

Abstract
Ventral tegmental area (VTA) GABA neurons appear to be critical substrates underlying the acute and chronic effects of ethanol on dopamine (DA) neurotransmission in the mesocorticolimbic system implicated in alcohol reward. The aim of this study was to examine the role of midbrain connexin-36 (Cx36) gap junctions (GJs) in ethanol’s rewarding effects. Using behavioral, molecular and electrophysiological methods we compared the effects of ethanol in mature Cx36 knockout (KO) mice and age-matched wild-type (WT) controls. Cx36 KO mice exhibited significantly more ethanol-induced ataxia in the open field test, but less disruption in motor coordination than their WT controls in the rotarod paradigm. Cx36 KO mice and WT mice treated with the Cx36 antagonist mefloquine (MFQ) consumed significantly less ethanol than their vehicle-treated WT controls in the drink-in-the-dark procedure. The firing rate of VTA GABA neurons in WT mice was inhibited by ethanol with an IC50 of 0.25 g/kg, while VTA GABA neurons in KO mice were significantly less sensitive to ethanol. Dopamine neuron sIPSC frequency was reduced by ethanol (30 mM) in WT mice, but not affected in KO mice. Cx36 KO mice evinced a significant up-regulation in DAT and D2 receptors in the VTA, as assessed by quantitative RT-PCR. These findings demonstrate the behavioral relevance of Cx36 GJ-mediated electrical coupling between GABA neurons in mature animals, and suggest that loss of coupling between VTA GABA neurons results in disinhibition of DA neurons, a hyper-DAergic state and lowered hedonic valence for ethanol consumption.
INTRODUCTION
The prevailing view is that enhancement of dopamine (DA) transmission in the mesolimbic system underlies the rewarding properties of drugs of abuse including alcohol (Wise, 2004). The primary reward centers in the mesolimbic reward system consist of DA neurons in the midbrain ventral tegmental area (VTA) that innervate the nucleus accumbens (NAcc). In support of a role for the meolimbic DA system in alcohol reward, ethanol increases the firing rate of DA neurons both in vivo and in vitro (Brodie and Appel, 1998; Gysling and Wang, 1983; Mereu et al., 1984), similar to what has been observed for many other drugs of abuse. In addition, animals will self-administer ethanol directly into the VTA (Gatto et al., 1994), and an ethanol-induced increase of DA release in the mesolimbic system, detected by microdialysis, has been reported extensively (Diana et al., 2008). Dopamine neuron activity is controlled by local-circuit midbrain GABAergic interneurons, and ethanol potently inhibits their activity (Gallegos et al., 1999; Ludlow et al., 2009; Mereu and Gessa, 1985; Steffensen et al., 2009; Stobbs et al., 2004; Xiao and Ye, 2008), which many have theorized results from disinhibition of DA neurons by ethanol.
Gap junctions (GJs) are pores formed by connexin (Cx) subunits which enable the passage of current and small molecules between cells, thereby providing a means for intercellular communication in both the developing and mature nervous system (Bennett and Zukin, 2004). Two types of Cxs are ubiquitously expressed in mammalian neurons: Cx36, found mostly in GABA interneurons of the mature CNS (Belluardo et al., 2000; Condorelli et al., 2000); and Cx45, expressed throughout the nervous system during development and less so in the adult (Maxeiner et al., 2003). Gap junctions have been linked to the generation of high-frequency oscillations, and GABA neuron networks may regulate these oscillations through GJs (Buzsaki and Chrobak, 1995; Galarreta and Hestrin, 2001; Tamas et al., 2000). Despite the fact that GJs are involved in neuronal oscillations and synchrony, evidence for their role in behavior has lagged behind the molecular and physiological evidence. Although most research on oscillatory patterns focus upon the limbic system, hippocampus, thalamus, and neocortex, the midbrain also exhibits rhythmic oscillations (Kitai et al., 1999). Indeed, GJs exist in critical areas of the mesolimbic reward pathway in mature animals, including the VTA (Allison et al., 2006; Lassen et al., 2007) and NAcc (Moore and Grace, 2002). The role of GJs in mediating or modulating reward is supported by only a few studies. For example, cocaine and methamphetamine self-administration produces region-specific and time-dependent changes in Cx36 expression in rats (McCracken et al., 2005a; McCracken et al., 2005b), suggesting at least a correlative involvement of Cx36 GJs in drug reward.
While the role of DA in regulating GJ-mediated electrical coupling is well-documented in the retina (Baldridge et al., 1998), the role of GJs in regulating DA release or vice versa in the mesolimbic or nigrostriatal DA systems has received only minimal attention. The first demonstration of GJ involvement in DA effects was in the striatum, where dye coupling was observed between striatal neurons following DA depletion (Onn and Grace, 1994). In the midbrain VTA, we have provided evidence in support of Cx36-mediated intercellular communication between mature VTA GABA neurons; including: 1) Electron micrographs showing GJs between GABA neurons (Steffensen et al., 1998); 2) Stimulation of the internal capsule evoking multiple spike discharges that are sensitive to GJ blockers including the Cx36 antagonist mefloquine (MFQ; (Allison et al., 2006)); 3) Dopamine coupling of spikes (Stobbs et al., 2004); 4) Dye coupling of neurons (Allison et al., 2006); and 5) High expression of Cx36 transcripts and protein (Allison et al., 2006; Lassen et al., 2007). While it is well-known that alcohols such as heptanol or octanol block GJ-communication in non-neuronal systems (Wentlandt et al., 2004), few studies have addressed the role of GJs in mediating ethanol’s effects on neuronal cells. Mainly, we have shown that ethanol blocks electrical coupling between VTA GABA neurons (Stobbs et al., 2004), and others have shown that GJs are implicated in ethanol withdrawal seizures (Perez-Velazquez et al., 1994). Given this evidence suggesting a role for Cx36 GJs in mediating ethanol effects on mature VTA GABA neurons, we hypothesize that adult mice lacking Cx36 GJs (i.e., Cx36 knock-out (KO) mice) would be less sensitive to the rewarding effects of ethanol. In addition, we hypothesize that Cx36 KO mice would not exhibit the typical ethanol inhibition of VTA GABA neuron activity or GABA input to DA neurons (Gallegos et al., 1999; Ludlow et al., 2009; Steffensen et al., 2009; Stobbs et al., 2004; Xiao and Ye, 2008).
MATERIALS AND METHODS
Animal Subjects
The care and use of mice and experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Brigham Young University which met or exceeded NIH guidelines. Only adult male mice (PND 60-120) were used in this study. Connexin-36 (Cx36) knock-out (KO) mice were originally created on a C57/B6 background in the laboratory of Dr. David Paul (Harvard Medical School, Cambridge, MA). In Cx36 KO mice, the Cx36 coding sequence was replaced by a LacZ-IRES-PLAP reporter cassette (Deans et al., 2001). Activation of the Cx36 promoter results in expression of the cytoplasmic protein β-gal, and cells that would normally generate Cx36 transcripts expressed the β-gal reporter throughout their cytoplasmic domain. Cx36 KO founders were a gift of Dr. Marla Feller (University of California, San Diego, CA), and were compared against age-matched C57/B6 wild-type (WT) mice (Charles River, Wilmington, MA). Once weaned at PND 21, all mice were housed in groups of four/cage and placed on a reverse light/dark cycle with lights ON from 8 PM to 8 AM. Mice were given ad libitum access to solid food and water except those that were evaluated for ethanol self-administration in the drink-in-the-dark procedure.
Open Field Assay for Motor Activity
To evaluate the role of Cx36 GJs in ethanol effects on locomotor activity, we compared WT and Cx36 KO mice in an open field assay. This was accomplished by measuring motor activity using a piezoelectric sensor cemented to the bottom of the suspended floor of 18” wide by 18” deep by 14” tall clear plexiglass chambers located in sound-attenuated boxes. Piezoelectric signals facilitated both amplitude and frequency measurements of motor activity (e.g., tremors, etc…; (Macey et al., 1996)). The signal from the piezoelectric transducers was amplified (10X) and filtered (0.1-100Hz) with a CyberAmp 820 amplifier (Axon Instruments), and digitized with a National Instruments (Austin, TX) multifunction data acquisition board at 250 samples/sec. Piezoelectric potentials were analyzed using a root mean square (rms) digital processing algorithm using Igor Pro Software (Oswego, OR). It is well-known that mice habituate to their environment, becoming less active after a period of intense exploratory activity. A normal mouse will have elevated locomotor activity when first put in the chamber with a diminution in activity over a 30 min session. Mice were habituated to the chamber for 5 daily sessions before testing with ethanol. Motor activity was evaluated for 30 min after an injection of varying doses of acute ethanol.
