N2) not expressing Aβ and transgenic Aβ worms expressing Aβ1–42 in the secretory system (5) and apparently in the cytosol were compared to discern whether PK treatment improved the ability of the kinetic aggregation assay to differentiate these samples, in comparison to our previously reported assay (5). PDS from N2 and Aβ worms was sonicated for 20 min, treated with PK for 2 h at 25 °C and then treated with Roche protease inhibitor cocktail. The mixed solution was sonicated for an additional 20 min, and then added to the Aβ1–40 readout peptide. While PK treatment (Figure 5a) did not increase the t50 differences noticeably relative to PK untreated (Figure 5b), this step clearly reduced the uncertainty between replicated results, thus improving the sensitivity of the kinetic aggregation assay to detect Aβ aggregates in PDS samples. We also evaluated whether thermal denaturation instead of PI treatment further improved the sensitivity. While thermal treatment reduced the t50 differences between WT and AD worms (Figure S6a), the reproducibility of the replicates improved (cf. Figure S6a to b), suggesting further optimization using a thermal denaturation step could prove useful.
Figure 5.

Aβ1–40 kinetic aggregation assay with added wild type (N2) or Aβ worm PDS with PK and PI pretreatment (a) or without PK and PI pretreatment (b). Triplicate results are shown in each figure. The assay was performed as described in Figure 2 except that the worm samples were sonicated for only 20 min before PK treatment. Insets: statistical analysis of results obtained in (a) (p < 0.005) and in (b) (p < 0.013), with data reported as mean ± SD.
We also estimated the quantity of amyloid in the Alzheimer's transgenic worm samples, again using the “synthetic fibril equivalents of amyloid” approach. As discussed in detail above, we normalized the t50 values obtained in kinetic aggregation assays run on different days to the t50 of wild type worm PDS without Aβ amyloid added as the control. The normalized t50 of the Aβ worm PDS sample corresponds to 9.6 ng/mL of “synthetic fibril equivalents of amyloid”. The estimated amount of “synthetic fibril equivalents of amyloid” in the original Aβ worm PDS sample is ~3.8 μg/mL, as the Aβ worm PDS sample is diluted 400 fold in the kinetic aggregation assay.
Detection of Aβ amyloid in mammalian cell culture media
We also explored the ability of the Aβ1–40 kinetic aggregation assay to detect Aβ amyloid outside of the cell, using stably transfected T-REx-293 cell lines expressing empty vector (EV), wild type APP (APPwt) or APPswe. Western blot analysis of the conditioned media revealed the presence of APP and Aβ peptide in the culture media of the tetracycline-induced T-REx-293 APPswe cell line, while only APP (no Aβ) was detected in the T-REx-293 APPwt conditioned media (Figure S7a). Endogenous background levels of APP, but not Aβ, were detected in the EV cell culture media. A significant difference was observed after PK and PI treatment between the seeding efficiency of EV cell media and APPswe cell media after 4 days of overexpression (p<0.002) (Figure 6). However, the difference between EV cell media and APPwt cell media is less distinct (p>0.09), as expected. These data demonstrate that the Aβ peptides in the APPswe cell culture media exist in an aggregated seeding-competent state. Without PK treatment, the distinction in the kinetic aggregation assay between APPswe and EV cells is much smaller (Figure S7b).
Figure 6.
