

Abstract
Medulloblastoma (MB) is the most common malignant pediatric brain tumor and is thought to arise from genetic anomalies in developmental pathways required for the normal maturation of the cerebellar cortex, notably developmental pathways for granule cell progenitor (GCP) neurogenesis. Over the past decade, a wide range of studies have identified genes and their regulators within signaling pathways, as well as non-coding RNAs, that have critical roles in both normal cerebellar development and pathogenesis. These include the Notch, Wnt/β-catenin, Bone Morphogenic Proteins (Bmp) and Sonic Hedgehog (Shh) pathways. In this review, we highlight the function of these pathways in the growth of the cerebellum and the formation of MB. A better understanding of the developmental origins of these tumors will have significant implications for enhancing the treatment of this important childhood cancer.
INTRODUCTION
In their classical treatise on brain tumors, Bailey and Cushing postulated that, “the histogenesis of the brain furnishes the indispensable background for an understanding of its tumors” [1]. Genetic and molecular analyses of normal cerebellar development and medulloblastomas (MB) have identified changes in the regulation of signaling pathways that are important for cerebellar progenitor cell neurogenesis. These pathways include those which promote growth, such as Sonic hedgehog (Shh), Wnt, Notch, Insulin growth factor (IGF)/ phosphatase and tensin homolog (PTEN)/mammalian target of Rapamycin (mTOR), as well as pathways that inhibit growth [eg. bone morphogenic protein (Bmp) and basic fibroblast growth factor (bFGF) signaling pathways]. Molecular changes in pathways associated with MB development include both loss and gain of function mutations, as well as alterations in the mRNA or protein levels of regulators within these pathways. Here, we review neurogenesis and cell type specification in the developing cerebellum, and then discuss the etiology of MBs and how pathways that function in normal development are involved in tumor formation and proliferation.
OVERVIEW OF CEREBELLAR DEVELOPMENT
The cerebellar cortex is a remarkably simple laminar structure [2]. The most prevalent neuronal subclass in the cerebellum, indeed within the entire mammalian central nervous system (CNS), is the cerebellar granule neuron. Granule neurons serve an essential role in coordinating afferent input to, and motor output from, the cerebellum through their excitatory connections with Purkinje neurons. While the role(s) of the cerebellum in sensorimotor functions, balance control and the vestibular ocular reflex have long been appreciated, recent studies have revealed a role for the cerebellum in a wide range of cognitive functions, including feed-forward sensory-motor learning, speech and spatial memory [3–9]. Notably, a loss of spatial memory and other cognitive functions have been reported in children after successful tumor resection [10]. Although the function of the cerebellum in learning and memory is complex, the remarkably simple architectonics of the cerebellum make it an attractive model system for studying CNS tumors, especially developmental tumors such as MB.
During embryogenesis, the cerebellar territory arises from rhombomere 1 in a zone bounded by the transcription factors Otx2 and Hoxa2 [11–13]. In addition to its important role in cerebellar development, Otx2 is amplified and over-expressed in MBs, suggesting that Otx2 is an oncogenic driver for MB [14]. Beginning on about embryonic day 10.25 (E10.25) in the mouse, a complex pattern of neurogenesis and cell movements generate the cerebellar cortex and cerebellar nuclei [15]. Classical neuroanatomical studies indicate that the dorsomedial ventricular zone (VZ) along the IVth ventricle gives rise to the principal output neuron of the cerebellar cortex, the Purkinje cell, neurons of the cerebellar nuclei and more than half a dozen types of cerebellar interneurons, including Golgi, basket and stellate cells [2, 16, 17]. A secondary germinal zone forms along the anterior aspect of the rhombic lip, which generates the cerebellar granule neuron, as well as a subpopulation of neurons of the cerebellar nuclei [18, 19] and neurons of several pre-cerebellar nuclei of the “cerebellar system” [13, 19–22] (Figure 1).
Figure 1. Neurogenic Zones in Embryonic Cerebellar Histogenesis.

In the embryonic cerebellar anlagen, the vast majority of cerebellar neurons, including Purkinje neurons (the major output neurons of the cerebellum), neurons of the cerebellar nuclei, more than half a dozen types of interneurons and cerebellar astroglia arise in the ventricular zone (VZ) lining the IVth ventricle (green). Different classes of cerebellar neurons are generated in a precise sequence, with neurons of the deep nuclei being generated first [18, 28], followed by Purkinje neurons and interneurons. Progenitors of the cerebellar nuclei migrate through the thickening wall of the anlage along the processes of radial glial cells to establish an external zone, which moves to a position deep to the Purkinje cells, as immature Purkinje neurons migrate away from the VZ and as GCPs migrate into the EGL, beginning at about E12 in the mouse [28]. GCPs emerge in a secondary neurogenic zone along the edge of the neuroepithelium in a zone called the rhombic lip (RL, purple). Proliferating precursors in this zone move onto the surface of the emerging anlage, where they form the EGL (purple), which gives rise to the GCPs, a subpopulation of neurons of the cerebellar nuclei [19, 21] and neurons of the lateral pontine nucleus in the brainstem [13]. Cells in the EGL (inset, purple cells) include mitotic figures and small cells with long migratory processes. Coronal view. BS, brainstem; CB, cerebellum; CP choroid plexus; EGL, external germinal layer; MB, midbrain; RL, rhombic lip.
Recent genetic experiments demonstrate that granule cell progenitors (GCPs) undergo predominantly symmetric divisions during early postnatal external germinal zone (EGL) development and that clonally related granule cells exit the cell cycle within a narrow time frame [23]. In agreement with earlier reports [24, 25], these studies show a progressive slowing of GCP proliferation just before birth, and rapid expansion of distinct clones of granule cells just prior to cell cycle exit in the postnatal period [23]. Evidence from the Gene Expression Nervous System Atlas (GENSAT) Project, which has generated transgenic mouse lines that express the enhanced green fluorescent protein (EGFP) reporter gene in a large variety of CNS cell types [26], demonstrates that some genes that mark the GCP lineage are not expressed in all GCPs at all developmental stages (see GENSAT website for details: www.gensat.org). These findings are consistent with the idea that there may be different subsets of GCPs, some of which are relevant to the development of specific subtypes of MBs [27] (BOX 1).
BOX 1. WHAT IS THE CELL OF ORIGIN OF MBs?