Fixed-Speed Rotarod for Balance Assay
To evaluate the role of Cx36 GJs in ethanol effects on motor coordination, we compared WT and Cx36 KO mice in the rotarod paradigm. To accomplish this, mice were studied on a Rota-Rod/RS LE8200 apparatus (Letica Scientific Instruments, Barcelona, Spain). Mice were first trained on the rotarod at a fixed speed of 14 rpm until they were able to remain on it for 300 sec. Mice were administered ethanol and replaced upon the rotarod to be measured for up to 300 second intervals at 30, 45, and 60 minutes after ethanol injection (the mice having been measured in the piezoelectric chambers for the first 30 minutes after injection). A 14 rpm rotarod speed and 300 second measurement interval remained the same through all experiments while injections varied in ethanol dose. In the 300 sec period in which the mice were tested on the rotarod, the longest time the mice remained on the rod was recorded. The effect of time of all doses is compared between WT and Cx36 KO mice.
Ethanol Drink-in-the-Dark Procedure
For ethanol self-administration studies in WT and Cx36 KO mice, a drink-in-the-dark method was employed (Rhodes et al., 2005). In order to control for ethanol history, separate groups of naïve male mice were used for ethanol self-administration studies. As opposed to rats, mice will self-administer ethanol, so no sucrose fading procedure is necessary, nor is food deprivation. Two hours after the beginning of the dark cycle, water bottles were replaced by a sipper tube filled with 20% ethanol. Mice were then allowed to drink for two hours a day, for 5-9 days, depending on the study. Alcohol drinking was measured using a graduated 10 mL pipette sipper tube, recording the ethanol level before and after the two-hour period. Two thirds of the bedding was removed from the cages before the drinking test to prevent potential bedding build-up that would cause sipper tube leakage. Bedding was replaced after the drinking test each day. To control for any physical characteristic that may affect drinking performance, mice were given 10% sucrose for five days to illuminate any significant differences between KO and WT mice (Gupta et al., 2008). One mouse drank significantly lower quantities of sucrose and was not included in the ethanol self-administration test.
Surgical Procedure and Single Cell Electrophysiology In Vivo
For acute electrophysiological recordings of VTA GABA neurons, mice were anesthetized using Isoflurane and placed in a stereotaxic apparatus. In order to control for ethanol history, separate groups of naïve male mice were used for electrophysiological studies. Anesthesia level was maintained at 1% throughout the experiments. Body temperature was maintained at 37.4 ± 0.4°C by a feedback regulated heating pad. With the skull exposed, holes were drilled for placement of stimulating and recording electrodes. Extracellular potentials were recorded by 3.0 M KCl-filled micropipettes (2-4 MΩ; 1 μm inside diameter). Potentials were amplified with an Axon Instruments Multiclamp 700A amplifier (Union City, CA). Microelectrodes were oriented, via stereotaxic coordinates, into the VTA (from bregma: 2.7 – 3.2 posterior (P), 0.25 - 1.0 lateral (L), 3.7 – 5.2 ventral (V)) with a piezoelectric microdrive (EXFO Burleigh 8200 controller and Inchworm, Victor, NY). Single cell activity was filtered at 0.3-10 kHz (-3dB) with the Multiclamp 700A amplifier and displayed on Tektronix (Beaverton, OR) digital oscilloscopes. Potentials were sampled at 20 kHz (12 bit resolution) with National Instruments data acquisition boards in Macintosh computers (Apple Computer, Cupertino, CA). Extracellularly-recorded action potentials were discriminated with a World Precision Instruments WP-121 Spike Discriminator (Sarasota, Fl) and converted to computer-level pulses. Single-unit potentials, discriminated spikes and stimulation events were captured by National Instruments NB-MIO-16 digital I/O and counter/timer data acquisition boards (Austin, TX) in Macintosh computers.
Preparation of Brain Slices
Midbrain slices were obtained as previously described (Steffensen et al., 2008). Briefly, mice were anesthetized with Ketamine (60 mg/kg), decapitated, and brains were quickly dissected and sectioned into 210 μm thick horizontal slices in ice-cold artificial cerebrospinal fluid (ACSF), bubbled with 95% O2 / 5% CO2. This cutting solution consisted of (in mM): 220 Sucrose, 3 KCl, 1.25 NaH2PO4, 25 NaH2CO3, 12 MgSO4, 10 Glucose, 0.2 CaCl2, and 0.4 Ketamine. These VTA-targeted horizontal slices were immediately placed into an incubation chamber containing normal ACSF bubbled with 95% O2 / 5% CO2 at 34-35° consisting of (in mM): 124 NaCl, 2 KCl, 1.25 NaH2PO4, 24 NaHCO3, 12 glucose, 1.2 MgSO4, 2 CaCl2, pH 7.3. Slices were incubated for at least 30 min prior to being transferred to a recording chamber. Once transferred to a recording chamber with continuous normal ACSF flow (2.0 ml/min), the temperature was maintained at 35°C throughout the experiment. Slices where allowed to stabilize for an additional 15 min before recordings were made. Cells were visualized with either a Nikon Eclipse FN1 or E600FN microscope in the transmitted de Sénarmont Differential Interference Contrast configuration (IR-DIC).
Whole-cell Recordings In Vitro
For recording DA neuron firing rate, electrodes were pulled from borosilicate glass capillary tubes on a horizontal Flaming/Brown Micropipette puller (Sutter Instrument Co, Novato, CA) and filled with a solution consisting of (in mM): 123 K-gluconate, 9 NaCl, 25 KCl, 0.2 EGTA, 10 Hepes, 1 MgCl2, 3 NaATP, and 1 Na3GTP. For recording sIPSCs in DA neurons, the pipette solution consisted of (in mM): 128 KCl, 20 NaCl, 0.3 CaCl2, 1.2 MgCl2, 10 HEPES, 1 EGTA, 2 Mg-ATP, 0.25 Na-GTP and 4.5 QX314 (pH 7.3). Pipettes had tip resistances of 2.5 - 4 MΩ, and series resistances typically ranging from 7 to 15 MΩ. Cells were recorded under voltage clamp conditions and with a holding potential of -70 mV. Access resistance (Ra, typically 100 to 300 MΩ) and membrane resistance (Rm), were continuously monitored with a 5 mV voltage step delivered every 10 seconds throughout experiments. Only experiments that maintained stable Ra and Rm (less than 15% change) were included in this study. Current clamp recordings (i.e., spikes) were filtered at 5 kHz and voltage clamp recordings (i.e., sIPSCs) were filtered at 2 kHz with an Axon Instruments Multiclamp 700B amplifier and digitized at 20 and 5 kHz, respectively, using an Axon Instruments 1440A digitizer and pClamp v10 software. For determination of spontaneous synaptic current rate, the amplitude threshold was set to 5X root-mean-square amplitude in a segment of the trace that had no events. Spontaneous events were automatically detected, verified, and corrected by visual inspection of the recordings. Firing rate and sIPSC frequency were calculated by comparing 5 min of control and test data.
Characterization of VTA GABA and Dopamine Neurons
VTA GABA neurons were identified in vivo by stereotaxic coordinates (see above) and by spontaneous electrophysiological criteria (Steffensen et al., 1998). They included: Relatively fast firing rate (>10Hz); ON-OFF phasic non-bursting activity; spike duration less than 200 μsec; and activation by generalized sensory stimulation (Ludlow et al., 2009). In some experiments, multiple spike discharges were evoked in putative GABA neurons by stimulation of the internal capsule, as previously reported (Steffensen et al., 1998). We evaluated only those spikes that had greater than 5:1 signal-to-noise ratio. Dopamine neurons were studied and characterized in horizontal brain slices in vitro in order to study both their firing rate and GABA-mediated inhibitory synaptic input. Neurons in the VTA of KO and WT mice that exhibited a modest non-cation specific inward rectifying current (Ih) in combination with low input resistance were assumed to be DA neurons (Allison et al., 2006; Steffensen et al., 2008).