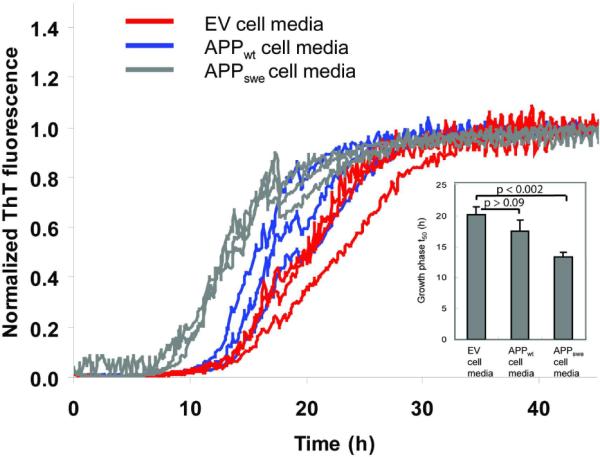
Aβ1–40 kinetic aggregation traces with added conditioned cell media after PK and PI pretreatment, results shown in triplicate. Cells were treated with tetracycline for 4 days to induce APP expression. Tetracycline-treated EV cells do not over express APP; whereas APPwt cells overexpress wild type APP; APPswe cells overexpress APPswe and Aβ. The assay was performed as described for the worm samples. Inset: Statistical analysis of results reported as mean ± SD. p > 0.09 between EV and APPwt cells; p < 0.002 between EV and APPswe cells.
Aβ amyloid isolation using an ion exchange resin to improve the kinetic aggregation assay
We also explored a complementary method that uses an ion exchange resin to bind amyloid from sonicated tissue extracts and cell lysates, allowing contaminating lipids, proteins, etc. that attenuate t50 differences in the kinetic aggregation assay to be washed away. Apart from making the assay easier to perform, this approach also has the capacity to concentrate the Aβ aggregates to enhance the sensitivity of the kinetic aggregation assay. First, we tested the ability of the ion exchange resin to bind amyloid fibrils by pre-incubating weak cation exchange beads (CM sepharose fast flow, GE) with preformed sonicated Aβ amyloid fibrils before adding the beads to the kinetic aggregation assay. Unlike the CM beads that were incubated with buffer only, CM beads pre-incubated with Aβ amyloid fibrils significantly decreased the t50 of the growth phase of the kinetic aggregation assay (Figure 7a), demonstrating that the CM beads bind amyloid and seed the kinetic aggregation assay. The seeding activity of the Aβ fibrils isolated using the CM beads is equivalent to simply adding the same quantity of sonicated fibrils directly into the kinetic aggregation assay (Figure 7b). The t50 of the seeded Aβ (10 μM) kinetic aggregation assay in buffer, where the seeds are presented directly, is identical to the t50 of the seeded kinetic aggregation assay where the seeds are absorbed to and presented on beads (cf. shorter green and gray bars in Figure 7b). Moreover, adding beads only or nothing to an Aβ (10 μM) kinetic aggregation assay in buffer affords identical kinetics, (cf. taller gray and green bars in Figure 7b), demonstrating that the beads have no effect on the unseeded t50.
Figure 7.

Ion exchange resin amyloid isolation approach to the Aβ1–40 kinetic aggregation assay. (a) CM cation exchange beads were incubated at 4 °C with shaking overnight without (red curve) or with (blue curve) Aβ1–40 amyloid seeds (13 ng), and then washed with H2O before being added to the Aβ1–40 kinetic aggregation assay. (b) Comparison (t50) of the Aβ1–40 sonicated fibrils added directly to the Aβ1–40 readout peptide (gray bar) vs. sonicated fibrils pre-incubated with CM beads and washed before being added to the Aβ1–40 readout peptide (green bar). Data are reported as the mean ± SD of triplicate experiments. (c) A comparison of the Aβ1–40 (10 μM) amyloidogenesis t50 using the CM resin amyloid isolation strategy vs. mouse brain homogenate doped with known fibril concentrations (sonication but no PK treatment). Data are reported as mean ± SD of triplicate results. CM beads (8 μL) were incubated with mouse brain homogenates doped with known quantities of Ab amyloid seeds at 4 °C with shaking overnight, then washed and added to the Aβ1–40 kinetic aggregation assay. A P value < 0.005 is realized when comparing the mean t50 values in the absence and presence (0.43 ng/mL) of Aμ seeds (indicated with a * in the figure) utilizing the CM ion exchange bead protocol (filled diamonds). In contrast, a P value of > 0.9 is observed when the t50 values obtained in the absence and in the presence (0.43 ng/mL) of Aβ seeds are compared without utilizing the CM bead approach (unfilled triangles, no PK treatment). (d) Differentiating WT mouse brain (red curves) from AD mouse brain (blue curves) using the CM bead amyloid isolation approach (triplicate results are reported). Before adding CM beads to the readout peptide in the kinetic aggregation assay, the beads were loaded with amyloid (if present) by incubating at 4 °C with WT or AD mouse brain homogenate (total protein concentration was 50 μg/mL) with shaking overnight, followed by a washing protocol. Inset: t50 of the growth phases reported as mean ± SD. A P value of < 0.002 is noted between WT and AD (indicated with a * in the figure).