One of the critical questions in both normal cerebellar development and MB formation is whether the vast population of GCPs includes subpopulations of progenitors with distinct genetic properties and whether these subpopulations contribute to different subgroups of MBs (27, 32). While MBs with a SHH signature originate from GCPs in the EGL that sustain mutations in the SHH signaling pathway, MB cells with an activated WNT signaling pathway instead arise from progenitors born in the lower rhombic lip of the cerebellum in the floor of the IVth ventricle. The cell of origin for the other two subgroups of MBs has yet to be elucidated. Studies from the GENSAT Project, a large-scale CNS gene expression atlas, which has generated more than 600 lines of transgenic lines of mice that express the enhanced green fluorescent protein (EGFP) reporter genes in a large variety of CNS cell types [26] demonstrates that some genes in a subset of GCPs at particular developmental stages, GENSAT website: www.gensat.org)These studies raise the intriguing possibility that early postnatal GCPs are not a homogenous population, but instead represent a subsets of progenitors possibly with different patterns of connectivity and/or different susceptibility to tumorigenesis. The precise identification of subsets of normal GCPs responsible for each subgroup of MBs will require genetic fate mapping studies and massive parallel sequencing analysis of single cells to define genetic differences/alterations. These studies will increase our understanding of cerebellar circuitry and function, and will advance the development of novel targeted therapies for MBs, each with a specific genetic signature.
Although the role of radial glia in patterning the cerebellar lamina has been described [28] , the neurogenic role of cerebellar radial glia has not been characterized in detail. Molecular genetic studies on transgenic lines of mice expressing the radial glial gene Blbp [29, 30] show that radial glia are neurogenic in all brain regions, including the cerebellum [29]. Biochemical and molecular genetic studies further demonstrate that Blbp is a downstream target of the Notch signaling pathway [31]. These studies raise the question as to whether neuron-glial interactions regulate cerebellar neurogenesis via neurogenic radial glia. As discussed below, recent studies identify Blbp-expressing cells originating in the cerebellar VZ as a cell of origin of classic MBs [32].
Mitogenic Pathways that Promote GCP Proliferation
In the early postnatal period, multiple mitogenic pathways drive the rapid expansion of the pool of GCPs in the EGL that generate the enormous population of granule neurons in the cerebellar cortex (Figure 2). The discovery of Shh, the first key mitogen that promotes GCP neurogenesis [33] followed the observation that mutations in the twelve transmembrane receptor Patched1 (Ptch1), a gene that binds Shh and functions as an antagonist of Shh signaling, promotes MB in mouse models of the disease and is associated with sporadic human MB (27, 33). The importance of Shh to cerebellar histogenesis is underscored by elegant genetic analyses demonstrating that levels of Shh signaling control the foliation patterning of the cerebellar cortex [34]. A large body of evidence now implicates defects in Shh signaling in MB (reviewed in [27]).
Figure 2. Changing Patterns of Gene Expression and Signaling Pathways in Cerebellar Granule Cell Progenitor Expansion.

(A) On E12.5 in the mouse, Atoh1/Math1 is expressed along the dorsal ridge of the spinal cord and in both the anterior and posterior rhombic lip. Adapted, with permission, from [76]. (B) In the postnatal cerebellar cortex, Atoh1/Math1 (green) is highly expressed in proliferating GCPS in the outer aspect of the EGL along the surface of the cerebellum (adapted, with permission, from [66])(C) As dividing GCPs (green) exit the cell cycle, they express p27Kip (red) (adapted, with permission, from [66]). (D) Four major pathways control GCP growth. The Shh pathway induces expression of the transcription factor Gli1, which up-regulates expression of cyclins (Ccnd D1, 2) and Mycn. Canonical Wnt signaling activates the b-catenin pathway, which up-regulates expression of both c-Myc and Mycn. Jag1 activates Notch2 signaling, which promotes proliferation and inhibits cell cycle exit, leading to a sustained expression of the bHLH transcription factor Atoh1/Math1. In contrast, Bmps are negative growth regulators. BMP2 and BMP4 induce expression of the transcription factor Id1,2, which inhibits Atoh1 expression, leading to cell cycle exit and neuronal differentiation.
Shh appears to regulate GCP proliferation by several mechanisms. In the absence of Shh, Ptch represses the function of Smoothened (Smo), a seven transmembrane G-protein-coupled receptor-like protein that activates the zinc finger protein transcription factors Gli1 and Gli2 and inactivates the transcriptional repressor Gli3. Together these proteins regulate the transcriptional program in the cell nucleus [33]. Studies on the inactivation of Gli alleles in Ptch1 heterozygous mouse models for MB suggest that Gli1 functions in the transformation of GCPs and highlights the key role of Shh pathway activation in MB [35]. Interestingly, the primary cilium, a cellular structure essential to cell proliferation and function, concentrates components of Shh signaling in cerebellar GCPs and is required for Shh-dependent GCP expansion [36]. In the absence of Shh stimulation, the three Gli transcription factors and their binding partner, Suppressor of Fuse (Sufu), are expressed in primary cilia together with Ptch. In the presence of Shh, Smo is recruited to the cilia to replace Ptch [37] and Gli proteins are activated.
Patients with Gorlin syndrome (also called Nevoid Basal Cell Carcinoma Syndrome) sustain germline mutations in PTCH1 and SUFU, predisposing them to multiple cancers, including MB [27]. Mouse models of MB with germline or conditional loss of Ptch1 develop MB with histopathological features characteristic of the human desmoplastic MB variant in half of the cases, whereas the other half express anaplastic features (Figure 3). Taken together, this SHH-subtype represents ~ 25–30% of all human MBs [38–42]. Molecular analysis of sporadic human MB primary samples with constitutive activation of the Shh signaling pathway revealed loss of PTCH1 and mutations in SUFU [43] and SMO [42]. Mice lacking Sufu develop MB in conjunction with the loss of tumor related protein gene (Trp53 also known as p53, or TP53 in humans), demonstrating that Sufu functions as a tumor suppressor gene [44].
Figure 3. Features of Human MB.