Quantitative RT-PCR
Bilateral tissue samples of the VTA, NAcc, and cortex were obtained from 1 mm thick horizontal brain slices from age-matched adult mice using an 18-gauge blunted needle. Tissue was homogenized and the mRNA was extracted using TriZOL (Invitrogen, Carlsbad, CA). Following extraction, mRNA was converted to cDNA using iScript RT mix (BioRad, Berkeley, CA) in a 20 μL total volume reaction and cycled in a C1000 Thermocycler (BioRad) using a reaction protocol of 25 °C for 8 minutes, 42 °C for 60 minutes, and 70 °C for 15 minutes. A preamplification reaction was performed using primers for 18S rRNA, DAT, TH and D2 receptor in a BioRad CFX96 qPCR machine using a hotstart step of 95 °C for 3 minutes, followed by a reaction protocol of 95 °C for 15 sec, 57°C for 20 sec, and 72 °C for 25 seconds, repeated 15 times. Prior to this, primers for 18S rRNA (Forward primer GTGCATGGCCGTTCTTAGTTG; Reverse primer GCCACTTGTCCCTGTAAGAAGTTG), DAT (Forward primer, CTGGTCATTGTTCTGCTCTACTTCA; Reverse primer, CCACACTGAGGTATGCTCTGATG), TH (Forward primer GGACAAGCTCAGGAACTATGC; Reverse primer GGTGTACGGGTCAAACTTCAC) and D2 (Forward primer ACCACTCAAGGGCAACTGTAC; Reverse primer CTCCCATTAGACTTCATGATAACG) were optimized to result in 94-95% amplification efficiency using serial dilutions of whole mouse brain cDNA. After preamplification, a real time quantitative PCR reaction was performed using iQ Supermix (BioRad), 0.5 μl of the preamplification multiplex reaction and primers for either 18S rRNA, DAT, TH or D2 with the appropriate FAM-TAMRA Taqman© probes (Applied Biosystems) designed to detect the amplified fragment (18S probe, TGGAGCGATTTGTCTGGTTAATTCCGATAAC; DAT probe, ATGCCCTATGTAGTCCTCACAGCCCTGCT; TH Probe, TCTCGTATCCAGCGCCCATTCTC; D2 Probe, CACCCTGAGGACATGAAACTCTGCA). The reaction was run on a BioRad CFX96 qPCR machine using a reaction protocol of a 3-minute 95 °C hotstart, followed by 50 cycles of 95 °C for 15 sec, 57 °C for 20 sec, and 72 °C for 25 sec. To ensure amplification of a single PCR product we confirmed that melt curves, performed using either primers for 18S, DAT, TH, and D2 resulted in a single peak. Melt curves were performed after a PCR reaction protocol of a 2-minute 98 °C hotstart step, followed by 45 cycles of 98 °C for 2 sec and 57 °C for 5 sec while using SsoFast EvaGreen Supermix (BioRad) in place of iQ supermix, and were measured from 65 °C to 95 °C in 0.2 °C increments for 10 seconds at each step. Cycle threshold (Ct) values and relative fold expression were calculated using CFX Manager Software (BioRad) and the 2-ΔΔCt method, as previously described (Ludlow et al., 2009). To verify that the WT, but not the KO mice, expressed Cx36, each sample was also processed as described above using primers for Cx36 (Forward Primer TGCAGCAGCACTCCACTATG; Reverse primer ATGGTCTGCTCATCATCGTACAC; Probe ATCCTGTTGACTGTGGTGGTGATCTTCC). Following the reverse transcription and preamplification, we performed PCR on the cDNA for each sample on a 2% agarose gel. PCR amplification products of 18S in both WT and Cx36 KO mice were run using 2% agarose gels, and compared with a 50 base pair ladder (Fermentas International). The amplification efficiencies of the PCR for both sequences have been optimized to the same level, allowing for relative expression calculations between different neurons by normalizing the expression of each subunit to the housekeeping gene 18S rRNA.
Drugs and Blood Alcohol Level Determination
For open field and rotarod testing, ethanol injections (0.75, 1.5, 2.5, and 4.0 g/kg; 16% w/v solution) were administered every other day after a previous low dose (i.e., 0.75 and 1.5 g/kg) and 5-7 days apart after a previous high dose (i.e., 2.5 and 4.0 g/kg) on a randomized schedule. Mefloquine (MFQ) was dissolved in 50% DMSO in saline, which served as vehicle (VEH). For the ethanol drink-in-the-dark procedure, mice were given 20% ethanol ad libitum from a sipper tube. The 20% ethanol drinking solution was prepared from 200 proof absolute anhydrous ethanol (Sigma Aldrich, St. Louis, MO) diluted to 20% (v/v) using tap water. The 10% sucrose solution was prepared using sucrose (Sigma Aldrich, St. Louis, MO) dissolved in tap water at 10% (w/v) concentration. For in vitro studies, sIPSCs were recorded in the presence of D- 2-amino-5-phosphonopentanoic acid (D-APV, 50 μM) and 6-cyano-23-dihydroxy-7-nitro-quinoxaline (CNQX, 30 μM) to block NMDA and AMPA-mediated synaptic currents, respectively. All sIPSCs were abolished by bicuculline (10 μM). The sodium channel blocker QX-314 (1.7 mM) was included in the patch pipette to prevent spiking and potential confounding with sIPSC inward currents. Blood samples (approximately 250 μl) were taken during decapitation of WT and KO mice 30 min after a 1.5 g/kg IP ethanol injection. They were spun in an Eppendorf microcentrifuge for 10 min at 3200 rpm and the serum extracted and assayed for ethanol content by the NAD/ADH enzyme-spectrophotometric method (Sigma Aldrich, St. Louis, MO).
Statistical Analyses
For repeated measures within subjects (e.g., ethanol dose response in motor tests and drinking), a mixed models ANOVA with post hoc t tests was performed using Statistical Analysis System (SAS Institute Inc, Cary, NC). For RT-PCR analysis, expression levels of DAT, D2 receptor and TH transcripts were normalized to WT mice averages for each brain region. Gene expression levels were compared using an unpaired two-sample t-test. Each sequence was amplified in triplicate reactions. For all other behavioral and electrophysiological comparisons, an unpaired, two-tailed t test was used with significance levels on graphs indicated with asterisks *,**,*** corresponding to significance levels P<0.05, 0.01 and 0.001, respectively. Values are expressed as means ± S.E.M. Analysis of sIPSCs was accomplished with MiniAnalysis (Synaptosoft: Decatur, GA) and Igor Pro (Wavemetrics: Oswego, OR) software packages. IGOR Pro Software was also used for the creation of figures.
RESULTS
Open Field Assay for Motor Activity: Role of Cx36 Gap Junctions in Ethanol Ataxia
The open field assay was used to measure the effects of ethanol on locomotor activity. There was no significant difference in baseline motor activity between KO and WT mice (P>0.05, t(2,13)=0.34; WT = 0.475 ± 0.04 Vrms, KO = 0.455 ± 0.04 Vrms; n=8,7 respectively; data not shown). Ethanol effects on motor activity in the open field assay were evaluated for 30 min 5 min after an ethanol injection. Intraperitoneal administration of ethanol (0.75-4.0 g/kg) significantly decreased motor activity in a dose-dependent manner in WT mice at all doses tested (P<0.05). Overall, ethanol-induced motor deficits were significantly greater in KO mice compared to WT mice (P=0.006, F(1,55)=8.2; Fig. 1). As KO mice were more sensitive than WT mice to the effects of ethanol in the open field assay for motor activity, we compared the effects of IP administration of the Cx36 antagonist mefloquine (MFQ; 40 mg/kg) and vehicle (VEH; isovolumic to MFQ) in WT mice in the open field assay to further evaluate the role of Cx36 gap junctions in ethanol’s behavioral effects. MFQ or VEH treatments were administered 30 min prior to testing in the open field assay. Intraperitoneal administration of ethanol (0.75-4.0 g/kg) did not significantly alter motor activity in MFQ mice compared to VEH controls (P=0.90, F(1,40)=0.02; data not shown).
Figure 1. Effects of acute ethanol, pentobarbital, MK-801 and methylphenidate on motor activity in Cx36 KO vs WT mice.
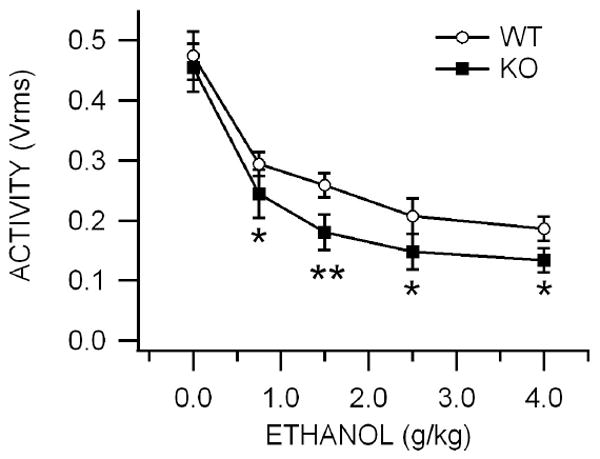
This graph compares the effects of IP administration of 0.75-4.0 g/kg ethanol on open field locomotor activity in KO vs WT mice. Values are expressed as percent baseline motor activity. Activity was recorded for 30 min, 5 min after IP administration of ethanol. Overall, ethanol-induced lowering of locomotor activity was significantly greater in KO than WT mice.