Furthermore, if the quantity of the bead slurry employed is increased relative to a fixed amount of Aβ seeds, the t50 remains unchanged (Figure S8), strongly demonstrating that the beads have no influence on the t50. Specifically, 5 μL, 8 μL, and 10 μL of CM bead slurry was employed with a fixed amount of Aβ fibrils (43 ng/mL) and statistically significant changes in t50 values in the kinetic aggregation assay in buffer were not observed, indicating that the beads were not overloaded with Aβ seeds. Thus, CM beads can pull down Aβ fibrils efficiently, very likely due to the electrostatic interactions between the CM beads and the positive charges lined up on the surface of the parallel in-register cross-β-sheet amyloid fibrils.
Next, we tested the ability of CM beads to bind to Aβ fibrils added into WT mouse brain homogenate. Analogous to the enhanced sensitivity obtained with PK treatment, CM bead amyloid isolation increases the sensitivity of the kinetic aggregation assay relative to that without CM bead amyloid isolation (Figure 7c). A P value < 0.005 is realized when comparing the mean t50 values in the absence and presence (0.43 ng/mL) of Aβ seeds (indicated with a * in Figure 7c) utilizing the CM bead amyloid isolation protocol (filled diamonds). In contrast, a P value of > 0.9 is observed without using the CM bead protocol (unfilled triangles), all else being equivalent.
Amyloid isolated from AD mouse brain homogenates using CM beads led to significantly faster aggregation kinetics compared to treating WT mouse brain homogenates with CM beads (Figure 7d). Taken together, these results suggest that the kinetic aggregation assay can be further optimized using ion exchange resins to isolate amyloid from complex biological samples.
The reason that the unseeded (0 ng/mL) Aβ (10 μM) aggregation t50 values in Figure 7c differ between untreated mouse brain postnuclear supernatant (PNS) and the CM resin treated mouse brain PNS, is that only in the latter case are the additional brain components removed by resin washing steps–slowing down the kinetics. In other words, there are effectively no brain components in the aggregation time course data depicted by the filled diamonds in Figure 7c. In contrast, in the open triangle data, brain PNS is added directly to the Aβ (10 μM) kinetic aggregation assay; hence it is faster, possibly because of the brain lipids.
Discussion
Quantification of the amyloid load in tissues remains challenging and is important to help us better understand the etiology of degenerative diseases (1–3, 42–51). Selective and sensitive detection of small quantities of specific amyloid fibrils formed in the early stages of disease is of special interest, as this offers diagnostic opportunities (37, 38, 52). Previously, we used an Aβ1–40 kinetic aggregation assay to detect amyloid in worm PDS (5), however this method was semiquantitative. We have demonstrated herein that we can selectively, sensitively and quantitatively detect Aβ amyloid load in a variety of cell and tissue homogenates using the optimized Aβ kinetic aggregation assay. A future goal is to render the kinetic aggregation assay rigorously quantitative in mouse and worm PDS, where accessory factors could complicate quantification relative to analogous samples where known quantities of amyloid were added. It will be very interesting in the future to discern how well the “synthetic fibril equivalents of amyloid” correlates with the absolute amount of amyloid in a transgenic AD model.