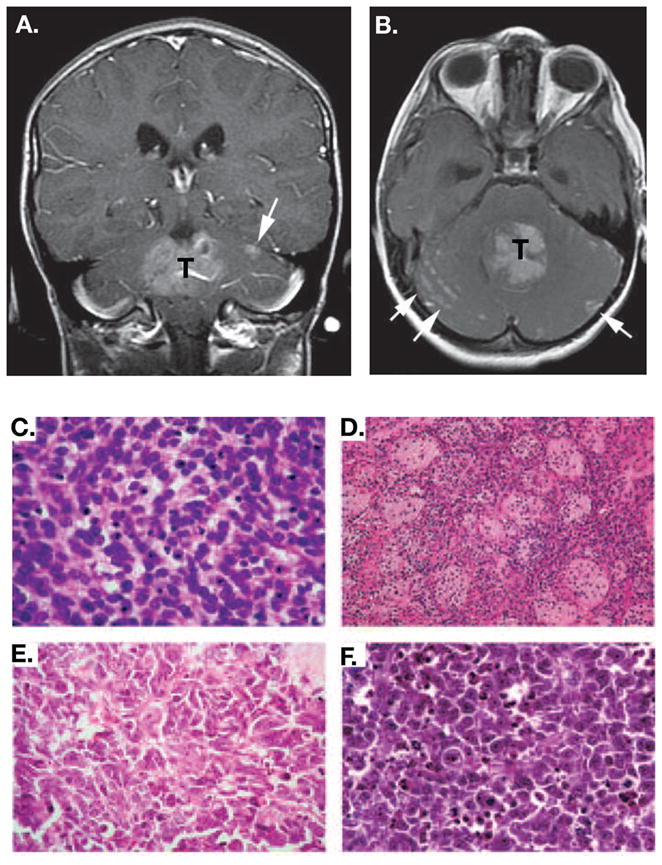
MBs are diagnosed by imaging studies (MRI and CT scans) and histopathology. An axial (A) and a coronal (B) contrast-enhanced T1 weighted image are shown in which the enhancing tumor is labeled with T and metastases are evident in the subarachnoid space (white arrows). Adapted, with permission, from [86]. Histological variants of MB include: (C) Classic MB, which is typified by a field of small uniform cells with large nuclei, (D) The nodular/desmoplastic MB, which combines nodules of differentiated neurocytic cells with a low growth fraction and desmoplastic internodular zones of pleomorphic cells with a high growth fraction, (E) The anaplastic MB, which contains polymorphic cells with a high growth fraction; extensive apoptosis is also evident, and (F) Large cell MB, which contains groups of large cells with vesicular nuclei and a single nucleolus. Anaplasia is evident in other regions of this variant. Adapted, with permission,from [27].
The Notch2 pathway also stimulates the expansion of GCPs during cerebellar development (Figure 2). Treatment of GCPs with Jagged 1 (Jag1), a ligand of Notch2, stimulates proliferation of GCPs 5–8 fold and inhibits granule neuron differentiation [45]. In addition, the basic helix-loop-helix (bHLH) transcription factor, Hes1, is up-regulated downstream of Notch2 activation by Jag1 and over-expression of Hes1 stimulates GCP proliferation [45]. Subsequent genetic studies demonstrate that one mechanism for the action of activated Notch2 involves antagonizing BMP signaling [46] and increasing Atoh1/Math1 expression [46]. This conclusion is consistent with gene expression studies of Ptch1 heterozygous mice with elevated Shh signaling, which have increased levels of expression of components of the Notch and Wnt pathways. Moreover, genetic studies on mice with reduced Shh signaling demonstrate a down-regulation of expression of Notch2, Jag1 and Hes1 [47], suggesting that Notch is downstream of Shh signaling.
Negative Regulators of GCP Proliferation
GCP development also depends on signals that negatively regulate proliferation and signals that promote GCP cell cycle exit and differentiation. Several molecules inhibit GCP growth, in part by antagonizing Shh-mediated proliferation of GCPs. These include bFGF [48] and members of the Bmp family (Figure 2). Earlier studies on cerebellar histogenesis showed that Bmps are initially required to specify granule neuron identity [49]. Later, in the early postnatal period, Bmp2 and Bmp4 are expressed in the inner portion of the EGL in postmitotic GCPs that are undergoing neuronal differentiation (50, 51). In vitro assays indicate that Bmp2 and Bmp4 inhibit Shh-induced GCP proliferation [50] via the Smad1 signaling pathway [50, 51]. Moreover, Bmp4 antagonizes Shh signaling and induces differentiation of GCPs by rapid post-transcriptional turnover of Atoh1/Math1 [52].
Differentiation of Embryonic Stem (ES) Cells to a Granule Cell Identity
To demonstrate the role of key developmental regulators in granule cell development [15], mouse ES cells that were treated with inducers of GCP identity in a stepwise manner differentiated into granule cells [53]. These treatments included culturing ES cells with “cerebellar organizers” [ie. Fgf8 and retinoic acid (RA)], followed by the local signals that “dorsalize” the cerebellar anlagen [ie. Wnt1, Wnt3a, Bmp6, Bmp7 and growth differentiation factor 7(Gdf7)], followed by BMPs that specify a GCP identity (ie. Bmp 6, 7 and GDF7 ) [49]. Subsequently, the cells were treated with mitogens (eg. Shh, Jag1) that expand the early postnatal GCP population [53]. Terminal differentiation was achieved by culturing the differentiating cells in medium that had been preconditioned with purified cerebellar glial cells. After these treatments, which parallel known steps of GCP differentiation in vivo [53], ES cells expressed multiple markers of GCPs, including the transcription factors En1, Atoh1/Math1, Zic1, Zic2, Pax6 and the α6 subunit of the GABAA receptor. To test the ability of these differentiated ES cells to functionally integrate into the cerebellar cortex in vivo, these cells were implanted into the cerebellar cortex of neonatal mice by stereotaxic injection. After 3 days to 2 weeks, cells migrated into the inner granular layer (IGL), the position where mature granule neurons undergo terminal differentiation and form synaptic connections with mossy fiber afferent axons. Thus, ES cell differentiation confirms the role of local signals in normal cerebellar neuron specification [28, 53] and provides an initial strategy for CNS cell replacement therapy.
CLASSIFICATION OF HUMAN MBS
Human MBs are diagnosed by a combination of imaging studies [magnetic resonance imaging (MRI) and X-ray computed tomography (CT) scans] and histopathology (Figure 3). Five subgroups of MBs have been classified by the World Health Organization (WHO) according to their severity (from best to worst prognosis) and histopathology: classic, desmoplastic, nodular, anaplastic and large cell anaplastic (LCA) [54] (reviewed in [27]) (Figure 3C–F). More recently, gene expression profiling has provided a second and more precise method for classifying subtypes of MBs [55, 56]. Transcriptional profiling of mRNAs in human MBs [55–58] has identified 4 subtypes of MB with distinct mRNA signatures. These molecular subtypes are distinct with respect to the underlying signaling pathway which is dysregulated in MBs, and include alterations in the (i) Wnt pathway, (ii) Shh pathway, (iii) Myc signaling and (iv) undefined genetic anomalies (Figure 4 and discussed further below). It is important to point out that there is incomplete overlap between the histopathologic and molecular subtypes of MBs. Thus although half of the SHH MBs, which arise from the constitutive activation of SHH signaling in GCPs [27, 39, 55, 56] have desmoplastic histology, the remaining SHH MBs have anaplastic histological features (Figure 4). Furthermore, in addition to genetic loss or gain of function, defects in epigenetic regulation and aberrant action of tumor suppressor genes have been reported in MBs (reviewed in [59]).
Figure 4. Subtypes of MB are Defined by Histopathology or Molecular Features.

Representation of two current classifications of MB subtypes, one based on histopathological features and the other on gene expression, and correlated outcome. The 5 subtypes of MBs defined by histopathology do not strictly overlap with the 4 subtypes defined by molecular analysis. At present, the most aggressive molecular subtype of MBs have undefined genetic anomalies.