Fixed-speed Rotarod: Role of Cx36 Gap Junctions in Ethanol-induced Motor Coordination
The rotarod apparatus was used to measure the effects of ethanol on motor coordination. Although there was a tendency for KO mice to perform better on the rotarod when first tested, and WT mice required more training sessions to reach criterion, there was no significant difference in baseline rotarod performance between KO and WT mice following five days of training (P<0.3, t(2,14)=1.1; WT = 265.13 ± 31.6 sec, KO = 300 ± 0.0 sec; n=8 each; Fig. 2A). Immediately following open-field testing, the fixed-speed rotarod apparatus was used to measure the ataxic effects of ethanol in KO vs WT mice. Wild-type mice were maximally affected by ethanol at 30 min after injection with partial recovery at 60 min (Fig. 2A; shown here at the 0.75 g/kg dose level). Cx36 KO mice were significantly less sensitive to ethanol than WT mice in the rotarod paradigm at dose levels 0.75-1.5 g/kg 30-60 min after injection (0.75 g/kg: P<0.01, t(2,46)=20.3; 1.5 g/kg: P<.001, t(2,46)=2.8; 2.5 g/kg: P>0.05, t(2,46)=0.83; and 4.0 g/kg: P>0.05,, t(2,46)=1.2; n=24 each; Fig. 2B; shown here at the 45 time point and shown as % of baseline rotarod performance). Ethanol ataxia was more pronounced in WT than KO mice across dose levels (P=0.0001, F(1,155)=19.71). When mice were evaluated three weeks after ethanol testing in the fixed-speed rotarod to determine if there might be latent changes in performance, there was a significant decrement in performance in WT compared to KO mice (P=0.0003, t(2,14)=4.64; WT = 72.9 ± 22.7 sec, KO = 229.9 ± 26.7 sec; n=8 each). As KO mice appeared to be less sensitive to the ataxic effects of ethanol than WT mice, we tested the effects of IP administration of MFQ and VEH in the fixed speed rotarod apparatus to further evaluate the effects of Cx36 GJs in ethanol ataxia. There was no significant difference in baseline rotarod performance between VEH-treated and MFQ-treated mice tested three days after intraperitoneal injections of VEH and MFQ and rotarod training (P=0.57, F(1,7)=0.36; VEH = 275 ± 25 sec, MFQ = 238.8 ± 54.8 sec; n=4 each). Intraperitoneal administration of ethanol (1.5-4.0 g/kg) significantly decreased rotarod performance in a dose-dependent manner at 30 min after injection in MFQ and VEH-treated mice (1.5 g/kg: P=0.0035, F(1,7)=22; 2.5 g/kg: P=9E-08, F(1,7)=898; and 4.0 g/kg: P=1E-08, F(1,7)=1831; n=4 each) and MFQ-treated mice (1.5 g/kg: P=0.62, F(1,15)=0.28; 2.5 g/kg: P=0.017, F(1,7)=10.7; and 4.0 g/kg: P=7.1E-09, F(1,7)=2112; n=4 each). However, rotarod performance was not significantly affected by ethanol in MFQ-treated mice compared to VEH-treated mice (P=0.089, F(1,23)=1.33).
Figure 2. Effects of acute ethanol, pentobarbital, MK-801 and methylphenidate on motor coordination and balance in KO vs WT mice.
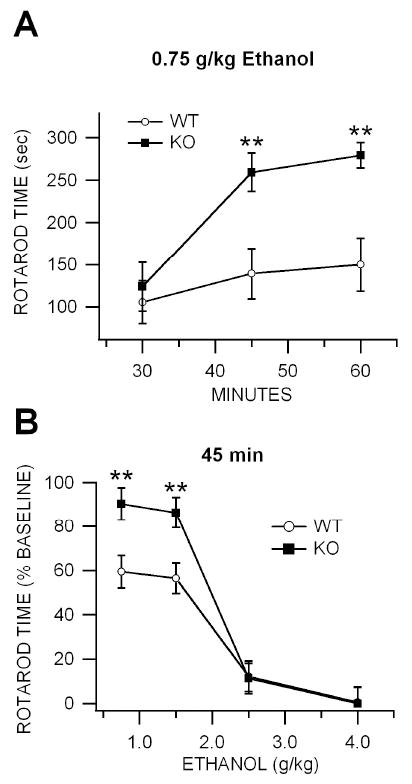
(A) This graph compares the time course of IP administration of 0.75g/kg ethanol on motor coordination in the fixed-speed rotarod paradigm in KO vs WT mice. Values are expressed as percent baseline rotarod time. Time on the rotarod was recorded for 30 min, 30 min after IP administration of ethanol, immediately following open field testing (Fig. 1). Ethanol-induced deficits in motor coordination, as well as recovery, were significantly more affected by 0.75 g/kg in WT than KO mice. (B) This graph compares the effects of IP administration of 0.75-4.0 g/kg ethanol on motor coordination in KO vs WT mice at the 45 min time point. Overall, ethanol decreased rotarod performance more in WT than KO mice.
Role of Cx36 Gap Junctions in Ethanol Consumption
We compared ethanol self-administration rates in six naïve KO vs eight naïve WT mice and in four naïve WT mice with the Cx36 blocker mefloquine (MFQ) vs four naïve WT mice with vehicle (VEH) in order to evaluate the role of Cx36 GJs in ethanol consumption. Compared to WT mice, KO mice consumed significantly less ethanol across the 9 days tested (Fig. 3A; P<0.0001, F(1,92)=40.66). Similarly, compared to VEH-treated mice, WT mice treated with MFQ (40 mg/kg) 3 hrs before testing consumed significantly less ethanol across the 5 days tested (Fig. 3B; P=0.002, F(1,31)=12.3; n=8). In order to determine the role of performance deficits in lowered ethanol consumption in the KO and MFQ-treated mice vs their controls, we compared them for sucrose consumption. There was no significant differences in sucrose consumption rates between KO vs WT mice (Fig. 3C; P=0.08, F(1,60)=3.27) or for MFQ vs VEH-treated mice (Fig. 3D; P=0.06, F(1,45)=3.1).
Figure 3. Ethanol consumption is reduced in Cx36 KO mice in the drink-in-the-dark procedure.
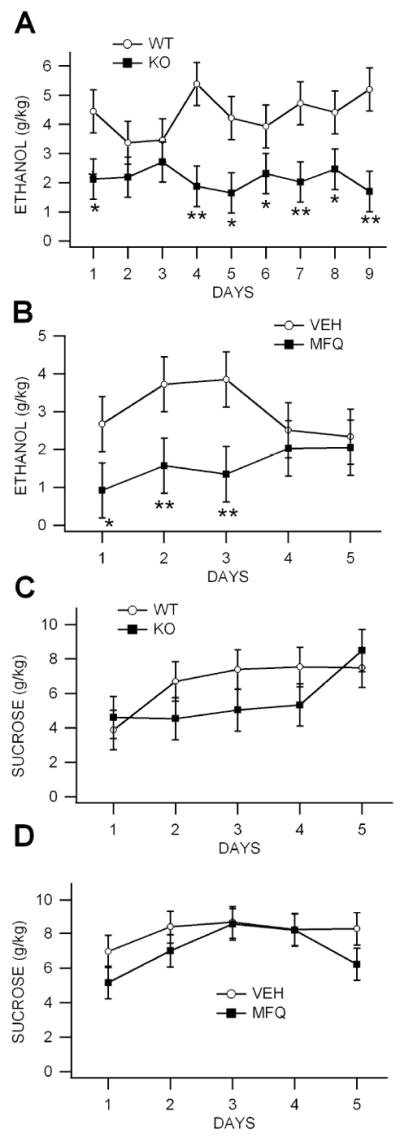
(A) Mice were allowed ad libitum access to a sipper tube containing 20% ethanol that was placed in their home cage for 2 hours 2 hours after the start of their dark phase. Knock-out mice consumed significantly less ethanol than WT mice from DAYS 1-9. (B) Wild-type mice were treated with 40 mg/kg MFQ 3 hours before testing each day. Mefloquine-treated mice consumed significantly less ethanol than vehicle (VEH)-treated WT controls. (C,D) To control for potential deficits in performance, mice were allowed ad libitum access to a sipper tube containing 10% sucrose. There was no significant difference in sucrose consumption between KO and WT mice (C) or between MFQ vs VEH WT controls (D).
Ethanol Effects on VTA GABA Neuron Firing Rate in Cx36 KO vs WT Mice
We have previously reported that GABA neurons in the VTA of mature rats express Cx36 GJs and dye-couple (Allison et al., 2006; Lassen et al., 2007). They are inhibited by ethanol in rats in vivo with an IC50 of 1.0 g/kg (Gallegos et al., 1999; Ludlow et al., 2009; Steffensen et al., 2009; Stobbs et al., 2004). We compared the effects of ethanol on VTA GABA neuron firing rate in adult age-matched, ethanol-naïve KO vs WT mice in vivo. There was no significant difference in baseline VTA GABA neuron firing rate between KO vs WT mice (P<0.05, t(2,40)=0.89; WT firing rate = 49.9 ± 3.93 Hz vs KO firing rate = 43.0 ± 7.9 Hz; n=27,15, respectively; data not shown). One dose of ethanol was tested on one neuron in each mouse. Figure 4 shows the effects of the same dose of ethanol (0.75 g/kg) on VTA GABA neurons recorded in WT (Fig. 4A,B), at dissimilar baseline firing rates,and a KO (Fig. 4C) mouse with a similar firing rate as one of the WT examples (Fig. 4C). Intraperitoneal administration of 0.75 g/kg ethanol markedly reduced firing rate in WT mice, while having little effect on VTA GABA neuron firing rate in KO mice at this dose in these mice. VTA GABA neurons in the VTA of WT mice were affected more by ethanol than KO mice across the doses tested (0.25 g/kg: P<0.01, t(2,7)=3.61; n=5,4; 0.75 g/kg: P<0.01, t(2,8)=12.5; n=5,5; 1.5 g/kg: P<0.05, t(2,8)=2.52; n=6,4; Fig 4D). Interestingly, mouse VTA GABA neurons were more sensitive to acute IP ethanol than rats, as the IC50 was 0.25 g/kg in mice vs 1.0 g/kg in rats (Gallegos et al., 1999; Ludlow et al., 2009; Steffensen et al., 2009; Stobbs et al., 2004).