A challenge in quantifying low levels of amyloid in biological samples is that amyloid fibrils are minor components of an environment densely packed with small molecule metabolites; macromolecules, including proteins, RNA, and DNA; as well as lipids and other membrane components. Moreover, the amyloid P component (53) and glycosaminoglycans (54) are known to selectively bind to the surface of amyloid fibrils, potentially masking their presence. These interactions could stabilize the amyloid fibrils and interfere with sonication to break up the fibrils into smaller seeds and/or sterically interfere with the seeding activity of sonicated amyloid seeds. In spite of not removing glycosaminoglycans or membrane components, removal of soluble proteins using PK treatment dramatically improves the sensitivity and the linear response of the kinetic aggregation assay. As does an ion exchange resin amyloid isolation strategy, which may be more practical to utilize as the beads with absorbed amyloid seeds can be added directly to the readout peptide. We envision that enzymatic degradation of sulfated glycosaminoglycans and membrane/lipid extraction will have the potential to further improve the Aβ1–40 kinetic aggregation assay, allowing us to get closer to our goal of rigorously quantifying the Aβ amyloid load in tissues and biological fluids.
One potential advantage of the ion exchange resin approach is that the amyloid in complex biological samples like cerebrospinal fluid (CSF) can be concentrated on the ion exchange resin surface, enhancing the capacity to detect amyloid using the kinetic aggregation assay. However, attempts to detect kinetic differences between Alzheimer's and age-matched control human CSF and blood plasma samples (obtained from David Holtzman at Washington University) using the kinetic aggregation assay have been unsuccessful to date. The substantial effort put into this suggests that the concentration of Aβ amyloid fibrils in Alzheimer's human CSF and blood plasma samples is very low, if present at all, and certainly below the detection limits of the current kinetic aggregation assay. Further optimization of the methodology to enhance the sensitivity may allow the assay to detect very small quantities of Aβ amyloid fibrils present in easily accessible human samples.
It should be noted that distinct biological samples required different optimized kinetic aggregation assays, presumably due to their unique molecular composition. For example, while PK treatment to remove interfering soluble proteins was generally useful, thermal treatment to inactivate PK and possibly other proteases significantly improved the mouse brain kinetic aggregation assay, but did not enhance the ability to detect Aβ amyloid in cell culture media, and only modestly improved detection of Aβ amyloid in C. elegans. From the perspective of practicality and ease of use, the ion exchange resin amyloid isolation approach is perhaps the most promising strategy for further enhancing the kinetic aggregation assay; however, future experiments are required to see if its promise can be realized experimentally.
Conclusion
Herein we outlined the basis for improved Aβ1–40 kinetic aggregation assays and demonstrated their general utility for the quantitative or semiquantitative, sensitive and selective detection of Aβ amyloid load in a variety of organismal fractions, including mouse brain homogenate, worm PDS, and cell culture media. Proteinase K treatment substantially improved the sensitivity and linearity of the Aβ1–40 kinetic aggregation assay in most organismal fractions. Thermal inactivation of proteinase K, and likely other proteases, further enhanced the utility of the Aβ1–40 kinetic aggregation assay, especially in mammalian tissue. Further optimization of the Aβ1–40 kinetic aggregation assay, perhaps using the ion exchange resin amyloid isolation strategy presented herein, should render it even more practical and quantitative, possibly enabling it to detect the production of Aβ amyloid fibrils in human CSF or other accessible fluids or tissues for diagnostic purposes. We envision that any amyloid protein that aggregates in vitro by a nucleated polymerization reaction can be used as a readout peptide in a kinetic aggregation assay to quantify the amyloid load of that peptide in a given tissue or body fluid.