Information about the cell type of origin of MB subtypes, diagnosed by histological or molecular criteria, would provide a major advance in understanding MBs (Box 1). A number of studies show GCPs are one cell of origin for some MBs [27], and recent studies identify Blbp-expressing cells originating in the cerebellar ventricular zone as a cell of origin of the classic WNT MB subtype [32], the MB variant with the best prognosis [60].
The Shh/ Ptch Signaling Pathway and Its Targets, Gli1, Gli2, Mycn and Cyclins, in MBs
Activation of the Shh pathway up-regulates cyclins (Ccnd1 and Ccnd2), which function in regulating the cell cycle, and expression of the proto-oncogene Mycn, a bHLH-transcription factor [63] (Figure 2d, Table 1). Conversely, Mycn suppresses the expression of two cyclin-dependent kinase (CdK) inhibitory proteins, p18Ink4c (Ink4c) and p27Kip1 (Figure 2C), which in turn induces the phosphorylation of the tumor suppressor proteins retinoblastoma (Rb) and p107, resulting in cell cycle progression [64, 65]. While Rb and p107 are required for normal cerebellar development and GCP survival, their loss in mice in conjunction with loss of Trp53 induces MBs [40, 61]. Studies in mice show that p18Ink4c is transiently expressed in GCPs to time their exit from the cell cycle, whereas the expression of p27Kip1 is induced in post-mitotic GCPs in the inner EGL and in the IGL [66]. Loss of Ink4c combined with loss of Ptch1 or Trp53 induces MB in mice [66]. Although mutations in Trp53 represent only ~10% of all human MBs and occur in all MB variants [58], Li-Fraumeni patients with familial TP53 mutations are prone to developing tumors, including MBs.
TABLE 1.
Important signaling pathways in cerebellar development and associated dysfunction in different subtypes of MB
| Signaling Pathway | Normal Pathway Function | Disease | Mutations/Abnormalities | MB Subtype | Mechanism of Oncogenesis | Refs. |
|---|---|---|---|---|---|---|
| Wnt | Cell proliferation | Turcot’s Syndrome1 MB | β-Catenin, APC, Monosomy 6 | WNT | Up-regulated transcription of proliferative genes | [27, 70] |
| Shh/Ptch | Cell proliferation | Gorlin Syndrome2 | Germline loss of function in Ptch1 and Sufu | SHH | Increased cell division, loss of cell cycle exit | [85] |
| Notch2 | Cell Proliferation | MB | Notch2, Jag1, Hes1 over expression | Unknown | Loss of cell cycle arrest | [71] |
| Bmp2,4 | Cell cycle arrest, neuronal differentiation | Gorlin Syndrome | BMP pathway genes down-regulated | SHH | Down regulation of expression, loss of cell cycle arrest, differentiation | [50, 51] |
Turcot’s syndrome is a rare syndrome considered to be an alternative form of familial adenomatous polyposis (FAP) that is associated with an increased risk of developing MB. FAP or APC-associated polyposis conditions include: FAP, attenuated FAP, Gardner Syndrome and Turcot Syndrome. FAP is also a colon cancer predisposition syndrome, beginning, on average, at age 16 years (range 7–16 years).
Gorlin syndrome (basal cell nevus syndrome or nevoid basal cell carcinoma syndrome) is a rare, autosomal dominant cancer syndrome with abnormalities in the skin, skeleton and nervous system.
Members of the MYC family are frequently overexpressed in MBs (reviewed in [62, 67]). Indeed, expression of Myc, or Mycn, in GCPs either from Trp53-null or Ptch1 heterozygous mice induces fully penetrant MB (100%) after transplantation into the cortices of naïve recipient animals [68]. These results are in agreement with a recent study [67] showing that targeted expression of MYCN functions in the initiation, progression, and maintenance of MB. Taken together, these studies suggest a central role for MYC family members in MB pathogenesis [67].
Wnt Signaling Pathways in MB
Deregulated WNT signaling occurs in ~ 10–15% of human MBs (27, 32). Mutations in adenomatous polyposis coli (APC) were first identified in patients with Turcot’s syndrome in combination with colon cancer and malignant neuroepithelial brain tumors, including MBs (reviewed in [27, 69]). Subsequently, mutations in BETA-CATENIN (CTNNB1), an integral component of the Wnt signaling pathway, were discovered in a subset of human MBs [58]. This particular MB subpopulation was found to also harbor a single copy loss of chromosome 6 (also called monosomy 6), which represents the most significant prognostic factor for the WNT-subtype of tumors that correlates with a good outcome [60]. Finally, the secreted frizzled-related protein (sFRP) family of Wnt inhibitors, sFRP-1, -2 and -3, that can limit both canonical and non-canonical WNT signaling, are often epigenetically silenced in MBs, which can contribute to excessive WNT signaling in these tumors. Overexpression of these SFRP proteins reduces the proliferation and anchorage-independent growth of MB cells, which limits tumor burden and prolongs survival in xenograft models of MB in mice [70].
Notch2 Signaling in MB
As mentioned earlier, Notch family members play important roles in regulating normal GCP expansion. In agreement with studies on developing GCPs[45], which showed that activated Notch2, but not Notch1, promotes GCP proliferation, studies of Notch protein levels in human medulloblastomas show enhanced levels of Notch2, but not Notch 1 or Notch 3 [71]. Importantly, the percentage of tumor cells immunopositive for NOTCH2 correlates with a higher tumor grade [71]. Expression of truncated, constitutively active forms of Notch2 also promote oncogenic growth [72], in support of the idea that activation of Notch2, but not Notch1, promotes tumor formation. Recent measurements of the mRNA levels of NOTCH2, and its downstream target HES1, mRNAs in 40 human embryonal brain tumors showed enhanced expression in 15% of the tumors [71]. Taken together, these findings suggest that activation of Notch2 promotes GCP neurogenesis and that defects in the control of Notch2 in GCP mitogenesis are associated with MB [72]. This conclusion is supported by the observation that Notch2, Hes1 and Jag1 are all expressed at high levels in mouse MB models [47, 73].
BMPs are Negative Regulators of MBs
In addition to mitogenic pathways, several pathways inhibit GCP expansion in the early postnatal period of development. Two studies report that BMPs oppose Shh-induced proliferation in MBs with a Shh-subtype and induce terminal neuronal differentiation. Bmp2 antagonizes Shh-dependent proliferation by expressing TIEG-1, which directly represses Mycn transcription [74]. In turn, TIEG-1 over expression promotes cell cycle arrest and apoptosis of GCPs in the absence of other differentiation signals (74). In addition, rapid post-transcriptional down regulation of Atoh1/Math1, a bHLH transcription factor required for cerebellar development [75, 76], functions in MB formation, since over expression of Math1/Atoh1 blocks differentiation [51]. This suggests that the Bmp pathway antagonizes Shh signaling by blocking cell division and inducing terminal neuronal differentiation by mechanisms that involve changes in the levels of Atoh1 [52]. Gene expression profiling of mouse and human MBs with a constitutively activated SHH pathway reveals that most genes in the BMP signaling pathway are down-regulated in these tumor cells compared with normally developing GCPs [51].