Figure 4. Inhibition of VTA GABA neuron firing rate is reduced in Cx36 KO mice.
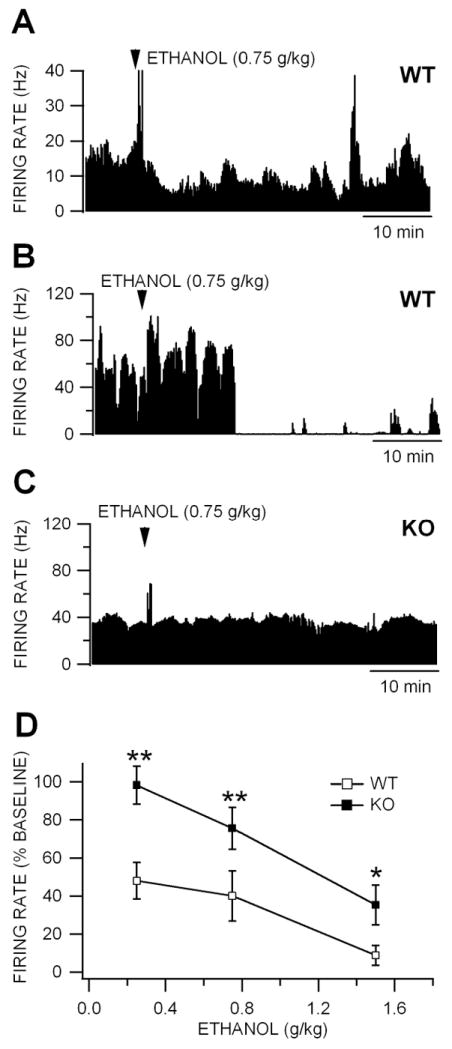
(A) This ratemeter record shows a representative VTA GABA neuron in a WT mouse recorded in vivo with a baseline firing rate of approximately 15 Hz before and after IP administration of 0.75 g/kg ethanol. Intraperitoneal administration of 1.0 g/kg ethanol reduced this neuron’s activity ~50% at approximately 10 min after injection. Transient increases in firing rate are often associated with systemic administration of ethanol (Steffensen et al., 2009). (B) This ratemeter record shows a representative VTA GABA neuron recorded in a separate WT mouse with a baseline firing rate of approximately 40 Hz before and after IP administration of 0.75 g/kg ethanol. Intraperitoneal administration of 1.0 g/kg ethanol suppressed this neuron’s activity at approximately 10 min after injection. (C) The ratemeter record shows a representative VTA GABA neuron in a KO mouse, with a baseline firing rate that was similar to that of the neuron in the WT mouse in (B), before and after IP administration of 0.75 g/kg ethanol. Ethanol did not alter the firing rate of this neuron in a KO mouse. (D) This graph compares the effects of IP administration of 0.75-4.0 g/kg ethanol on the firing rate of VTA GABA neurons recorded in KO vs WT mice. Ethanol reduced firing rate in both WT and KO mice. However, the IC50 for ethanol inhibition was 0.25 g/kg in WT mice and 1.4 g/kg in KO mice. Wild-type mice were significantly more sensitive to ethanol than KO mice at all doses tested.
Ethanol Effects on VTA Dopamine Neuron Responses in Cx36 KO vs WT Mice
We evaluated the baseline firing rate of VTA DA neurons and their GABA inhibitory input using patch clamp electrophysiological methods in the horizontal slice preparation of adult age-matched, ethanol-naïve KO vs WT mice. There was no significant difference in baseline VTA DA neuron firing rate between KO and WT mice (P=0.53, F(1,28)=0.404; WT firing rate = 2.09 ± 0.26 Hz vs KO firing rate = 1.88 ± 0.2 Hz; n=15,14, respectively; data not shown). We compared the effects of ethanol on GABA-mediated input to VTA DA neurons in adult age-matched, ethanol-naïve KO vs WT mice in vitro by studying sIPSCs in the presence of the GLU blockers APV and CNQX and the sodium channel blocker QX-314 in the patch pipette to prevent DA neuron spiking. Since ethanol reduces VTA GABA neuron firing rate with an IC50 of 0.25 g/kg, we chose a dose of 30 mM, which in the horizontal slice reduces DA neuron sIPSCs approximately 50%. While there was no difference in baseline DA neuron sIPSC frequency in KO and WT mice (P>0.05, t(2,24)=0.35; WT sIPSC rate = 5.44 ± 0.64 Hz vs KO sIPSC rate = 5.62 ± 0.53 Hz; n=16,11, respectively), superfusion of ethanol (30 mM) reduced sIPSC frequency (Fig. 5A) and shifted the averaged inter-event interval distribution plot to the right (Fig. 5B) in WT mice (n=8), but had little effect in KO mice (Fig. 5C,D; n=7). While ethanol significantly reduced DA neuron sIPSC frequency in WT mice (P<0.001, t(2,14)=7.9; n=8), it did not alter them in KO mice. There was a significant difference in ethanol effects on DA neuron sIPSC frequency between KO and WT mice (P<0.001, t(2,13)=6.2; n=8,7, respectively; Fig. 5E).
Figure 5. Lack of GABA-mediated inhibition to dopamine neurons in Cx36 KO mice.
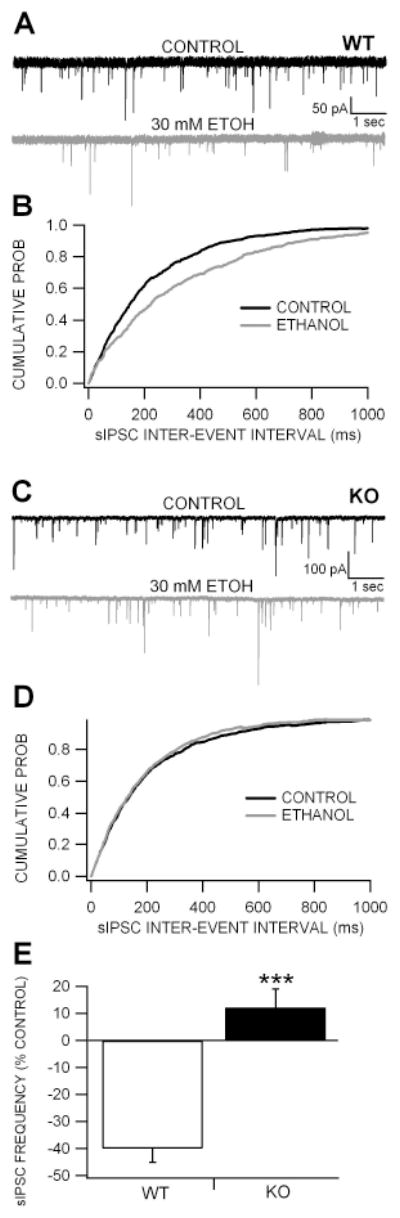
(A) These are representative 10 sec traces of DA neuron sIPSCs recorded in a WT mouse in the presence of APV and CNQX with QX-314 in the pipette to prevent spike events. Superfusion of 30% ethanol reduced sIPSCs approximately 50%. (B) This graph shows the averaged cumulated probability inter-event interval plots for ethanol effects on sIPSCs in WT mice. Note that ethanol shifts the curve to right confirming a slowing in frequency. (C) These are representative traces of DA neuron sIPSCs recorded in a KO mouse. Superfusion of 30 mM ethanol did not affect sIPSC frequency. (D) This graph shows the averaged cumulated probability inter-event interval plots for ethanol effects on sIPSCs in Cx36 KO mice. Note that ethanol has little effect on DA neuron sIPSC frequency. (E) This graph compares the effects of 30 mM ethanol on DA neuron sIPSC frequency in KO vs WT mice. While ethanol significantly reduced sIPSC frequency in WT mice, DA neurons in KO mice were significantly less sensitive to ethanol than WT mice.
DAT Expression in Cx36 KO vs WT mice: Quantitative RTPCR
Since KO mice consumed less ethanol and VTA GABA neuron firing rate and DA neuron GABA inhibition in KO mice were less sensitive to ethanol than WT mice, we postulated that KO mice were in a baseline hyper-DA state due to loss of coordinated GJ-mediated GABA inhibition of DA transmission. Thus, we confirmed the genotype of WT and KO mice for Cx36 and compared the mRNA expression of the three DA-related gene products DAT, D2 receptor, and tyrosine hydroxylase (TH) in the VTA, NAcc and cortex of KO mice compared to WT mice. Blood alcohol levels (BALs) were also evaluated in WT and KO mice in order to determine if lack of Cx36 GJs might modify the kinetics of ethanol elimination and contribute to the reported behavioral, electrophysiological and molecular effects. There was no significant difference between BALs obtained in WT and KO mice when sampled one hour after an IP injection of 1.5 g/kg ethanol (P>0.05, t(1,10)=0.64; WT BAL = 150 ± 11.5 mg% vs KO BAL = 140 ± 10.6 mg%; n=6 each). We then performed PCR of the cDNA for VTA tissue samples on 2% agarose gels and confirmed a Cx36 amplicon of the correct size (112 base pairs) in all WT mice, which was absent in all KO animals (Fig. 6A; n=6 each). Quantitative RT-PCR of tissue punches from the VTA confirmed that there was no expression of Cx36 expression in KO mice (Fig. 6B; n=6 each). Using quantitative RT-PCR of tissue punches from the VTA, NAcc and cortex, we detected expression of DAT, D2 and TH transcripts in these three brain areas; however, their expression in the VTA was up to 1,000X greater than cortical expression. In the VTA, DAT (P<0.05, t(1,8)= 2.3; n=5 each) and D2 receptor (P<0.05, t(1,10)=2.5; n=6 each), but not TH (P>0.05), expression were significantly elevated in KO vs WT mice (Fig. 6C). In the NAcc, although DAT expression was elevated in KO mice, it was not significantly different between KO and WT mice (P>0.05, t(1,10)=0.23; n=6 each; Fig. 6D). Moreover, neither D2 nor TH expression in the NAcc was significantly different in KO vs WT mice (P>0.05; n=6 each). In the cortex, the expression of DAT, D2 or TH were not significantly different between KO vs WT mice (P>0.05; n=6 each; Fig. 6E). The cycle threshold (Ct) RT-PCR values for the reference gene 18S were not significantly different (P > 0.05) between WT and KO preparations for any brain region.
