

Abstract
Gold nanoparticles (GNP) provide many opportunities in imaging, diagnostics, and therapies of nanomedicine. Hence, their biokinetics in the body are prerequisites for specific tailoring of nanomedicinal applications and for a comprehensive risk assessment.
We administered 198Au-radio-labelled monodisperse, negatively charged GNP of five different sizes (1.4, 5, 18, 80, 200nm) and 2.8nm GNP with opposite surface charges by intravenous injection into rats. After 24 h the biodistribution of the GNP was quantitatively measured by gamma-spectrometry.
The size and surface charge of GNP strongly determine the biodistribution. Most GNP accumulated in the liver increased from 50% of 1.4nm GNP to > 99% of 200nm GNP. In contrast, there was little size dependent accumulation of 18nm to 200nm GNP in most other organs. However, for GNP between 1.4nm and 5nm the accumulation increased sharply with decreasing size; i.e. a linear increase with the volumetric specific surface area. The differently charged 2.8nm GNP led to significantly different accumulations in several organs.
We conclude that the alterations of accumulation in the various organs and tissues, depending on GNP size and surface charge, are mediated by dynamic protein binding and exchange. A better understanding of these mechanisms will improve drug delivery and dose estimates used in risk assessment.
Keywords: gold nanoparticles, intravenous injection, female Wystar-Kyoto rat, organ accumulation, in vivo biodistribution, nanoparticle size, nanoparticle surface charge, hepato-biliary clearance, renal clearance
Introduction
The use of engineered nanoparticles (NP) offers a huge potential in the field of nanomedicine and nanotechnologies in the future. Especially gold nanoparticles (GNP) provide several opportunities in imaging, diagnostics, and therapies [1–3]. Their widespread use, their relatively simple generation and surface modification, and their special physico-chemical characteristics make GNP attractive candidates for the detection of cancerous cells as well as in tumor targeting [4–6]. There are, therefore, numerous studies concerning the properties of GNP as drug carriers [1, 2, 7]. Moreover, different administration routes such as inhalation, ingestion, or intravenous (i.v.) injection are possible. The advantage of this latter administration route is the direct access to the blood circulation and thereby a rapid distribution throughout the entire body.
Nevertheless, the investigation of cytotoxic effects is crucial. Based upon in vivo studies no toxic effects of 12.5nm GNP are found in the liver, lungs, kidneys, spleen or brain [8, 9]. Also, toxicity assessments of GNP of different sizes (3nm, 10nm, 50nm, and 100nm) in zebrafish embryos showed only minimal sub-lethal toxic effects [10]. A size-dependent toxic impact of GNP was investigated in vitro which occurred for 1.4nm GNP, but not for 15nm, or 0.8nm GNP [11]. The findings of the extraordinary cytotoxicity of the 1.4nm GNP were explained by the perfect fitting of these GNP in the major grooves of the DNA causing its immobility [9]. In addition, the cellular uptake of GNP varies with size and shape causing effects which need to be considered in determining the design of the nanomaterial [12].
Moreover, an important focus in order to achieve safe and well characterized tools for nanomedicine is the in vivo biodistribution of candidate NP as this will allow for determining the GNP dose to secondary organs which may eventually cause adverse health effects. Interestingly, commercially available 1.9nm GNP used as X-ray contrast agents, were surface modified “with a highly water soluble organic shell” (but not explicitly disclosed) such that they were excreted almost quantitatively via urine [13] making them ideal diagnostic tools without long-lasting residence time in the body. Similarly, 4 h after i.v. injection the renal clearance of 99mTc-labeled, cysteine-surface modified quantum dots (QD) with hydrodynamic diameters of < 5.5nm were shown by a strong QD increase of up to 80% in the urine. While for QD, with a hydrodynamic diameter of 8.65nm, the uptake in the liver reached 26.5% of injected dose (ID) and it was 6.3% ID [14] for the spleen.
Also, surface charge affects the biodistribution of GNP so that positive charged particles accumulate more in the kidneys while negative and non-charged particles showed a higher accumulation in the liver [15].
Once retained in the liver, the hepato-biliary pathway allows for clearance through the biliary duct into the duodenum. The daily excretion of 20nm low-density lipoprotein-GNP through the biliary duct was almost 5% from 4 to 12 days after i.v. administration [16]. For 50 – 100nm sized mesoporous silica nanoparticles, a rapid hepato-biliary transport (within < 30min was shown for highly charged particles) while less charged NP remained in the liver [17] for up to over 90 days.
The aim of the present study was to evaluate the biodistribution of monodisperse, spherical GNP of different sizes with well defined surface ligands of different surface charges. Therefore, a large size range of five different sizes (1.4nm, 5nm, 18nm, 80nm and 200nm) of mono-sulfonated triphenylphosphine (TPPMS) stabilized GNP, as well as opposite surface charge, with either positive cysteamine (CA) charge or negative thioglycolic acid (TGA) charge of equal size (2.8nm) were intravenously injected into healthy adult female rats. All GNP were radioactively labeled with 198Au allowing for a 100% balanced quantitative determination of the GNP biodistribution after 24 h. Our study aims to serve as a proof-of-principle for the validity of a systematic investigation of size and surface charge of GNP. Therefore, in this study we present the 24 h body distribution after i.v. injection of GNP. These initial studies need to be followed up by a complete kinetic study with more time points which are however, currently under scrutiny.
Methods
Animals
Healthy, female Wistar-Kyoto rats (WKY/Kyo@Rj rats, Janvier, Le Genest Saint Isle, France), 8 to 10 weeks old (approx. 250g body weight) were housed in pairs in humidity- and temperature-controlled ventilated cages on a 12 h day/night cycle.
A rodent diet and water were provided ad libitum. All experiments were conducted under German Federal guidelines for the use and care of laboratory animals and were approved by the Regierung von Oberbayern (Government of District of Upper Bavaria, Approval No. 211-2531-94/04) and by the Institutional Animal Care and Use Committee of the Helmholtz Center Munich.
Nanoparticle preparation and characterization
Mono-sulfonated triphenylphosphine (TPPMS) stabilized GNP (Table 1) (1.4nm and 18nm) were synthesized following previously published procedures [11]. Citrate stabilized, spherical GNP (5nm, 80nm, 200nm) were purchased from Plano (Wetzlar, Germany). Ligand exchange (citrate to TPPMS) was accomplished as previously described [11].
Table 1.