Epigenetic Silencing in MBs
In addition to the loss of key gene functions by gene deletions or loss of function mutations, epigenetic silencing plays an important role in the development of MBs. A survey of the methylation status of tumor suppressors or oncogenes in human MBs has revealed several epigenetic silencers: sFRPs that negatively regulate the WNT pathway [70], the S100 calcium protein gene family [59], the Kruppel-like factor 4 transcription factor [78], and hypermethylated in cancer-1 (HIC1), a tumor suppressor protein that regulates ATOH1/MATH1 gene expression [77]. HIC1 suppresses Atoh1/Math1 mRNA levels in normal GCPs, however, hypermethylation of the promoter of HIC1 in human MBs results in a suppression of its expression, which subsequently results in elevated levels of ATOH1/MATH1 [77]. A recent study has also revealed the presence of inactivating mutations in the histone-lysine N-methyltransferase genes MLL2 and MLL3 in a subset of human MBs [91]. The underlying genetic and cellular mechanism through which MLL genes contribute to tumorigenesis are not known, however, they have been shown to be important for the transcriptional regulation of a number of pathways during normal cerebellar development, including in the regulation of Hox genes [92], as well as potentially regulating genes involved in the Wnt pathway [93]. Thus, epigenetic modifications are likely to be important during development of the cerebellum as well being responsible for initiating some MBs.
MicroRNAs and MB
In the last 5 years, microRNAs (miRNAs) have emerged as novel regulators of many aspects of cell biology and developmental processes, including in the CNS. MicroRNAs are small non-coding 22 nucleotides long RNAs with sequences complementary to those in the 3’-UTR of targeted messenger RNAs. MicroRNAs are thought to regulate hundreds of mRNA targets. Several miRNAs have been demonstrated to be overexpressed in MBs, suggesting they may act as oncogenes, whereas others are down-regulated, suggesting they may function as tumor suppressors (79, 80). Two independent studies have identified the miRNA miR-17-92 cluster as an oncogene in MBs with a SHH signature [79, 80]. The miR-17-92 cluster belongs to a family of three clusters encoded by different chromosomes in mouse and human [81]. It encodes 6 unique microRNAs (miR-17a,b, 18, 19, 20 and 92) that share common seed sequences with miRNAs encoded by the two other clusters of the family (80, 81). MiR-17-92 was found to be a direct target of c-Myc [82]. Although several important targets of the miR-17-92 cluster have been identified in B cells and B cell lymphoma (81), direct targets have not yet been identified in cerebellar neurons or MBs. An interesting feature of miR-17-92 is that its over-expression drives tumorigenesis in collaboration with Ptch1 [80], suggesting that it is part of the SHH signaling pathway. If this cluster is required for MB tumorigenesis, anti-miRNAs to this cluster may provide a novel therapeutic approach for MBs, at least for tumors of the SHH-subtype.
Mouse Models of MB
Considering the large diversity of human MBs, it is remarkable that most current mouse models of MB recapitulate MBs of the SHH-subtype [39]. This is true for mice with a range of genetic anomalies, including mutations in Sufu, Smo, loss of one copy of Ptch1 alone or together with loss of p18Ink4c or p27Kip1, loss of Rb, hypermethylation of HIC1, loss of Ligase 4, Xrcc4, Brca2 and Parp, enzymes involved in DNA repair, often in combination with the loss of Trp53 (reviewed in [61]). Expression of Myc or ,Mycn ,or miR-17-92, induced MB development only in combination with loss of Ptch1 or loss of Trp53 [65, 80].
One exception, reported recently, involves the generation of MB of the WNT-subtype by the conditional expression of activated Beta-catenin in collaboration with the loss of Trp53 in cells expressing a Blbp transgene that is expressed in all cells of the VZ of both the cerebellar and hindbrain territories [32]. This finding is of interest as WNT-subtypes MBs arise from neural progenitors in the VZ (the primary neurogenic zone,), rather than in the EGL, a secondary neurogenic zone, and the anterior rhombic lip (RL), which generate GCPs.
Another interesting point of note is that most of the mouse models for MB require a loss of Trp53 for MB formation, even though mutations in TP53 are not common in human MBs (occurring in only ~ 10% of cases (58)). This may reflect a key role for the Trp53 checkpoint in normal mouse GCP development, which may not necessarily be the case in humans. Mouse models that faithfully recapitulate these human WNT- and SHH-MB subtypes have been developed [61]. However, mouse models for the MYC subgroup and the unspecified genetic subgroup [56, 58, 62] have not been generated and are urgently needed.
SUMMARY
Comparative studies on the development of the cerebellar cortex, especially on the molecular control of GCP neurogenesis, and MB formation have identified key positive and negative regulators of GCP proliferation and genetic pathways involved in cerebellar cancers. Although some features of mouse development, such as an apparently critical role for Trp53 in mouse tumorigenesis, are not as prominent in human MBs, the vast majority of primary signaling pathways implicated in normal mouse and human cerebellar development are aberrant in human and mouse models of MBs. Genetic and molecular analyses of potential subsets of normal GCPs and of the cell of origin of subgroups of human MBs are very likely to lead to a more rational approach for the discovery of critical pathways and novel therapeutic targets (Box 2). These ongoing investigations will be enhanced by more extensive gene expression data for GCPs, as well as other cerebellar neural progenitors. The analysis of transformed neurons in human MBs using next generation technologies, such as translating ribosomal affinity purification (TRAP) [83, 84] and massively parallel sequencing can be expected to identify novel genes that are involved in these signaling pathways. It is hoped that improved understanding of the underlying neurobiological pathways that are aberrant in MB will lead to improved therapeutic treatments for patients diagnosed with MB.
BOX 2. CURRENT AND FUTURE THERAPIES FOR MBs.
Current therapies to treat MBs include surgery, chemotherapy and radiation [87,88]. However, the increasing understanding of the signaling pathways and genetic anomalies associated with the different subtypes of human MB has led to the discovery of the first small molecule inhibitors of the SHH signaling pathway [89]. Remarkably, small molecule inhibitors of SMO have been shown to completely suppress the development of Shh-subtype MBs in genetic mouse models of MB, as well as in allografts in mice[89]. These compounds have recently entered phase I clinical trials [90]. Although they are very effective in reducing and eliminating tumor cells in mice, recent preliminary results from these trials have revealed that one patient who had displayed a remarkable remission of the disease had an aggressive recurrence due to a mutation in SMO that rendered the tumor cells insensitive to the drug [42]. Thus, although small molecular inhibitors of the SHH pathway show immense promise as future therapeutics for treating MBs with a SHH signature, other novel targeted therapies are still in critical need.