Figure 6. Expression of dopamine-related genes is up-regulated in the VTA of Cx36 KO mice.
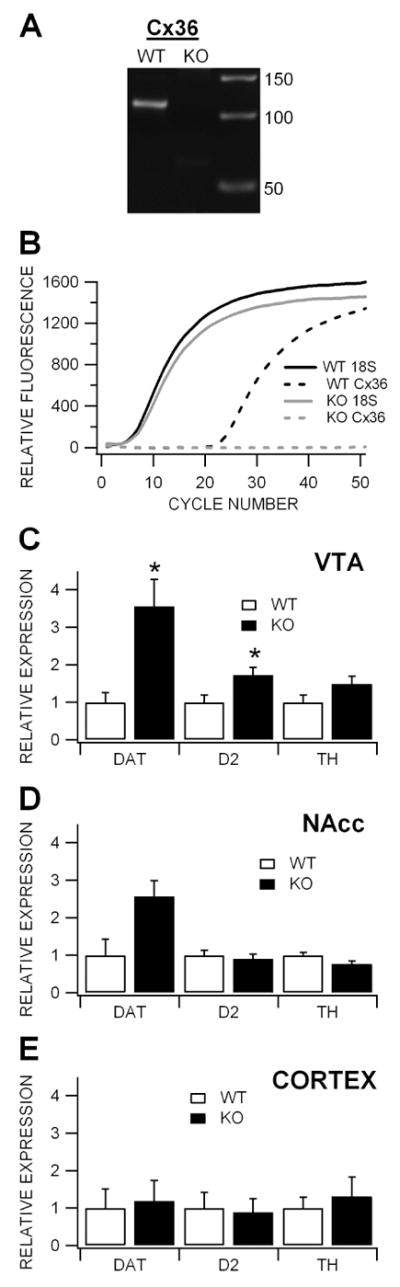
(A) Agarose gel showing Cx36 expression in the VTA of WT, but not KO mice. The amplified fragment of Cx36 is 112 base pairs long, as extrapolated from the amplicon ladder at right of the gel. (B) This graph shows the averaged quantitative RT-PCR analysis of 18S and Cx36 mRNA expression in the VTA of WT compared to KO mice. There was no significant difference in expression of the housekeeper gene 18S between KO and WT mice. Quantitative determinations can be made when Cx36 expression is compared to expression of the housekeeper gene. While Cx36 transcripts were expressed in the VTA of WT mice, there was no expression evident in KO mice. (C) This graph shows normalized (KO vs WT) expression of the DA-related transcripts DAT, D2 and TH in the VTA. Relative expression was determined by quantitative RT-PCR. Both DAT and D2 gene expression was significantly greater in the VTA of KO mice than WT mice. (D) While DAT expression was slightly elevated in the NAcc of KO mice, it was not significantly elevated. Neither D2 nor TH expression in the NAcc was significantly different in KO vs WT mice. (E) Expression of DAT, D2 and TH were not different in the cortex of WT and KO mice.
DISCUSSION
Cx36 KO mice were less sensitive to ethanol than WT mice in the open field test, but more sensitive in the rotarod paradigm. While both paradigms assess motor activity, the rotarod paradigm indexes motor coordination, while the open-field test measures locomotor activity. Thus, the absence of Cx36 GJs produces mixed effects on ethanol-induced motor deficits, likely due to the dissociation between the specific motor behaviors being tested in the open field test vs the rotarod paradigm, as well as the specific brain areas involved in motor activity. The cerebellum is one of the primary brain areas implicated in ethanol-induced ataxia (Criswell et al., 2008). Cx36 GJs are found in the molecular layer of the cerebellum (Belluardo et al., 2000), and many studies have shown that Cx36 GJs are responsible for cerebellar Purkinje cell synchrony (Long et al., 2002; Placantonakis et al., 2006; Van Der Giessen et al., 2008). It has been postulated that GJ synchrony within the cerebellum provides for motor function stabilization through feedback loops with the inferior olive (Van Der Giessen et al., 2008). Notwithstanding the physiological evidence supporting a role for Cx36 GJs in regulating motor activity, Cx36 KO mice do not exhibit overt ataxia (Bennett and Zukin, 2004). In a more recent study, it was confirmed that Cx36 KO mice possess the same motor performance as WT mice, but have impaired motor learning ability (Van Der Giessen et al., 2008). Our findings support these previous studies, as no overt signs of ataxia were observed in Cx36 KO mice, and baseline motor activity, as well as balance and coordination, were not significantly affected in KO mice when compared to WT mice in the open field and rotarod motor paradigms. Interestingly, despite the lack of significant differences in baseline motor responses between KO and WT mice, KO mice exhibited significantly better performance in the rotarod paradigm when retested three weeks after ethanol exposure, suggesting that loss of Cx36 GJs affects some aspects of motor learning. Notwithstanding the subtle differences in baseline motor activity, KO mice exhibited robust, but differential, sensitivity to ethanol’s effects in the rotarod paradigm and open-field tests, suggesting that a challenge to motor systems by ethanol overwhelms the compensations that may have occurred due to the lack of GJ regulation of motor activity. Although there were significant differences in ethanol effects on motor activity in WT and KO mice, pretreatment with the Cx36 GJ antagonist MFQ did not significantly affect the motor effects of ethanol in the open-field or rotarod paradigms compared to VEH controls. We did test the effects of MFQ in Cx36 KO mice (see comment to Reviewer #3 for more details). Mefloquine is not specific for gap junctions. It has effects other than being a selective Cx36 GJ blocker, such as increasing intracellular Ca2+ (Caridha et al., 2008; Dow et al., 2003; Zhou et al., 2006) and acting as a 5HT3 receptor antagonist (Thompson and Lummis, 2008). The non-specific effects of MFQ may be confounding its GJ blocking effects on motor responses.
One of the most important findings of this study was that Cx36 KO mice drank significantly less ethanol than WT mice, which consistently consumed approximately 4 g/kg over the two hour drink-in-the-dark procedure. Similar decreases in ethanol consumption were found in MFQ-treated mice compared to vehicle controls. This was not due to any performance deficit in KO mice or MFQ-treated mice, as there was no significant difference in drinking sucrose between KO vs WT or MFQ-treated vs vehicle-treated mice. One possible explanation is that the hedonic valence of consuming ethanol was lowered due to a hyper-DA state, which could result from the lack of coordinated inhibition of DA neurons in KO and MFQ-treated mice. The majority of evidence suggests that ethanol reward plays a role in the drink-in-the-dark procedure, but other features relevant to alcoholism such as stress, craving or withdrawal may contribute to the benavior. Indeed, it was somewhat surprising that ethanol intakes in WT and KO mice were relatively high on the first session of the procedure. Consumption rates are typically low on the first day and progressively increase over about 8-10 days reaching levels about 4 g/kg (Gupta et al., 2008). While WT mice in our study reached similar levels of responding they were at that level from the first day of the procedure and stabilized at that level for the 9 days of study. KO mice had a qualitatively similar intake profile, albeit at significantly lower consumption rates than the WT mice.