Physico-chemical parameters and dose metrics of administered 198Au-NP and their radioactive labels; additionally does metrics of 198Au-NP mass, surface area, number, and doses delivered to each rat by i.v. injection.
| Gold NP, core diameter (nm) | 1.4 | 5 | 18 | 80 | 200 | 2.8 | 2.8 |
|---|---|---|---|---|---|---|---|
| Surface modification, ligand | TPPMS | TPPMS | TPPMS | TPPMS | TPPMS | TGA (COO−) | CA (NH3+) |
| Hydrodynamic diameter (nm) | 2.9 # | 12.1§ | 21§ | 85$ | 205$ | NDa | NDa |
| Polydispersity index (PdI) | ND | 0.19 | 0.10 | 0.12 | 0.05 | NDa | NDa |
| pH Value of suspension | 5.6 | 5.8 | 6.4 | 5.4 | 5.5 | ND | ND |
| Zeta potential (mV) | −20.6 ± 0.5 | − 21.1 ± 1.4 | −22.8 ± 3.1 | − 22.3 ± 1.6 | − 41.3 ± 4.5 | negative | positive |
| Specific 198Au radioactivity (GBq/g) | 72 | 6.7 | 31 | 14.9 | 11.5 | 15.8 | 5.5 |
| Isotope ratio of 198Au to stable 197Au | 8.0 × 10−6 | 7.4 × 10−7 | 3.4 × 10−6 | 1.7 × 1−6 | 1.3 × 10−6 | 1.7 × 10−6 | 6.1 × 10−7 |
| Ratio of 198Au per NP | 6.6 × 10−4 | 2.8 × 10−3 | 0.60 | 26 | 307 | 1.2 × 10−3 | 4.1 × 10−4 |
| Admin. 198Au radioactivity (kBq) per rat | 0.22 ± 0.05 | 0.21 ± 0.3 | 0.09 ± 0.05 | 0.28 ± 0.03 | 0.23 ± 0.02 | 0.025 ± 0.003 | 0.16 ± 0.02 |
| Admin. mass of gold NP (μg) per rat | 3.1 ± 0.6 | 43.7 ± 5.3 | 2.9 ± 1.5 | 18.5 ± 2.3 | 19.8 ± 1.7 | 1.6 ± 0.2 | 29.0 ± 3.4 |
| Admin. number of gold NP per rat | 1.1 ± 0.2 × 1014 | 3.5 ± 0.4 × 1013 | 5.1 ± 2.6 × 1010 | 3.6 ± 0.5 × 109 | 3.9 ± 0.3 × 109 | 7.3 ± 0.9 × 1012 | 1.3 ± 0.2 x1014 |
| Admin. Surface Area (cm2) | 7.0 ± 1.4 | 27.6 ± 3.4 | 0.56 ± 0.27 | 0.73 ± 0.09 | 0.78 ± 0.07 | 1.4 ± 0.2 | 26.0 ± 3.0 |
TPPMS Triphenylphosphine mono-sulfonate; TGA (COO−) thioglycolic acid (mercaptoacetic acid); CA (NH3+) cysteamine (2-aminoethanethiol)
as determined earlier [47]
DLS measurement using Malvern HPPS5001, Herrenberg, Germany; in addition, TEM and UV/vis analyses confirmed the core size of the gold NP [21]
DLS measurement using Malvern Zetasizer Nano ZS, Herrenberg, Germany
Hydrodynamic diameter and PdI were not determined but the mean core size ± STD of both batches of GNP already surface modified with either the carboxyl or the amino groups used were determined to be 2.8 ± 0.4 nm
ND not determined
The fabrication of the 2.8nm GNP followed the same procedure as described in [11] by performing ligand exchange of initially 1.4nm GNP stabilized with triphenylphosphine (TPP). Via phase transfer from a dichloromethane phase to an aqueous phase TPP is replaced by thioglycolic acid (TGA) for carboxylated, negatively charged GNP and by cysteamine (CA) for aminated, positively charged particles, respectively. The driving force for this exchange reaction is the higher binding affinity of thiol groups to gold compared to phosphine groups accompanied with the change in polarity, since the GNP pass from the dichloromethane phase into the aqueous phase, when TPP is replaced by TGA or CA, respectively. Hence, the solubility/dispersion of the GNP in water gives immediate evidence that the ligand exchange was successful.
As has already been observed by others, the replacement of TPP by thiol ligands is associated with a growth of the gold core [18, 19]. This also holds for our ligand exchange procedure, where the introduction of thiol ligands TGA and CA resulted in an increase of the mean core particle size, which was determined by TEM analyses to be 2.8 ± 0.4 nm for both ligands.
All GNP were activated by neutron irradiation (197Au (n, γ) 198Au). For this, the GNP were activated at a neutron flux of 1014 cm−2 s−1 at the research reactor of the Helmholtz Center Berlin (formerly Hahn-Meitner Institute, Berlin), Germany. Gold amounts and irradiation times were adjusted to provide sufficient 198Au radioactivity for the subsequent in vivo studies. Specific 198Au radioactivity and the isotope ratio of 198Au to stable 197Au are given in Table 1. Note that this ratio is extremely low so that statistically, only one 198Au isotope can be found in all GNP from 1.4nm to 18nm and most of the GNP do not contain any 198Au atom at all; but in the 80nm and 200nm GNP an average number of 20 and 300 198Au atoms, respectively, are contained in the NP matrix, Table 1.
After the neutron irradiation immediately prior to rat application, the 1.4nm and the 2.8nm 198Au-NP solution was filtered through a 10cm column of Celite to remove agglomerates; losses determined by 198Au radioactivity accounted for about 10%. All other 198Au-NP suspensions from 5nm to 200nm were visually controlled for precipitates and their correct pink translucent colour of the colloidal suspension immediately prior to the application in rats; no changes were found when compared to the suspension prior to irradiation.
The hydrodynamic diameters (HD) and the polydispersity index (PdI) of the all NP from 5nm to 200nm were measured in duplicate by photon correlation spectroscopy (PCS; Malvern HPPS5001 or Nano Zetasizer ZS, Herrenberg, Germany). The HD were slightly increased compared to the core GNP diameters according to the surface modifications and a minute fraction of agglomerates was sometimes observed (however, when NP volume and not the intensity, was plotted the fraction of agglomerates disappeared; data not shown). The aqueous GNP solutions of 1.4nm to 18nm NP suspension remained stable during at least two weeks without any detectable precipitation or change of color. Due to gravitational sedimentation, 80nm and 200nm GNP settled during the two weeks but could, by vortexing, be dispersed into the same pink suspension as before.