Supplementary Material
Acknowledgments
This research was supported by NIH Grant AG031097, The Skaggs Institute for Chemical Biology, and the Lita Annenberg Hazen Foundation
Abbreviations
- Aβ
amyloid-β peptide
- AD
Alzheimer's disease
- AFM
atomic force microscopy
- APP
amyloid precursor protein
- C. elegans
Caenorhabditis elegans
- CSF
cerebrospinal fluid
- EV
empty vector
- PBS
phosphate buffered saline
- PDS
post debris supernatant
- PK
proteinase K
- PI
complete protease inhibitor cocktail
- RT
room temperature
- ThT
thioflavin T
- WT
wild type
Footnotes
Supplemental information Supplemental information contains experimental procedures for mice genotyping, APP-overexpressing lines, and AFM. Supplementary Figures are available, as referred to in the text. This material may be accessed free of charge online at http://pubs.acs.org
References
- 1.Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: A genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 2.Hardy J, Selkoe DJ. Medicine - The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 3.Golde TE, Estus S, Younkin LH, Selkoe DJ, Younkin SG. Processing of the amyloid protein precursor to potentially amyloidogenic derivatives. Science. 1992;255:728–730. doi: 10.1126/science.1738847. [DOI] [PubMed] [Google Scholar]
- 4.Amaducci L, Tesco G. Aging as a major risk for degenerative diseases of the central nervous system. Curr Opin Neurol. 1994;7:283–286. doi: 10.1097/00019052-199408000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 6.Cohen E, Paulsson JF, Blinder P, Burstyn-Cohen T, Du D, Estepa G, Adame A, Pham HM, Holzenberger M, Kelly JW, Masliah E, Dillin A. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell. 2009;139:1157–1169. doi: 10.1016/j.cell.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braak E, Braak H, Mandelkow EM. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 1994;87:554–567. doi: 10.1007/BF00293315. [DOI] [PubMed] [Google Scholar]
- 8.Lee VM, Balin BJ, Otvos LJ, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 1991;251:675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 9.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 10.Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 11.Gidalevitz T, Krupinski T, Garcia S, Morimoto RI. Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity. PLoS Genet. 2009;5:e1000399. doi: 10.1371/journal.pgen.1000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–1474. doi: 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- 13.Page LJ, Suk JY, Bazhenova L, Fleming SM, Wood M, Jiang Y, Guo LT, Mizisin AP, Kisilevsky R, Shelton GD, Balch WE, Kelly JW. Secretion of amyloidogenic gelsolin progressively compromises protein homeostasis leading to the intracellular aggregation of proteins. Proc Natl Acad Sci U S A. 2009;106:11125–11130. doi: 10.1073/pnas.0811753106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 15.Kayed R, Glabe CG. Conformation-dependent anti-amyloid oligomer antibodies. Methods Enzymol. 2006;413:326–344. doi: 10.1016/S0076-6879(06)13017-7. [DOI] [PubMed] [Google Scholar]
- 16.O'Nuallain B, Wetzel R. Conformational Abs recognizing a generic amyloid fibril epitope. Proc. Natl. Acad. Sci. U. S. A. 2002;99:1485–1490. doi: 10.1073/pnas.022662599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edison P, Archer HA, Gerhard A, Hinz R, Pavese N, Turkheimer FE, Hammers A, Tai YF, Fox N, Kennedy A, Rossor M, Brooks DJ. Microglia, amyloid, and cognition in Alzheimer's disease: An [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis. 2008;32:412–419. doi: 10.1016/j.nbd.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 18.Lockhart A, Lamb JR, Osredkar T, Sue LI, Joyce JN, Ye L, Libri V, Leppert D, Beach TG. PIB is a non-specific imaging marker of amyloid-beta (Abeta) peptide-related cerebral amyloidosis. Brain. 2007;130:2607–2615. doi: 10.1093/brain/awm191. [DOI] [PubMed] [Google Scholar]