While there were no significant differences in the baseline firing rate of VTA GABA neurons between KO and WT mice, there was a marked difference in the sensitivity of VTA GABA neurons in KO and WT mice to ethanol. Mainly, VTA GABA neurons in Cx36 KO mice were considerably less sensitive to ethanol inhibition of their firing rate than WT mice. Ethanol consistently inhibited VTA GABA neuron firing rate in WT mice with an IC50 of 0.25 g/kg, which is even lower than it is in rats (i.e., 1.0 g/kg; (Gallegos et al., 1999; Steffensen et al., 2009; Stobbs et al., 2004)). Indeed, the inhibition of GABA neuron firing rate by ethanol is an order of magnitude more sensitive than ethanol’s enhancement of VTA DA neuron firing rate (Brodie and Appel, 1998), providing further evidence for the sensitivity of VTA GABA neurons to ethanol, and highlighting their importance in mediating ethanol’s effects in the mesolimbic DA system. The prevailing dogma is that DA neurons in the VTA are inhibited by GABA input. However, the source of that GABA input is controversial, as direct evidence is lacking for direct inhibition of DA neurons by local circuit GABA neurons. Notwithstanding these concerns, DA neurons evince well-known inhibitory synaptic input, as measured by GABA-mediated sIPSCs in the slice preparation. While there were no significant differences in the baseline firing rate of VTA DA neurons between KO and WT mice, there was a marked difference in ethanol effects on GABA inhibition to DA neurons. Mainly, KO mice were relatively insensitive to ethanol effects on DA neuron sIPSCs. In WT mice, ethanol inhibited sIPSC frequency in DA neurons, consistent with what some have reported (Xiao and Ye, 2008), but not all (Theile et al., 2008). There was little affect of ethanol on DA neuron sIPSCs in KO mice. In WT mice, ethanol inhibition of VTA GABA neuron firing rate might lead to disinhibition of DA neurons, causing an increase in DA release and reward. One logical explanation for the lack of effect of ethanol on inhibitory drive to DA neurons is that it would have little effect on DA neurons in KO mice due to the loss of ethanol-sensitive Cx36 GJs on GABA neurons that regulate them. We have previously demonstrated that ethanol inhibits Cx36 GJ-mediated electrical coupling between VTA GABA neurons (Stobbs et al., 2004), which could disinhibit DA neurons, increase DA neurotransmission, and cause euphoria in WT mice. Since Cx36 KO lack functional GJs, ethanol could not disinhibit DA neurons, which could result in less ethanol euphoria and, consequently, lowered ethanol consumption. Conceivably, a hyper-DA state in KO mice might result from diminished GABA inhibitory drive to VTA DA neurons, due to the lack of coordinated inhibition from electrically-coupled GABA neurons.
Since baseline GABA and DA neuron firing rate and GABA inhibition to DA neurons was not significantly different in KO vs WT mice, we postulated that adaptations in DA regulation might have compensated in KO mice for the loss of coordinated GABA inhibition. Indeed, both DAT and D2 genes were significantly up-regulated in Cx36 KO mice. However, the up-regulation was specific to the VTA, suggesting that not having Cx36 GJs results in a hyper-DA state. This might explain the better rotarod performance in the KO mice and our observation that they exhibit paroxysmal running behavior against the walls of their cages. The DAT and D2 up-regulation in the VTA may result from reduced GABA input. The lowered ethanol consumption combined with the up-regulation of DA-related genes would suggest that KO mice are in a hyper-DA state, and have compensated developmentally by over-expressing DA-related gene products in the VTA. Upregulation of DAT and D2 receptors is a hallmark of a hyper-DA state, as these feedback regulatory proteins sense the buildup of DA and increase their number, and perhaps activity, to try to lower DA release. A hyper-DA state could occlude detection of rewarding events, should DA neurons fail to respond to phasic environmental or pharmacological stimuli.
Despite the fact that GJs are clearly involved in neuronal synchrony, even in mature animals, their physiological and behavioral relevancy remains understudied and perhaps under-appreciated. Thus, our findings provide additional evidence that GJs are physiologically and behaviorally relevant. Mainly, that loss of GJs produces a hyper-DA state and lowered hedonic valence, likely through loss of GABA inhibition to DA neurons, as supported by our electrophysiological findings in GABA and DA neurons in the VTA. Our findings support the few studies that have implicated Cx36 GJs in drug reward. For example, cocaine and methamphetamine self-administration produces region-specific and time-dependent changes in Cx36 expression in rats (McCracken et al., 2005a; McCracken et al., 2005b). Of particular relevance to this study, activation of DA receptors differentially modulates the frequency of GABA sIPSCs in Cx36 KO mice compared with WTs, suggesting that DA plays a role in altering the coupling of Cx36-expressing GABA interneurons in the striatum (Cummings et al., 2008). Indeed, striatal neurons in Cx36 KO mice show reduced baseline sIPSCs compared to WT mice, supporting our view that KO mice can’t recruit the full complement of inhibition to principal cells, like striatal or DA neurons, from the GABAergic electrically-coupled networks that subserve them. Indeed, Cx36 GJs exist in several structures of the reward pathway in mature animals, including the VTA (Allison et al., 2006; Lassen et al., 2007) and striatum (Moore and Grace, 2002). This study joins that of a few others in demonstrating evidence that electrical synapses between GABA neurons have physiological relevancy, which strengthens the argument that GJs serve as critical modulators of brain activity and behavior, and may be strategic targets for the clinical treatment of many disorders involving the DA system including alcoholism.
Acknowledgments
This work was funded by PHS Grant AA13666 to SCS
References
- Allison DW, Ohran AJ, Stobbs SH, Mameli M, Valenzuela CF, Sudweeks SN, Ray AP, Henriksen SH, Steffensen SC. Synapse. 1. Vol. 60. New York, NY: 2006. Connexin-36 gap junctions mediate electrical coupling between ventral tegmental area GABA neurons; pp. 20–31. [DOI] [PubMed] [Google Scholar]
- Baldridge WH, Vaney DI, Weiler R. The modulation of intercellular coupling in the retina. Semin Cell Dev Biol. 1998;9(3):311–318. doi: 10.1006/scdb.1998.0235. [DOI] [PubMed] [Google Scholar]
- Belluardo N, Mudo G, Trovato-Salinaro A, Le Gurun S, Charollais A, Serre-Beinier V, Amato G, Haefliger JA, Meda P, Condorelli DF. Expression of connexin36 in the adult and developing rat brain. Brain Res. 2000;865(1):121–138. doi: 10.1016/s0006-8993(00)02300-3. [DOI] [PubMed] [Google Scholar]
- Bennett MV, Zukin RS. Electrical coupling and neuronal synchronization in the Mammalian brain. Neuron. 2004;41(4):495–511. doi: 10.1016/s0896-6273(04)00043-1. [DOI] [PubMed] [Google Scholar]
- Brodie MS, Appel SB. The effects of ethanol on dopaminergic neurons of the ventral tegmental area studied with intracellular recording in brain slices. Alcoholism, clinical and experimental research. 1998;22(1):236–244. [PubMed] [Google Scholar]
- Buzsaki G, Chrobak JJ. Temporal structure in spatially organized neuronal ensembles: a role for interneuronal networks. Curr Opin Neurobiol. 1995;5(4):504–510. doi: 10.1016/0959-4388(95)80012-3. [DOI] [PubMed] [Google Scholar]
- Caridha D, Yourick D, Cabezas M, Wolf L, Hudson TH, Dow GS. Mefloquine-induced disruption of calcium homeostasis in mammalian cells is similar to that induced by ionomycin. Antimicrob Agents Chemother. 2008;52(2):684–693. doi: 10.1128/AAC.00874-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condorelli DF, Belluardo N, Trovato-Salinaro A, Mudo G. Expression of Cx36 in mammalian neurons. Brain Res Brain Res Rev. 2000;32(1):72–85. doi: 10.1016/s0165-0173(99)00068-5. [DOI] [PubMed] [Google Scholar]
- Criswell HE, Ming Z, Kelm MK, Breese GR. Brain regional differences in the effect of ethanol on GABA release from presynaptic terminals. J Pharmacol Exp Ther. 2008;326(2):596–603. doi: 10.1124/jpet.107.135418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings DM, Yamazaki I, Cepeda C, Paul DL, Levine MS. Neuronal coupling via connexin36 contributes to spontaneous synaptic currents of striatal medium-sized spiny neurons. J Neurosci Res. 2008;86(10):2147–2158. doi: 10.1002/jnr.21674. [DOI] [PubMed] [Google Scholar]
- Deans MR, Gibson JR, Sellitto C, Connors BW, Paul DL. Synchronous activity of inhibitory networks in neocortex requires electrical synapses containing connexin36. Neuron. 2001;31(3):477–485. doi: 10.1016/s0896-6273(01)00373-7. [DOI] [PubMed] [Google Scholar]