Zeta-potential measurements were made in duplicate using appropriate working dilutions in a Zetapalssystem (Brookhaven Instruments Corporation, Holtsville and later using Nano Zetasizer, Malvern, Herrenberg). The NP suspensions were diluted with purified water to adjust the concentration level (ratio 1:1). Measurements were made in duplicates (10 cycles each run).
Nanoparticle administration and animal maintenance in metabolic cages
Nanoparticle suspensions were administered to the animals via i.v. injection. The rats were anesthetized by inhalation of 5% isoflurane until muscular tonus relaxed. The rats were kept under isoflurane anesthesia and a flexible i.v. catheter (24 G, ¾ in) was placed into the tail vein. Subsequently, 120μl of the suspension was slowly injected using a 1-ml-insulin-syringe (dead space of syringe connector and catheter was determined to vary between 50 and 70μl) resulting in an effectively injected GNP suspension volume of 70 to 50μl.
Retention and excretion
After the i.v. injection and recovery from anesthesia, each rat was kept isolated in metabolic cages for the next 24 h and the urine and feces were collected separately and quantitatively.
Sample preparation
24 h after administration, the rats were anesthetized (5% isoflurane inhalation) and euthanized by exsanguination via the abdominal aorta. Approximately 70% of the total blood volume was withdrawn based on an estimation of the total blood volume of the body weight [20]. For radio-analysis, organs – each in toto - and tissues listed below, as well as total excretion and the entire remaining carcass, were collected as previously described [21].
Organs: lungs, liver, spleen, kidneys, brain, heart, uterus, gastro-intestinal tract (GIT): esophagus, stomach, small and large intestine;
Tissues: the injected site of the tail was separated;
Remainder: total remaining carcass (skin, skeleton, soft tissue) beyond the listed organs and tissues;
Excretion: total urine and feces, collected separately
Total exsanguinated blood
During dissection, no organs were cut and all fluids were cannulated where necessary in order to avoid cross contaminations.
The hepato-biliary GNP clearance was calculated summing up the GNP percentages determined by the small intestine, colon and total fecal excretion; it describes the biliary pathway from the liver into the small intestine and subsequently into fecal excretion.
Radio analysis
The 198Au radioactivity of all samples were measured without any further physico-chemical preparation by γ-spectroscopy in either a lead-shielded 10 ml or a lead-shielded 1 l well type NaI(Tl) scintillation detector. The count rates were adjusted for physical decay and background radiation. Additionally, count rates were calibrated to a 198Au reference source at a reference date in order to correlate 198Au radioactivities to the numbers, surface areas, and masses of the GNP. Samples yielding net counts (i.e. background-corrected counts) in the photo-peak region-of-interest of the 198Au gamma spectrum less than three standard deviations of the totally measured counts were defined to be below the detection limit. For a complete balance of the administered 198Au radioactivity within each rat, 198Au radioactivities of all samples were summed for each rat and used as a denominator for the calculation of the 198Au percentage of each sample.
Blood correction adjustment
Blood contents of organs and the remaining body were calculated according to the findings of Oeff et al. [20]. The NP content of the remaining blood volume of each sample was estimated and subtracted from the measured 198Au radioactivity to obtain the absolute 198Au activity of the tissue or organ. In the case of the carcass, the difference between the total blood volume of the animal (6.7% of the body weight [20]), the sum of all organ blood contents, and the collected blood sample was calculated to be the blood volume of the carcass.
Calculations and statistical analysis
All calculated values are given as a percentage of the relevant integral radioactivity of all samples in each animal with the standard deviation (SD).
All calculated significances are based on a one-way analysis of variance (ANOVA) followed by a post hoc Tukey test. In the case of an individual two group comparison, the unpaired t-test was used. The criterion for statistical significance was p ≤ 0.05 if not otherwise stated. Outliers were identified by the Grubbs test and excluded.
Results
Particle characterization and administration
Physico-chemical parameters of all GNP are given in Table 1 indicating that the GNP were single and showing a narrow size distribution prior to in vivo injection; in addition, the mean administered GNP mass, surface, and number doses per rat are listed in Table 1. Note that in the following, all GNP percentages of the administered GNP dose provided for organs and tissues represent exclusive GNP retention in the tissue without GNP of the blood volume in these organs and tissues – see Methods.
Effect of TPPMS surface modified GNP size from 1.4nm to 200nm
Our studies showed a strong size dependency of the distribution and accumulation of GNP in all organs, tissues and excretions. GNP percentages per organ or tissue are given in Figs. 1A–J and 2A; GNP percentages of the ID per gram of organ or tissue are given in Table 2.
Figure 1.