Acknowledgments
We thank Dr. James Barkovich (UCSF) for kindly providing the MRI images in Figure 3. We are grateful to David Murphy for editing the manuscript and to Drs. Hourinaz Behesti, Jane Johnson and Linda Van Aelst for critically reading the manuscript. Supported by the Starr Cancer Consortium (MEH), National Institutes of Health (NIH) R01-NS051778 (MEH), New York State Stem Cell Science (NYSTEM) contract number CO24345 (MEH), NIH CA-096732 (MFR), the Children’s Brain Tumor Foundation (CBTF)(MFR), the Pediatric Brain Tumor Foundation (PBTF)(MFR), the American Brain Tumor Foundation (ABTA)(MFR) and the American Lebanese-Syrian Associated Charities (ALSAC) of St. Jude Children’s Research Hospital (MFR).
Footnotes
Disclosure statement
The authors have no conflict of interest related to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Mary E. Hatten, Email: hatten@rockefeller.edu.
Martine F. Roussel, Email: martine.roussel@stjude.org.
References
- 1.Bailey P, Cushing H. A Classification of the Tumors of the Glioma Group on a Histogenetic Basis With a Correlated Study of Prognosis. Lippincott; 1926. [Google Scholar]
- 2.Palay SL, Chan-Palay S. The Cerebellar Cortex: cytology and organization. Springer-Verlag; 1974. [Google Scholar]
- 3.Boyden ES, et al. Cerebellum-dependent learning: The Role of Multiple Plasticity Mechanisms. Annu Rev Neurosci. 2004;27:581–609. doi: 10.1146/annurev.neuro.27.070203.144238. [DOI] [PubMed] [Google Scholar]
- 4.De Zeeuw CI, Yeo CH. Time and tide in cerebellar memory formation. Current opinion in neurobiology. 2005;15:667–674. doi: 10.1016/j.conb.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 5.du Lac S, et al. Learning and memory in the vestibulo-ocular reflex. Annu Rev Neurosci. 1995;18:409–441. doi: 10.1146/annurev.ne.18.030195.002205. [DOI] [PubMed] [Google Scholar]
- 6.Fiez JA. Cerebellar contributions to cognition. Neuron. 1996;16:13–15. doi: 10.1016/s0896-6273(00)80018-5. [DOI] [PubMed] [Google Scholar]
- 7.Fiez JA, Petersen SE. Neuroimaging studies of word reading. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:914–921. doi: 10.1073/pnas.95.3.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmahmann JD, Caplan D. Cognition, emotion and the cerebellum. Brain. 2006;129:290–292. doi: 10.1093/brain/awh729. [DOI] [PubMed] [Google Scholar]
- 9.Timmann D, et al. Failure of cerebellar patients to time finger opening precisely causes ball high-low inaccuracy in overarm throws. J Neurophysiol. 1999;82:103–114. doi: 10.1152/jn.1999.82.1.103. [DOI] [PubMed] [Google Scholar]
- 10.Levisohn L, et al. Neuropsychological consequences of cerebellar tumour resection in children: cerebellar cognitive affective syndrome in a paediatric population. Brain. 2000;123 ( Pt 5):1041–1050. doi: 10.1093/brain/123.5.1041. [DOI] [PubMed] [Google Scholar]
- 11.Joyner AL. Engrailed, Wnt and Pax genes regulate midbrain--hindbrain development. Trends Genet. 1996;12:15–20. doi: 10.1016/0168-9525(96)81383-7. [DOI] [PubMed] [Google Scholar]
- 12.Joyner AL, et al. Otx2, Gbx2 and Fgf8 interact to position and maintain a mid-hindbrain organizer. Curr Opin Cell Biol. 2000;12:736–741. doi: 10.1016/s0955-0674(00)00161-7. [DOI] [PubMed] [Google Scholar]
- 13.Wingate RJ, Hatten ME. The role of the rhombic lip in avian cerebellum development. Development (Cambridge, England) 1999;126:4395–4404. doi: 10.1242/dev.126.20.4395. [DOI] [PubMed] [Google Scholar]
- 14.Adamson DC, et al. OTX2 is critical for the maintenance and progression of Shh-independent medulloblastomas. Cancer Res. 2010;70:181–191. doi: 10.1158/0008-5472.CAN-09-2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hatten ME, Heintz N. Mechanisms of neural patterning and specification in the developing cerebellum. Annu Rev Neurosci. 1995;18:385–408. doi: 10.1146/annurev.ne.18.030195.002125. [DOI] [PubMed] [Google Scholar]
- 16.Dino MR, et al. Unipolar brush cell: a potential feedforward excitatory interneuron of the cerebellum. Neuroscience. 2000;98:625–636. doi: 10.1016/s0306-4522(00)00123-8. [DOI] [PubMed] [Google Scholar]
- 17.Laine J, Axelrad H. Extending the cerebellar Lugaro cell class. Neuroscience. 2002;115:363–374. doi: 10.1016/s0306-4522(02)00421-9. [DOI] [PubMed] [Google Scholar]
- 18.Fink AJ, et al. Development of the deep cerebellar nuclei: transcription factors and cell migration from the rhombic lip. J Neurosci. 2006;26:3066–3076. doi: 10.1523/JNEUROSCI.5203-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang VY, et al. Math1 expression redefines the rhombic lip derivatives and reveals novel lineages within the brainstem and cerebellum. Neuron. 2005;48:31–43. doi: 10.1016/j.neuron.2005.08.024. [DOI] [PubMed] [Google Scholar]
- 20.Dymecki SM, Tomasiewicz H. Using Flp-recombinase to characterize expansion of Wnt1-expressing neural progenitors in the mouse. Developmental biology. 1998;201:57–65. doi: 10.1006/dbio.1998.8971. [DOI] [PubMed] [Google Scholar]
- 21.Machold R, Fishell G. Math1 is expressed in temporally discrete pools of cerebellar rhombic-lip neural progenitors. Neuron. 2005;48:17–24. doi: 10.1016/j.neuron.2005.08.028. [DOI] [PubMed] [Google Scholar]
- 22.Wingate RJ. The rhombic lip and early cerebellar development. Current opinion in neurobiology. 2001;11:82–88. doi: 10.1016/s0959-4388(00)00177-x. [DOI] [PubMed] [Google Scholar]
- 23.Espinosa JS, Luo L. Timing neurogenesis and differentiation: insights from quantitative clonal analyses of cerebellar granule cells. J Neurosci. 2008;28:2301–2312. doi: 10.1523/JNEUROSCI.5157-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujita S. Quantitative analysis of cell proliferation and differentiation in the cortex of the postnatal mouse cerebellum. J Cell Biol. 1967;32:277–287. doi: 10.1083/jcb.32.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujita S, et al. 3H-thymidine autoradiographic studies on the cell proliferation and differentiation in the external and internal granular layers of the mouse cerebellum. J Comp Neurol. 1966;128:191–209. doi: 10.1002/cne.901280206. [DOI] [PubMed] [Google Scholar]
- 26.Gong S, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- 27.Gilbertson RJ, Ellison DW. The origins of medulloblastoma subtypes. Annual review of pathology. 2008;3:341–365. doi: 10.1146/annurev.pathmechdis.3.121806.151518. [DOI] [PubMed] [Google Scholar]