- 19.Naiki H, Hasegawa K, Yamaguchi I, Nakamura H, Gejyo F, Nakakuki K. Apolipoprotein E and antioxidants have different mechanisms of inhibiting Alzheimer's beta-amyloid fibril formation in vitro. Biochemistry. 1998;37:17882–17889. doi: 10.1021/bi980550y. [DOI] [PubMed] [Google Scholar]
- 20.Teplow DB. Structural and kinetic features of amyloid beta-protein fibrillogenesis. Amyloid. 1998;5:121–142. doi: 10.3109/13506129808995290. [DOI] [PubMed] [Google Scholar]
- 21.Harper JD, Lansbury PT. Models of amyloid seeding in Alzheimer's disease and scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Ann Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 22.Ferrone FA. Nucleation: The connections between equilibrium and kinetic behavior. Methods Enzymol. 2006;412:285–299. doi: 10.1016/S0076-6879(06)12017-0. [DOI] [PubMed] [Google Scholar]
- 23.Powers ET, Powers DL. Mechanisms of protein fibril formation: nucleated polymerization with competing off-pathway aggregation. Biophys J. 2008;94:379–391. doi: 10.1529/biophysj.107.117168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wellings DA, Atherton E. Standard Fmoc protocols. Methods Enzymol. 1997;289:44–67. doi: 10.1016/s0076-6879(97)89043-x. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Q, Powers ET, Nieva J, Huff ME, Dendle MA, Bieschke J, Glabe CG, Eschenmoser A, Wentworth P, Jr., Lerner RA, Kelly JW. Metabolite-initiated protein misfolding may trigger Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:4752–4757. doi: 10.1073/pnas.0400924101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, Cervera P, Le Bouc Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- 27.Ban T, Hoshino M, Takahashi S, Hamada D, Hasegawa K, Naiki H, Goto Y. Direct observation of Abeta amyloid fibril growth and inhibition. J Mol Biol. 2004;344:757–767. doi: 10.1016/j.jmb.2004.09.078. [DOI] [PubMed] [Google Scholar]
- 28.Andersen CB, Yagi H, Manno M, Martorana V, Ban T, Christiansen G, Otzen DE, Goto Y, Rischel C. Branching in amyloid fibril growth. Biophys J. 2009;96:1529–1536. doi: 10.1016/j.bpj.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LeVine H., III Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999;309:274–284. doi: 10.1016/s0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- 30.O'Nuallain B, Williams AD, Westermark P, Wetzel R. Seeding specificity in amyloid growth induced by heterologous fibrils. J Biol Chem. 2004;279:17490–17499. doi: 10.1074/jbc.M311300200. [DOI] [PubMed] [Google Scholar]
- 31.Krebs MR, Morozova-Roche LA, Daniel K, Robinson CV, Dobson CM. Observation of sequence specificity in the seeding of protein amyloid fibrils. Protein Sci. 2004;13:1933–1938. doi: 10.1110/ps.04707004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmidt M, Sachse C, Richter W, Xu C, Fandrich M, Grigorieff N. Comparison of Alzheimer Abeta(1–40) and Abeta(1–42) amyloid fibrils reveals similar protofilament structures. Proc Natl Acad Sci U S A. 2009;106:19813–19818. doi: 10.1073/pnas.0905007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paravastu AK, Leapman RD, Yau WM, Tycko R. Molecular structural basis for polymorphism in Alzheimer's beta-amyloid fibrils. Proc Natl Acad Sci U S A. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R. 3D structure of Alzheimer's amyloid-beta(1–42) fibrils. Proc Natl Acad Sci U S A. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jarrett JT, Berger EP, Lansbury PT., Jr. The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 36.Toeroek M, Milton S, Kayed R, Wu P, McIntire T, Glabe CG, Langen R. Structural and Dynamic Features of Alzheimer's Abeta Peptide in Amyloid Fibrils Studied by Site-Directed Spin Labeling. J Biol Chem. 2002;277:40810–40815. doi: 10.1074/jbc.M205659200. [DOI] [PubMed] [Google Scholar]
- 37.Atarashi R, Moore Roger A, Sim Valerie L, Hughson Andrew G, Dorward David W, Onwubiko Henry A, Priola Suzette A, Caughey B. Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat Methods. 2007;4:645–650. doi: 10.1038/nmeth1066. [DOI] [PubMed] [Google Scholar]