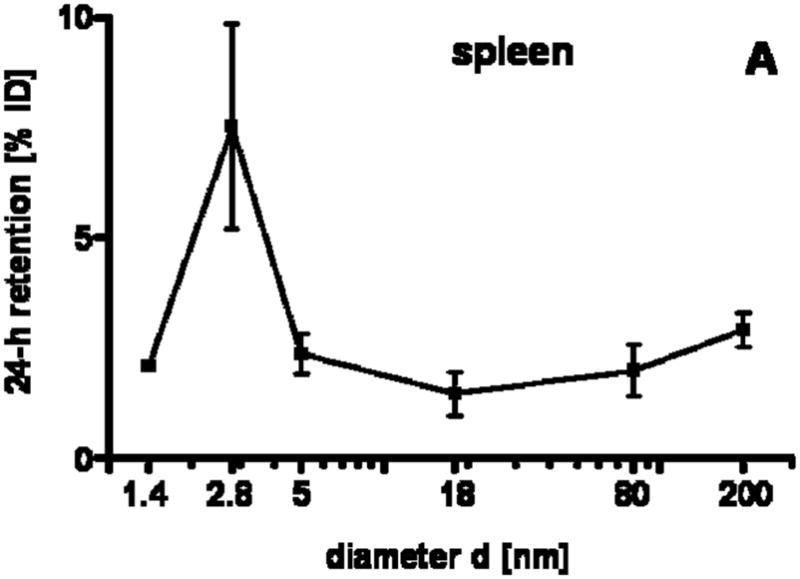
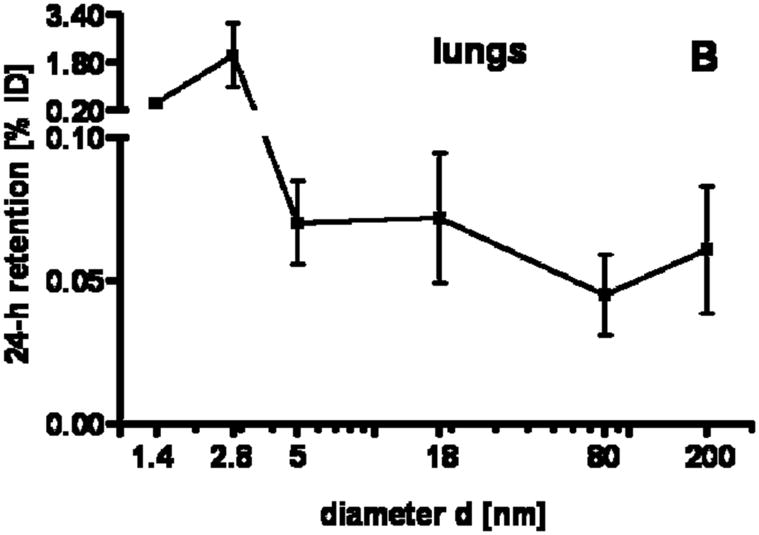
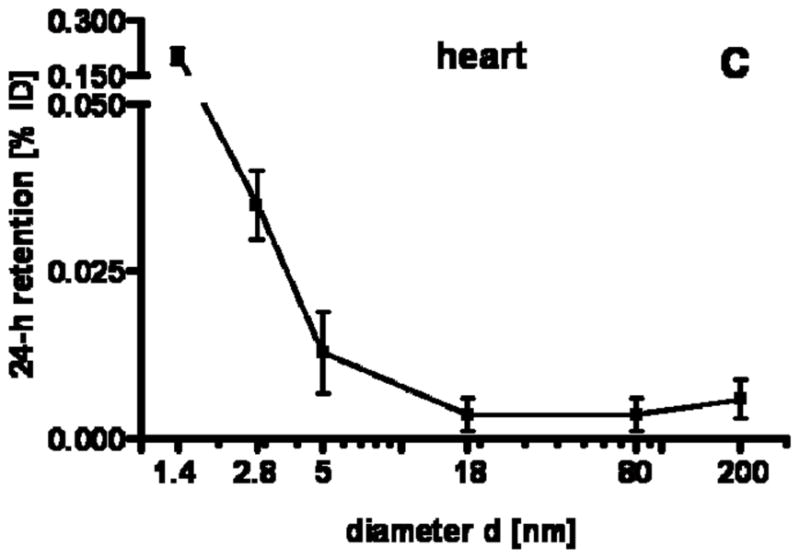
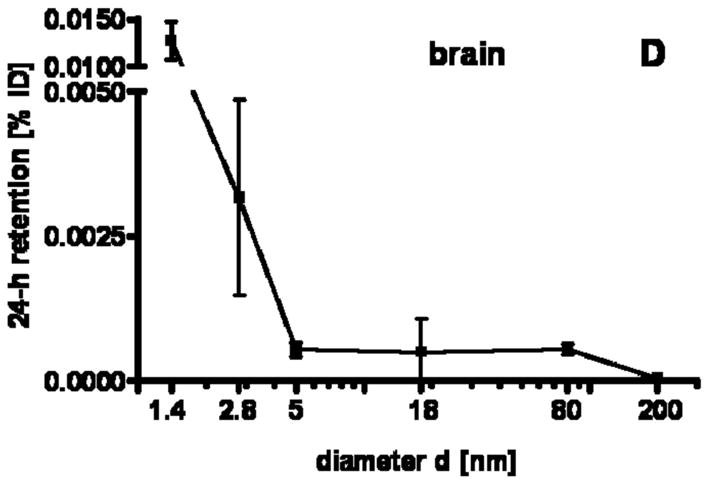
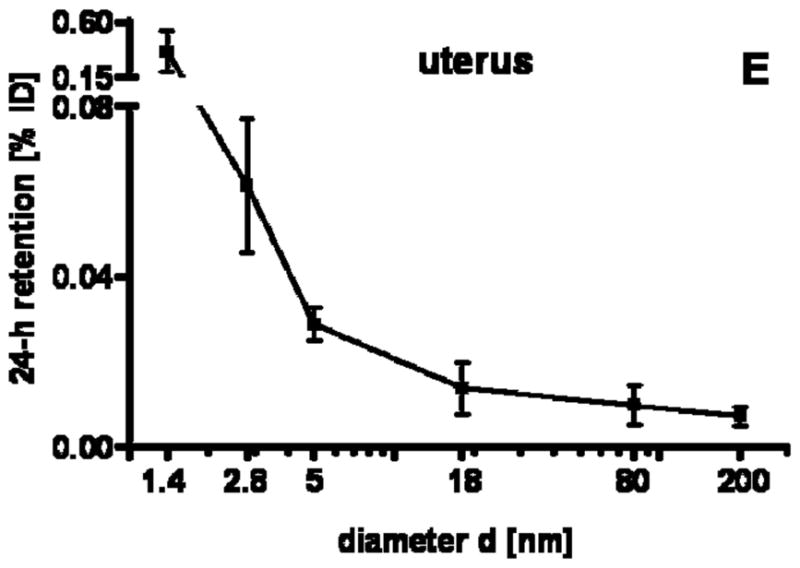
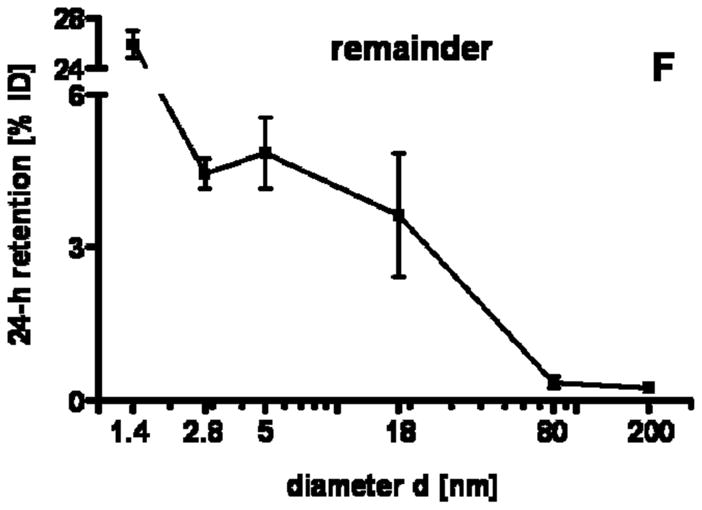
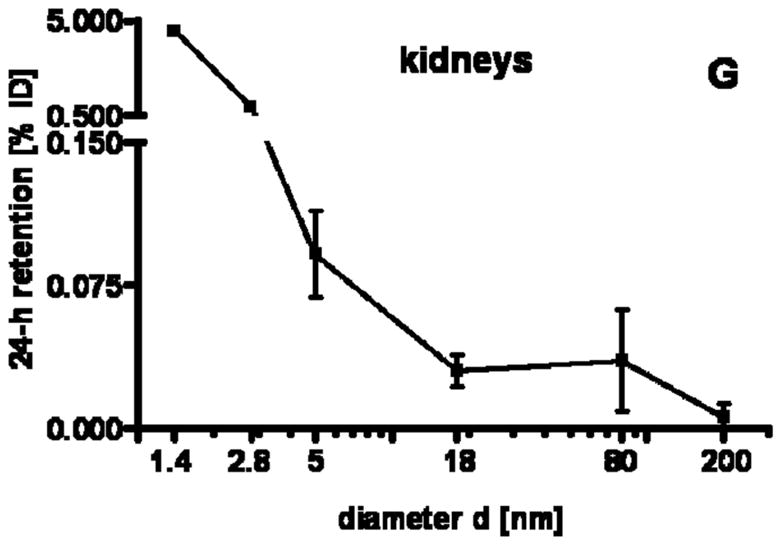
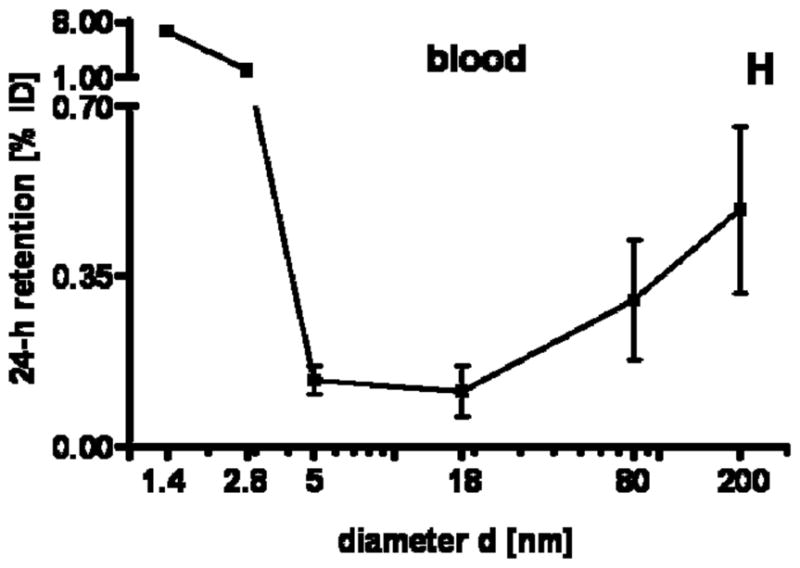
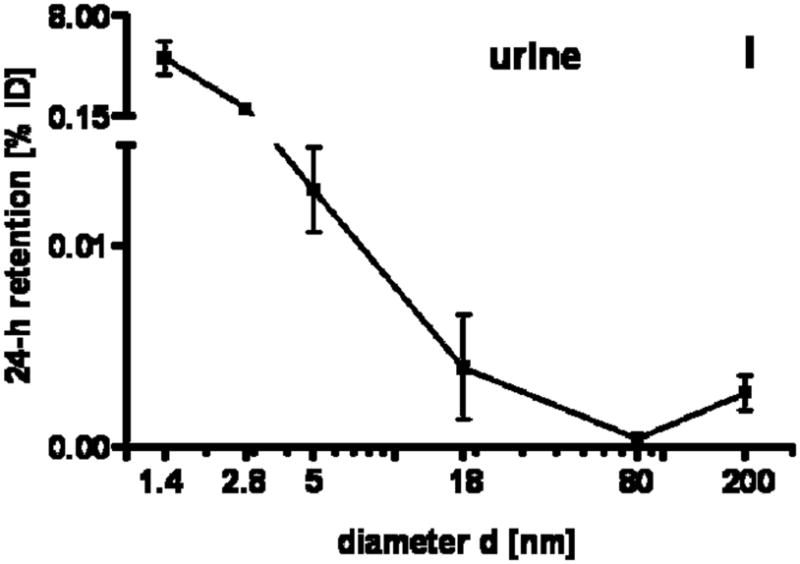

24 h retention/accumulation of intravenously injected, negatively charged, spherical GNP (1.4nm, 5nm, 18nm, 80nm, 200mm coated with TPPMS; 2.8nm carboxyl-coated); GNP percentages of initial dose are given for whole organs, the entire remaining carcass and total blood and urine; in each panel the respective organ, tissue or body fluid is indicated. Data are mean + SD, n = 4 rats. Note log scale of x-axis. In the table panel significant changes of accumulation of given GNP sizes versus those of all other GNP sizes are listed as determined by the ANOVA test. (* p<0.05, + p<0.01; α p<0.001).
Figure 2.