- Diana M, Peana AT, Sirca D, Lintas A, Melis M, Enrico P. Crucial role of acetaldehyde in alcohol activation of the mesolimbic dopamine system. Ann N Y Acad Sci. 2008;1139:307–317. doi: 10.1196/annals.1432.009. [DOI] [PubMed] [Google Scholar]
- Dow GS, Hudson TH, Vahey M, Koenig ML. The acute neurotoxicity of mefloquine may be mediated through a disruption of calcium homeostasis and ER function in vitro. Malar J. 2003;2:14. doi: 10.1186/1475-2875-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarreta M, Hestrin S. Electrical synapses between GABA-releasing interneurons. Nat Rev Neurosci. 2001;2(6):425–433. doi: 10.1038/35077566. [DOI] [PubMed] [Google Scholar]
- Gallegos RA, Criado JR, Lee RS, Henriksen SJ, Steffensen SC. Adaptive responses of GABAergic neurons in the ventral tegmental area to chronic ethanol. J Pharmacol Exp Ther. 1999;291:1045–1053. [PubMed] [Google Scholar]
- Gatto GJ, McBride WJ, Murphy JM, Lumeng L, Li T-K. Ethanol self-infusion into the ventral tegmental area by alcohol-preferring (P) rats. Alcohol. 1994;11(6):557–564. doi: 10.1016/0741-8329(94)90083-3. [DOI] [PubMed] [Google Scholar]
- Gupta T, Syed YM, Revis AA, Miller SA, Martinez M, Cohn KA, Demeyer MR, Patel KY, Brzezinska WJ, Rhodes JS. Acute effects of acamprosate and MPEP on ethanol Drinking-in-the-Dark in male C57BL/6J mice. Alcoholism, clinical and experimental research. 2008;32(11):1992–1998. doi: 10.1111/j.1530-0277.2008.00787.x. [DOI] [PubMed] [Google Scholar]
- Gysling K, Wang RY. Morphine-induced activation of A10 dopamine neurons in the rat. Brain research. 1983;277:119–127. doi: 10.1016/0006-8993(83)90913-7. [DOI] [PubMed] [Google Scholar]
- Kitai ST, Shepard PD, Callaway JC, Scroggs R. Afferent modulation of dopamine neuron firing patterns. Curr Opin Neurobiol. 1999;9(6):690–697. doi: 10.1016/s0959-4388(99)00040-9. [DOI] [PubMed] [Google Scholar]
- Lassen MB, Brown JE, Stobbs SH, Gunderson SH, Maes L, Valenzuela CF, Ray AP, Henriksen SJ, Steffensen SC. Brain stimulation reward is integrated by a network of electrically coupled GABA neurons. Brain Res. 2007;1156:46–58. doi: 10.1016/j.brainres.2007.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long MA, Deans MR, Paul DL, Connors BW. Rhythmicity without Synchrony in the Electrically Uncoupled Inferior Olive. The Journal of Neuroscience. 2002;22(24):10898–10905. doi: 10.1523/JNEUROSCI.22-24-10898.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludlow KH, Bradley KD, Allison DW, Taylor SR, Yorgason JT, Hansen DM, Walton CH, Sudweeks SN, Steffensen SC. Acute and chronic ethanol modulate dopamine D2-subtype receptor responses in ventral tegmental area GABA neurons. Alcoholism, clinical and experimental research. 2009;33(5):804–811. doi: 10.1111/j.1530-0277.2009.00899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macey DJ, Schulteis G, Heinrichs SC, Koob GF. Alcohol. 2. Vol. 13. Fayetteville, NY: 1996. Time-dependent quantifiable withdrawal from ethanol in the rat: effect of method of dependence induction; pp. 163–170. [DOI] [PubMed] [Google Scholar]
- Maxeiner S, Kruger O, Schilling K, Traub O, Urschel S, Willecke K. Spatiotemporal transcription of connexin45 during brain development results in neuronal expression in adult mice. Neuroscience. 2003;119(3):689–700. doi: 10.1016/s0306-4522(03)00077-0. [DOI] [PubMed] [Google Scholar]
- McCracken CB, Hamby SM, Patel KM, Morgan D, Vrana KE, Roberts DCS. Synapse. I3. Vol. 58. New York, NY: 2005a. Extended cocaine self-administration and deprivation produces region-specific and time-dependent changes in connexin36 expression in rat; pp. 141–150. [DOI] [PubMed] [Google Scholar]
- McCracken CB, Patel KM, Vrana KE, Paul DL, Roberts DCS. Synapse. 1. Vol. 56. New York, NY: 2005b. Amphetamine withdrawal procedures region-specific and time-dependent changes in connexin36 expression in rat; pp. 39–44. [DOI] [PubMed] [Google Scholar]
- Mereu G, Fadda F, Gessa GL. Ethanol stimulates the firing rate of nigral dopaminergic neurons in unanesthetized rats. Brain research. 1984;292:63–69. doi: 10.1016/0006-8993(84)90890-4. [DOI] [PubMed] [Google Scholar]
- Mereu G, Gessa GL. Low doses of ethanol inhibit the firing of neurons in the substantia nigra, pars reticulata: a GABAergic effect? Brain research. 1985;348:201–203. doi: 10.1016/0006-8993(85)91249-1. [DOI] [PubMed] [Google Scholar]
- Moore H, Grace AA. A role for electrotonic coupling in the striatum in the expression of dopamine receptor-mediated stereotypies. Neuropsychopharmacology. 2002;27(6):980–992. doi: 10.1016/S0893-133X(02)00383-4. [DOI] [PubMed] [Google Scholar]
- Onn SP, Grace AA. Dye coupling between rat striatal neurons recorded in vivo: compartmental organization and modulation by dopamine. J Neurophysiol. 1994;71(5):1917–1934. doi: 10.1152/jn.1994.71.5.1917. [DOI] [PubMed] [Google Scholar]
- Perez-Velazquez JL, Valiante TA, Carlen PL. Modulation of gap junctional mechanisms during calcium-free induced field burst activity: a possible role for electrotonic coupling in epileptogenesis. J Neurosci. 1994;14(7):4308–4317. doi: 10.1523/JNEUROSCI.14-07-04308.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Placantonakis DG, Bukovsky AA, Aicher SA, Kiem H-P, Welsh JP. Continuous Electrical Oscillations Emerge from a Coupled Network: A Study of the Inferior Olive using Lentiviral Knockdown of Connexin36. 26The Journal of Neuroscience. 2006;19:5008–5016. doi: 10.1523/JNEUROSCI.0146-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiology & Behavior. 2005;84:53–63. doi: 10.1016/j.physbeh.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Steffensen SC, Svingos AL, Pickel VM, Henriksen SJ. Electrophysiological characterization of GABAergic neurons in the ventral tegmental area. J Neurosci. 1998;18(19):8003–8015. doi: 10.1523/JNEUROSCI.18-19-08003.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffensen SC, Taylor SR, Horton ML, Barber EN, Lyle LT, Stobbs SH, Allison DW. Cocaine disinhibits dopamine neurons in the ventral tegmental area via use-dependent blockade of GABA neuron voltage-sensitive sodium channels. Eur J Neurosci. 2008;28:2028–2040. doi: 10.1111/j.1460-9568.2008.06479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffensen SC, Walton CH, Hansen DM, Yorgason JT, Gallegos RA, Criado JR. Contingent and non-contingent effects of low-dose ethanol on GABA neuron activity in the ventral tegmental area. Pharmacol Biochem Behav. 2009;92(1):68–75. doi: 10.1016/j.pbb.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stobbs SH, Ohran AJ, Lassen MB, Allison DW, Brown JE, Steffensen SC. Ethanol suppression of ventral tegmental area GABA neuron electrical transmission involves NMDA receptors. J Pharmacol Exp Ther. 2004;311(1):282–289. doi: 10.1124/jpet.104.071860. [DOI] [PubMed] [Google Scholar]
- Tamas G, Buhl EH, Lorincz A, Somogyi P. Proximally targeted GABAergic synapses and gap junctions synchronize cortical interneurons. Nature neuroscience. 2000;3(4):366–371. doi: 10.1038/73936. [DOI] [PubMed] [Google Scholar]
- Theile JW, Morikawa H, Gonzales RA, Morrisett RA. Ethanol enhances GABAergic transmission onto dopamine neurons in the ventral tegmental area of the rat. Alcoholism, clinical and experimental research. 2008;32(6):1040–1048. doi: 10.1111/j.1530-0277.2008.00665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AJ, Lummis SC. Antimalarial drugs inhibit human 5-HT(3) and GABA(A) but not GABA(C) receptors. Br J Pharmacol. 2008;153(8):1686–1696. doi: 10.1038/bjp.2008.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Giessen RS, Koekkoek SKE, van Dorp S, De Gruijl JR, Cupido A, Khosrovani S, Dortland B, Wellershaus K, Degen J, Deuchars J, Fuchs EC, Monyer H, Willecke K, De Jeu MT, De Zeeuw CI. Role of Olivary Electrical Coupling in Cerebellar Motor Learning. Neuron. 2008;58:599–612. doi: 10.1016/j.neuron.2008.03.016. [DOI] [PubMed] [Google Scholar]
- Wentlandt K, Kushnir M, Naus CC, Carlen PL. Ethanol inhibits gap-junctional coupling between P19 cells. Alcoholism, clinical and experimental research. 2004;28(9):1284–1290. doi: 10.1097/01.alc.0000139705.17646.ba. [DOI] [PubMed] [Google Scholar]
- Wise RA. Dopamine, learning and motivation. Nat Rev Neurosci. 2004;5(6):483–494. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]
- Xiao C, Ye JH. Ethanol dually modulates GABAergic synaptic transmission onto dopaminergic neurons in ventral tegmental area: role of mu-opioid receptors. Neuroscience. 2008;153(1):240–248. doi: 10.1016/j.neuroscience.2008.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Xiao C, McArdle JJ, Ye JH. Mefloquine enhances nigral gamma-aminobutyric acid release via inhibition of cholinesterase. J Pharmacol Exp Ther. 2006;317(3):1155–1160. doi: 10.1124/jpet.106.101923. [DOI] [PubMed] [Google Scholar]