- 38.Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411:810–813. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 39.Soderberg L, Dahlqvist C, Kakuyama H, Thyberg J, Ito A, Winblad B, Naslund J, Tjernberg Lars O. Collagenous Alzheimer amyloid plaque component assembles amyloid fibrils into protease resistant aggregates. FEBS J. 2005;272:2231–2236. doi: 10.1111/j.1742-4658.2005.04647.x. [DOI] [PubMed] [Google Scholar]
- 40.Tjernberg LO, Naaslund J, Thyberg J, Gandy SE, Terenius L, Nordstedt C. Generation of Alzheimer amyloid beta peptide through nonspecific proteolysis. J Biol Chem. 1997;272:1870–1875. doi: 10.1074/jbc.272.3.1870. [DOI] [PubMed] [Google Scholar]
- 41.Jankowsky JL, Savonenko A, Schilling G, Wang J, Xu G, Borchelt DR. Transgenic mouse models of neurodegenerative disease: opportunities for therapeutic development. Curr Neurol Neurosci Rep. 2002;2:457–464. doi: 10.1007/s11910-002-0073-7. [DOI] [PubMed] [Google Scholar]
- 42.Reilly JF, Games D, Rydel RE, Freedman S, Schenk D, Young WG, Morrison JH, Bloom FE. Amyloid deposition in the hippocampus and entorhinal cortex: quantitative analysis of a transgenic mouse model. Proc Natl Acad Sci U S A. 2003;100:4837–4842. doi: 10.1073/pnas.0330745100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmidt SD, Nixon RA, Mathews PM. ELISA method for measurement of amyloid-beta levels. Meths Mol Biol. 2005;299:279–297. doi: 10.1385/1-59259-874-9:279. [DOI] [PubMed] [Google Scholar]
- 44.Bellotti V, Mangione P, Merlini G. Review: Immunoglobulin Light Chain Amyloidosis-The Archetype of Structural and Pathogenic Variability. J Struct Biol. 2000;130:280–289. doi: 10.1006/jsbi.2000.4248. [DOI] [PubMed] [Google Scholar]
- 45.Comenzo RL. Systemic immunoglobulin light-chain amyloidosis. Clin Lymphoma Myeloma. 2006;7:182–185. doi: 10.3816/CLM.2006.n.056. [DOI] [PubMed] [Google Scholar]
- 46.Golde TE, Dickson D, Hutton M. Filling the gaps in the Abeta cascade hypothesis of Alzheimer's disease. Curr Alzheimer Res. 2006;3:421–430. doi: 10.2174/156720506779025189. [DOI] [PubMed] [Google Scholar]
- 47.Hammarstrom P, Wiseman RL, Powers ET, Kelly JW. Prevention of Transthyretin Amyloid Disease by Changing Protein Misfolding Energetics. Science. 2003;299:713–716. doi: 10.1126/science.1079589. [DOI] [PubMed] [Google Scholar]
- 48.Johnson SM, Wiseman RL, Sekijima Y, Green NS, Adamski-Werner SL, Kelly JW. Native State Kinetic Stabilization as a Strategy To Ameliorate Protein Misfolding Diseases: A Focus on the Transthyretin Amyloidoses. Acc Chem Res. 2005;38:911–921. doi: 10.1021/ar020073i. [DOI] [PubMed] [Google Scholar]
- 49.Sekijima Y, Wiseman RL, Matteson J, Hammarstrom P, Miller SR, Sawkar AR, Balch WE, Kelly JW. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121:73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 50.Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 51.Wahrle SE, Holtzman DM. Neurobiol. Alzheimer's Dis. 3rd Ed. 2007. Apolipoprotein E, amyloid beta peptide, and Alzheimer's disease; pp. 161–172. [Google Scholar]
- 52.Colby DW, Zhang Q, Wang S, Groth D, Legname G, Riesner D, Prusiner SB. Prion detection by an amyloid seeding assay. Proc Natl Acad Sci U S A. 2007;104:20914–20919. doi: 10.1073/pnas.0710152105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coria F, Castano E, Prelli F, Larrondo-Lillo M, Van Duinen S, Shelanski ML, Frangione B. Isolation and characterization of amyloid P component from Alzheimer's disease and other types of cerebral amyloidosis. Lab Invest. 1988;58:454–458. [PubMed] [Google Scholar]
- 54.Suk Ji Y, Zhang F, Balch William E, Linhardt Robert J, Kelly Jeffery W. Heparin accelerates gelsolin amyloidogenesis. Biochemistry. 2006;45:2234–2242. doi: 10.1021/bi0519295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.