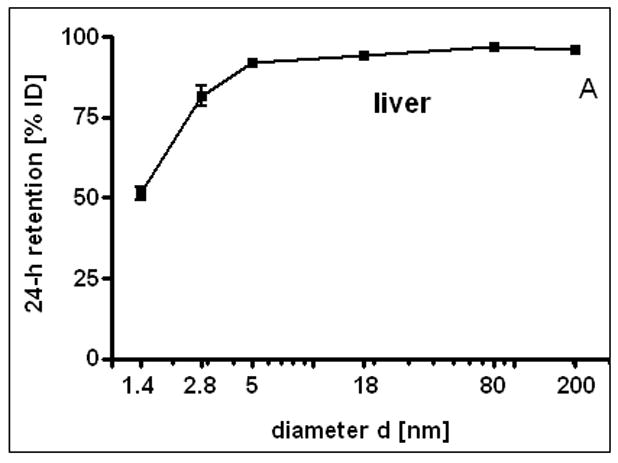
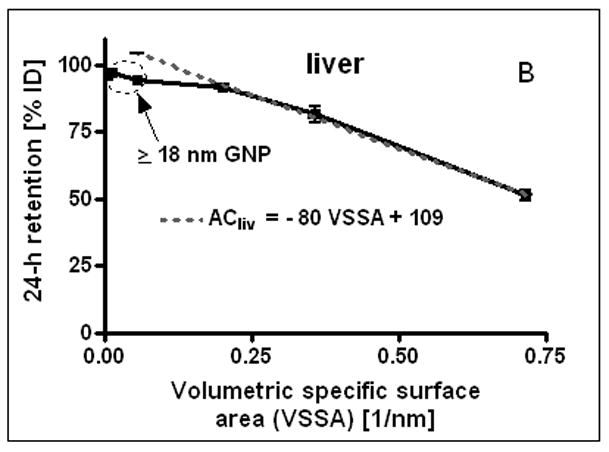
24 h liver accumulation of intravenously injected, negatively charged, spherical GNP (1.4nm, 5nm, 18nm, 80nm, 200mm coated with TPPMS; 2.8nm carboxyl-coated); GNP percentages of initial dose in whole liver. Panel A: Percentages plotted over GNP diameter in log-scale. Panel B: Percentages plotted over the volumetric specific surface area (VSSA 1/d) of GNP. Data are mean + SD, n = 4 rats. Note log scale of x-axis. Significant changes of accumulation of given GNP sizes versus those of all other GNP sizes are indicated as determined by the ANOVA test. (* p<0.05, + p<0.01; α p<0.001).
Table 2.
GNP concentrations per gram of organ, tissue or blood: 24 h retention/accumulation of intravenously injected GNP (1.4nm, 5nm, 18nm, 80nm, 200mm coated with TPPMS; 2.8nm carboxyl- and amino-coated); GNP concentrations are given as percentages of initial dose. Data are mean ± SD, n = 4 rats.
| 198Au-GNP | 1.4 nm | 5 nm | 18 nm | 80 nm | 200 nm | 2.8 nm COO− | 2.8 nm NH3+ |
|---|---|---|---|---|---|---|---|
| Liver | 5.1 × 100 ± 4.8 × 10−1 | 1.0 × 10+1 ± 2.2 × 10−1 | 1.0 × 10+1 ± 1.1 × 100 | 1.1 × 10+1 ± 1.3 × 100 | 1.2 × 10+1 ± 1.3 × 100 | 9.2 × 100 ± 2.7 × 10−1 | 7.9 × 100 ± 4.8 × 10−1 |
| Spleen | 2.3 × 100 ± 2.4 × 10−1 | 2.7 × 100 ± 1.2 × 100 | 1.6 × 100 ± 3.8 × 10−1 | 1.9 × 100 ± 5.2 × 10−1 | 3.6 × 100 ± 5.6 × 10−1 | 8.9 × 100 ± 1.9 × 100 | 1.3 × 101 ± 1.4 × 100 |
| Lungs | 2.0 × 10−1 ± 1.4 × 10−1 | 3.5 × 10−2 ± 5.1 × 10−3 | 3.8 × 10−2 ± 1.3 × 10−2 | 2.7 × 10−2 ± 1.1 × 10−2 | 3.7 × 10−2 ± 1.4 × 10−2 | 1.2 × 100 ± 3.4 × 10−1 | 1.2 × 100 ± 9.2 × 10−2 |
| Kidneys | 1.9 × 100 ± 2.8 × 10−1 | 3.7 × 10−2 ± 1.0 × 10−2 | 1.3 × 10−2 ± 4.5 × 10−3 | 1.6 × 10−2 ± 1.3 × 10−2 | 2.5 × 10−3 ± 3.2 × 10−3 | 4.0 × 10−1 ± 2.8 × 10−2 | 4.8 × 10−1 ± 2.9 × 10−2 |
| Heart | 1.8 × 10−1 ± 5.4 × 10−3 | 6.9 × 10−3 ± 2.3 × 10−3 | 9.4 × 10−3 ± 3.5 × 10−3 | 3.4 × 10−3 ± 2.0 × 10−3 | 5.6 × 10−3 ± 2.2 × 10−3 | 3.9 × 10−2 ± 1.6 × 10−3 | 5.2 × 10−2 ± 1.6 × 10−3 |
| Brain | 6.4 × 10−3 ± 1.2 × 10−3 | 6.0 × 10−5 ± 4.3 × 10−5 | 3.1 × 10−5 ± 3.7 × 10−5 | 2.9 × 10−4 ± 6.8 × 10−5 | 2.1 × 10−5 ± 2.3 × 10−5 | 1.8 × 10−3 ± 4.0 × 10−4 | 2.8 × 10−3 ± 2.2 × 10−4 |
| Uterus | 1.8 × 10−1 ± 5.1 × 10−2 | 7.9 × 10−3 ± 1.2 × 10−3 | 3.7 × 10−3 ± 1.8 × 10−3 | 1.5 × 10−3 ± 7.5 × 10−4 | 1.2 × 10−3 ± 3.8 × 10−4 | 2.6 × 10−2 ± 2.8 × 10−3 | 3.2 × 10−2 ± 5.1 × 10−3 |
| Remainder | 3.5 × 10−1 ± 2.1 × 10−2 | 7.7 × 10−3 ± 1.6 × 10−3 | 5.9 × 10−3 ± 3.0 × 10−3 | 1.6 × 10−2 ± 6.6 × 10−3 | 2.6 × 10−2 ± 9.4 × 10−3 | 2.2 × 10−2 ± 1.1 × 10−3 | 4.4 × 10−2 ± 2.6 × 10−3 |
| Blood | 1.1 × 10−1 ± 6.0 × 10−3 | 2.4 × 10−2 ± 3.6 × 10−3 | 1.6 × 10−2 ± 6.6 × 10−3 | 1.6 × 10−3 ± 5.0 × 10−4 | 8.6 × 10−4 ± 6.0 × 10−4 | 1.1 × 10−1 ± 3.0 × 10−3 | 9.5 × 10−2 ± 5.2 × 10−3 |
GNP retention in blood
Surprisingly, we found small but detectable percentages > 0.1% of the administered GNP of all sizes in the blood which were still circulating 24 h after administration, Fig. 1H. These percentages had a minimum at 5–18nm and increased slightly to 200nm. However, they increased even more sharply from 5nm to 2.8nm and 1.4nm up to about 8%. This increasing percentage with decreasing GNP in the size ranged between 1.4nm and 5nm was proportional to the volumetric specific surface area (VSSA) [22] which corresponds to the inverse diameter 1/d of the GNP.