- 28.Morales D, Hatten ME. Molecular markers of neuronal progenitors in the embryonic cerebellar anlage. J Neurosci. 2006;26:12226–12236. doi: 10.1523/JNEUROSCI.3493-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anthony TE, et al. Radial glia serve as neuronal progenitors in all regions of the central nervous system. Neuron. 2004;41:881–890. doi: 10.1016/s0896-6273(04)00140-0. [DOI] [PubMed] [Google Scholar]
- 30.Feng L, et al. Brain lipid-binding protein (BLBP): a novel signaling system in the developing mammalian CNS. Neuron. 1994;12:895–908. doi: 10.1016/0896-6273(94)90341-7. [DOI] [PubMed] [Google Scholar]
- 31.Anthony TE, et al. Brain lipid-binding protein is a direct target of Notch signaling in radial glial cells. Genes Dev. 2005;19:1028–1033. doi: 10.1101/gad.1302105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gibson P, et al. Subtypes of medulloblastoma are different diseases. Nature. 2010;468:1095–1099. doi: 10.1038/nature09587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- 34.Corrales JD, et al. The level of sonic hedgehog signaling regulates the complexity of cerebellar foliation. Development (Cambridge, England) 2006;133:1811–1821. doi: 10.1242/dev.02351. [DOI] [PubMed] [Google Scholar]
- 35.Kimura H, et al. Gli1 is important for medulloblastoma formation in Ptc1+/− mice. Oncogene. 2005;24:4026–4036. doi: 10.1038/sj.onc.1208567. [DOI] [PubMed] [Google Scholar]
- 36.Spassky N, et al. Primary cilia are required for cerebellar development and Shh-dependent expansion of progenitor pool. Developmental biology. 2008;317:246–259. doi: 10.1016/j.ydbio.2008.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wen X, et al. Kinetics of hedgehog-dependent full-length Gli3 accumulation in primary cilia and subsequent degradation. Mol Cell Biol. 2010;30:1910–1922. doi: 10.1128/MCB.01089-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goodrich LV, et al. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science (New York), N Y. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 39.Lee Y, et al. A molecular fingerprint for medulloblastoma. Cancer Res. 2003;63:5428–5437. [PubMed] [Google Scholar]
- 40.Marino S, et al. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14:994–1004. [PMC free article] [PubMed] [Google Scholar]
- 41.Oliver TG, et al. Loss of patched and disruption of granule cell development in a pre-neoplastic stage of medulloblastoma. Development (Cambridge, England) 2005;132:2425–2439. doi: 10.1242/dev.01793. [DOI] [PubMed] [Google Scholar]
- 42.Yauch RL, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science (New York), N Y. 2009;326:572–574. doi: 10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brugieres L, et al. Incomplete penetrance of the predisposition to medulloblastoma associated with germ-line SUFU mutations. J Med Genet. 2010;47:142–144. doi: 10.1136/jmg.2009.067751. [DOI] [PubMed] [Google Scholar]
- 44.Lee Y, et al. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene. 2007;26:6442–6447. doi: 10.1038/sj.onc.1210467. [DOI] [PubMed] [Google Scholar]
- 45.Solecki DJ, et al. Activated Notch2 signaling inhibits differentiation of cerebellar granule neuron precursors by maintaining proliferation. Neuron. 2001;31:557–568. doi: 10.1016/s0896-6273(01)00395-6. [DOI] [PubMed] [Google Scholar]
- 46.Machold RP, et al. Antagonism between Notch and bone morphogenetic protein receptor signaling regulates neurogenesis in the cerebellar rhombic lip. Neural development. 2007;2:5. doi: 10.1186/1749-8104-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dakubo GD, et al. Expression of Notch and Wnt pathway components and activation of Notch signaling in medulloblastomas from heterozygous patched mice. Journal of neuro-oncology. 2006;79:221–227. doi: 10.1007/s11060-006-9132-2. [DOI] [PubMed] [Google Scholar]
- 48.Fogarty MP, et al. Fibroblast growth factor blocks Sonic hedgehog signaling in neuronal precursors and tumor cells. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:2973–2978. doi: 10.1073/pnas.0605770104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alder J, et al. Generation of cerebellar granule neurons in vivo by transplantation of BMP-treated neural progenitor cells. Nature neuroscience. 1999;2:535–540. doi: 10.1038/9189. [DOI] [PubMed] [Google Scholar]
- 50.Rios I, et al. Bmp2 antagonizes sonic hedgehog-mediated proliferation of cerebellar granule neurones through Smad5 signalling. Development (Cambridge, England) 2004;131:3159–3168. doi: 10.1242/dev.01188. [DOI] [PubMed] [Google Scholar]
- 51.Zhao H, et al. Post-transcriptional down-regulation of Atoh1/Math1 by bone morphogenic proteins suppresses medulloblastoma development. Genes Dev. 2008;22:722–727. doi: 10.1101/gad.1636408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ayrault O, et al. Atoh1 inhibits neuronal differentiation and collaborates with Gli1 to generate medulloblastoma-initiating cells. Cancer Res. 2010;70:5618–5627. doi: 10.1158/0008-5472.CAN-09-3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Salero E, Hatten ME. Differentiation of ES cells into cerebellar neurons. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:2997–3002. doi: 10.1073/pnas.0610879104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Louis DN, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kool M, et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PloS one. 2008;3:e3088. doi: 10.1371/journal.pone.0003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Northcott PA, et al. Medulloblastoma Comprises Four Distinct Molecular Variants. J Clin Oncol. 2010 doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ferretti E, et al. Hedgehog checkpoints in medulloblastoma: the chromosome 17p deletion paradigm. Trends Mol Med. 2005;11:537–545. doi: 10.1016/j.molmed.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 58.Thompson MC, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24:1924–1931. doi: 10.1200/JCO.2005.04.4974. [DOI] [PubMed] [Google Scholar]
- 59.Lindsey JC, et al. Identification of tumour-specific epigenetic events in medulloblastoma development by hypermethylation profiling. Carcinogenesis. 2004;25:661–668. doi: 10.1093/carcin/bgh055. [DOI] [PubMed] [Google Scholar]
- 60.Ellison DW, et al. beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children's Cancer Study Group Brain Tumour Committee. J Clin Oncol. 2005;23:7951–7957. doi: 10.1200/JCO.2005.01.5479. [DOI] [PubMed] [Google Scholar]