By distinguishing GNP retention in serum versus blood cells by means of centrifugation there was a ten-fold higher retention of 18nm GNP in the cell fraction compared to the serum, indicating their binding, in part, to blood cells (since 18nm GNP did not settle in an aqueous suspension under the same centrifugation condition). The same was also found for the 80nm and 200nm GNP. However, while 18nm GNP did not settle in an aqueous suspension, detectable fractions of 80nm and 200nm GNP were spun down under the same centrifugation conditions indicating that the GNP may also have settled as free GNP. Fractions of 5nm GNP were equally found in the serum and blood cells. The 1.4nm GNP percentage in the serum was as high as 75% compared to 25% in blood cells.
GNP accumulation in the liver
After the i.v. injection, most GNP were rapidly eliminated from circulation and accumulated predominantly in the liver, Fig. 2A. However, while GNP accumulation was in the range from 91.9 to 96.9% for 5, 18, 80 and 200nm GNP, the distribution of 1.4nm and carboxylated 2.8nm GNP showed significantly lower percentages of 51.3% and 81.6%, respectively. Hence, for 5, 18, 80 and 200nm GNP accumulation in any other organ or tissue was only a few percent and even lower than 4% for 200nm GNP, Fig. 1A–I. In contrast, almost 50% of the 1.4nm GNP escaped being trapped in the liver and thus accumulated in the other organs and tissues as well as excretion. Interestingly, the negatively charged 2.8nm GNP with a carboxyl surface coating fits very well into this size dependency pattern with 81.6 in the liver and hence almost 20% in the other organs and tissues as well as excretion, (Fig. 1A–I and Fig. 2A). When plotting liver accumulation ACliv between 1.4nm and 5nm over the volume specific surface area (VSSA), it declined linearly with increasing VSSA (in 1/nm) of the GNP. This can be described with a linear equation:
I.e. with increasing specific surface area fewer GNP are trapped in the liver, Fig. 2B.
For morphologic GNP identification in the liver we localized 18nm GNP using transmission electron microscopy (TEM) (Fig. 3). These GNP were predominantly found in Kupffer cells, (panel A) but also, to a smaller extent, in endothelial cells (panel B) and in hepatocytes (panel C). In the endothelial cells and hepatocytes the 18nm GNP appeared mostly isolated while in Kupffer cells we found both small and large agglomerates of GNP.
Figure 3.
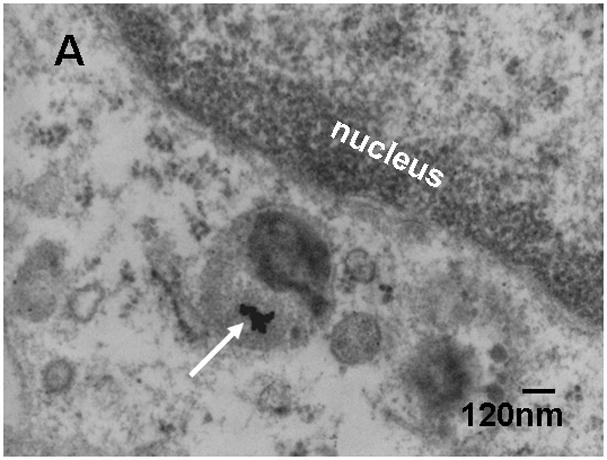
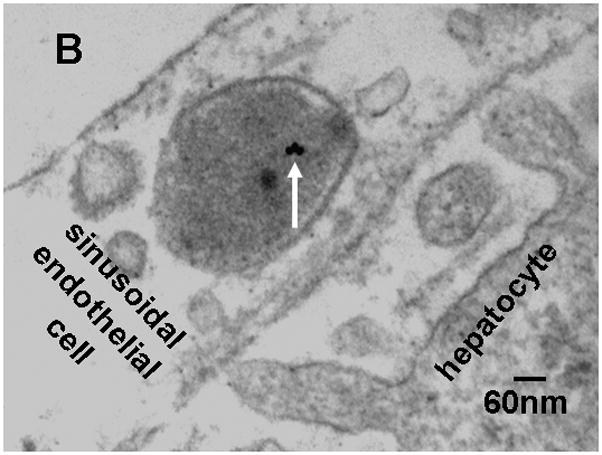
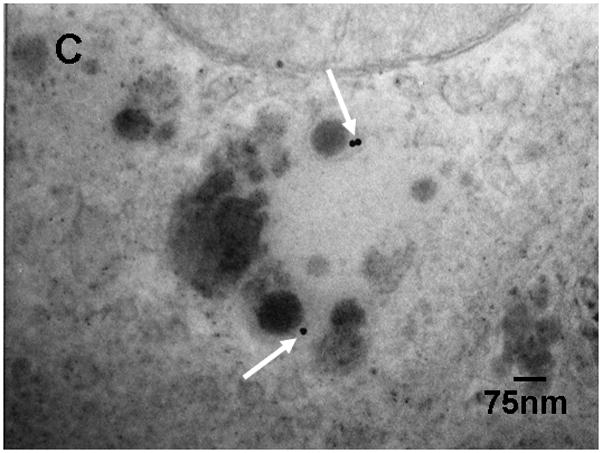
Transmission electron micrographs of TPPMS-coated 18nm GNP in a Kupffer cell (GNP agglomerate) panel A), an endothelial cell (panel B) and a hepatocyte (panel C); arrows point towards GNP.
GNP accumulation in spleen
The spleen, which exhibits macrophages in its vasculature like the liver, does not show a GNP size dependent pattern, but the accumulated GNP percentages of around 2% were quite constant for all TPPMS coated GNP ranging from 1.4nm to 200nm, Fig. 1A. However, the carboxyl-coated 2.8nm GNP showed a significantly higher accumulation of 8.6% compared to all TPPMS coated GNP. A significantly higher accumulation of 11.4% than those of all TPPMS coated GNP was observed for the positively charged amino coated 2.8nm GNP, see Fig. 5A.
Figure 5.