- 61.Behesti H, Marino S. Cerebellar granule cells: insights into proliferation, differentiation, and role in medulloblastoma pathogenesis. Int J Biochem Cell Biol. 2009;41:435–445. doi: 10.1016/j.biocel.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 62.Eberhart CG, et al. Histopathological and molecular prognostic markers in medulloblastoma: c-myc, N-myc, TrkC, and anaplasia. Journal of neuropathology and experimental neurology. 2004;63:441–449. doi: 10.1093/jnen/63.5.441. [DOI] [PubMed] [Google Scholar]
- 63.Kenney AM, et al. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development (Cambridge, England) 2003;130:15–28. doi: 10.1242/dev.00182. [DOI] [PubMed] [Google Scholar]
- 64.Knoepfler PS, et al. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002;16:2699–2712. doi: 10.1101/gad.1021202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zindy F, et al. Genetic alterations in mouse medulloblastomas and generation of tumors de novo from primary cerebellar granule neuron precursors. Cancer Res. 2007;67:2676–2684. doi: 10.1158/0008-5472.CAN-06-3418. [DOI] [PubMed] [Google Scholar]
- 66.Uziel T, et al. The tumor suppressors Ink4c and p53 collaborate independently with Patched to suppress medulloblastoma formation. Genes Dev. 2005;19:2656–2667. doi: 10.1101/gad.1368605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Swartling FJ, et al. Pleiotropic role for MYCN in medulloblastoma. Genes Dev. 2010;24:1059–1072. doi: 10.1101/gad.1907510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zindy F, et al. N-Myc and the cyclin-dependent kinase inhibitors p18Ink4c and p27Kip1 coordinately regulate cerebellar development. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:11579–11583. doi: 10.1073/pnas.0604727103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marino S. Medulloblastoma: developmental mechanisms out of control. Trends Mol Med. 2005;11:17–22. doi: 10.1016/j.molmed.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 70.Kongkham PN, et al. The SFRP family of WNT inhibitors function as novel tumor suppressor genes epigenetically silenced in medulloblastoma. Oncogene. 2010;29:3017–3024. doi: 10.1038/onc.2010.32. [DOI] [PubMed] [Google Scholar]
- 71.Xu P, et al. Differential expression of Notch family members in astrocytomas and medulloblastomas. Pathol Oncol Res. 2009;15:703–710. doi: 10.1007/s12253-009-9173-x. [DOI] [PubMed] [Google Scholar]
- 72.Fan X, et al. Notch1 and notch2 have opposite effects on embryonal brain tumor growth. Cancer Res. 2004;64:7787–7793. doi: 10.1158/0008-5472.CAN-04-1446. [DOI] [PubMed] [Google Scholar]
- 73.Hallahan AR, et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res. 2004;64:7794–7800. doi: 10.1158/0008-5472.CAN-04-1813. [DOI] [PubMed] [Google Scholar]
- 74.Alvarez-Rodriguez R, et al. Bone morphogenetic protein 2 opposes Shh-mediated proliferation in cerebellar granule cells through a TIEG-1-based regulation of Nmyc. The Journal of biological chemistry. 2007;282:37170–37180. doi: 10.1074/jbc.M705414200. [DOI] [PubMed] [Google Scholar]
- 75.Ben-Arie N, et al. Math1 is essential for genesis of cerebellar granule neurons. Nature. 1997;390:169–172. doi: 10.1038/36579. [DOI] [PubMed] [Google Scholar]
- 76.Helms AW, et al. Autoregulation and multiple enhancers control Math1 expression in the developing nervous system. Development (Cambridge, England) 2000;127:1185–1196. doi: 10.1242/dev.127.6.1185. [DOI] [PubMed] [Google Scholar]
- 77.Briggs KJ, et al. Cooperation between the Hic1 and Ptch1 tumor suppressors in medulloblastoma. Genes Dev. 2008;22:770–785. doi: 10.1101/gad.1640908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nakahara Y, et al. Genetic and epigenetic inactivation of Kruppel-like factor 4 in medulloblastoma. Neoplasia (New York, NY) 12:20–27. doi: 10.1593/neo.91122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Northcott PA, et al. The miR-17/92 polycistron is up-regulated in sonic hedgehog-driven medulloblastomas and induced by N-myc in sonic hedgehog-treated cerebellar neural precursors. Cancer Res. 2009;69:3249–3255. doi: 10.1158/0008-5472.CAN-08-4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Uziel T, et al. The miR-17~92 cluster collaborates with the Sonic Hedgehog pathway in medulloblastoma. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:2812–2817. doi: 10.1073/pnas.0809579106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ventura A, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell. 2008;132:875–886. doi: 10.1016/j.cell.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.O'Donnell KA, et al. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 83.Doyle JP, et al. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. 2008;135:749–762. doi: 10.1016/j.cell.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Heiman M, et al. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang ZJ, et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell. 2008;14:135–145. doi: 10.1016/j.ccr.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Barkovich AJ. Pediatric Neuroimaging. Lippincott Williams and Wilkins; 2005. [Google Scholar]
- 87.Gajjar A, Pizer B. Role of high-dose chemotherapy for recurrent medulloblastoma and other CNS primitive neuroectodermal tumors. Pediatr Blood Cancer. 2010;54:649–651. doi: 10.1002/pbc.22378. [DOI] [PubMed] [Google Scholar]
- 88.Merchant TE, et al. Multi-institution prospective trial of reduced-dose craniospinal irradiation (23.4 Gy) followed by conformal posterior fossa (36 Gy) and primary site irradiation (55.8 Gy) and dose-intensive chemotherapy for average-risk medulloblastoma. Int J Radiat Oncol Biol Phys. 2008;70:782–787. doi: 10.1016/j.ijrobp.2007.07.2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Romer J, Curran T. Targeting medulloblastoma: small-molecule inhibitors of the Sonic Hedgehog pathway as potential cancer therapeutics. Cancer Res. 2005;65:4975–4978. doi: 10.1158/0008-5472.CAN-05-0481. [DOI] [PubMed] [Google Scholar]
- 90.Rudin CM, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. The New England journal of medicine. 2009;361:1173–1178. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Parsons DW, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2010 doi: 10.1126/science.1198056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Agger K, et al. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449:731–734. doi: 10.1038/nature06145. [DOI] [PubMed] [Google Scholar]
- 93.Sierra J, et al. The APC tumor suppressor counteracts beta-catenin activation and H3K4 methylation at Wnt target genes. Genes Dev. 2006;20:586–600. doi: 10.1101/gad.1385806. [DOI] [PMC free article] [PubMed] [Google Scholar]