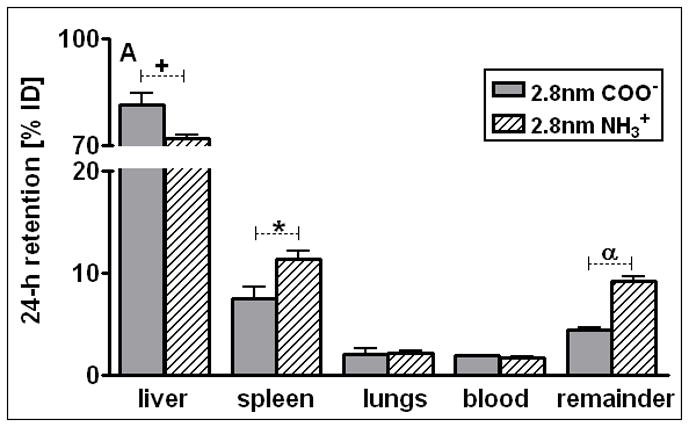
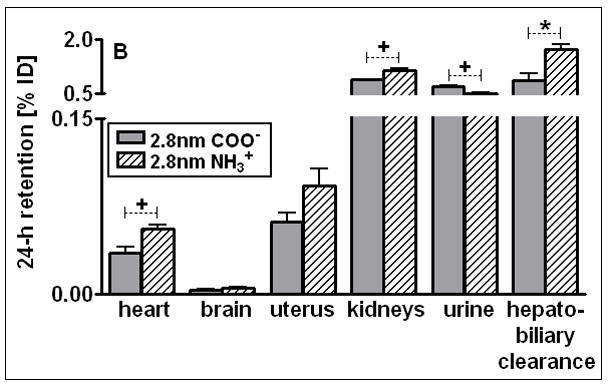
Percentages of 24 h retention/accumulation of intravenously injected, 2.8nm GNP (coated either thioglycolic acid or with cysteamine); panel A: data obtained from liver, spleen, lungs, blood, remaining carcass; panel B: data obtained from heart, brain, uterus, kidneys urine and hepato-biliary clearance are given. Data are mean ± SD, n = 4 rats. Significant changes of accumulation of given GNP sizes versus those of the other GNP sizes are indicated as determined by the ANOVA test. (* p<0.05, + p<0.01; α p<0.001).
GNP accumulation in other organs and tissues
Regarding GNP size dependency, the liver is the only organ which showed increased accumulation with increasing GNP size. In contrast, most of the other organs, tissues, and blood showed quite a low and similar size independent accumulation of GNP ranging from 18nm to 200nm size: the kidneys, heart, brain, uterus, remainder (Fig. 1C–G). Yet, this size independent accumulation of 18nm, 80nm, and 200nm GNP varies considerably among these organs and tissues. In contrast, from 5nm to 2.8nm and 1.4nm GNP the accumulation rose sharply with decreasing GNP size (Fig. 1C–G). Interestingly, the accumulated percentages of the small GNP were proportional to the volumetric specific surface area (VSSA) which is inversely related to the diameter 1/d. Slopes of the linear equation were determined by data fitting for each organ, remaining carcass, and blood, (see Table 3).
Table 3.
Slope m of organ accumulation AC over VSSA for GNP of 1.4nm to 2.8nm to 5nm diameter; Slopes were determined by data fitting obtained from each organ, or remaining carcass or blood by the linear equation ACorg = m * VSSA + b; R2 is coeffiecient of determination.
| Organ, tissue or body fluid | Slope m, [%/d−1] | R2 |
|---|---|---|
| Kidneys | 8.8 | 0.98 |
| heart | 0.39 | 0.96 |
| brain | 0.024 | 0.99 |
| uterus | 0.67 | 0.96 |
| remainder | 44 | 0.90 |
| blood | 13 | 0.99 |
| urine | 9.5 | 0.97 |
This described pattern was found for all TPPMS coated GNP accumulated in the lungs, Fig. 1B; i.e. a constant percentage from 18nm to 200nm of about 0.06% and a significant increase from 0.07% for 5nm GNP to 0.43% of 1.4nm was observed. However, the carboxyl-coated 2.8nm GNP showed a much higher accumulation than all TPPMS coated GNP. This pattern corresponded to the pattern in spleen, Fig. 1A.
Interestingly, the remaining carcass, consisting of the skeleton and soft tissue (muscle, fat, skin), showed significantly higher percentages of the ID compared to all organs except the liver, Fig. 1F. These high percentages relate to the high mass of the remaining carcass since the GNP percentages per gram of carcass are quite low, Table 2. GNP retention in the carcass followed the same size dependency pattern as the blood and other organs, Fig. 1F. Additionally, the collected urine during the 24 h after GNP application also showed a two-phase pattern with size independent percentages of about 0.004% for 18nm to 200nm GNP, and a size dependent VSSA proportional increase with the significantly highest percentage of 4.7% for the 1.4nm GNP.
Hepato-biliary GNP clearance
The hepato-biliary GNP clearance describes the biliary pathways from the liver into the small intestine and subsequently into fecal excretion. Hepato-biliary clearance of the TPPMS coated GNP showed an inverse linear relationship to the GNP diameter over the size range of 5nm to 200nm. The linear equation is given in Fig. 4. The clearance of the carboxylated 2.8nm GNP also fit into this equation. Only the smallest GNP of 1.4nm did not follow this size relationship and were cleared with the highest percentage of 4.6 ± 0.4% which was significantly higher than for all other GNP.
Figure 4.
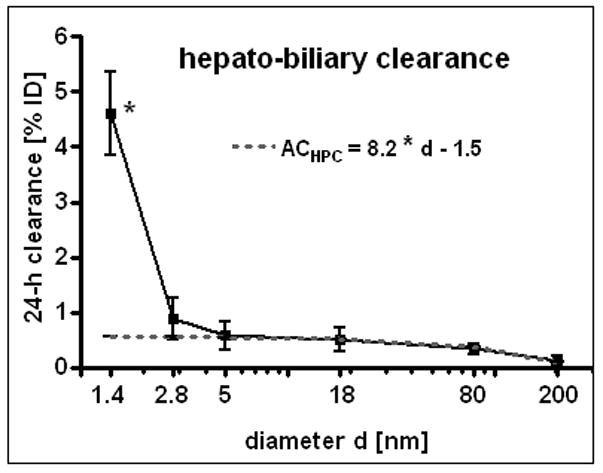
Hepato-biliary clearance of GNP from the liver to the small intestine and fecal excretion; percentages of initial GNP dose of intravenously injected, negatively charged GNP (1.4nm, 5nm, 18nm, 80nm, 200mm coated with TPPMS; 2.8nm carboxyl-coated). Data are mean ± SD, n = 4 rats. Linear equation is fitted for data from 2.8nm to 200nm GNP. Note the log scale of x-axis bends the linear line. Significant change of accumulation of 1.4nm GNP versus those of all other GNP sizes is indicated as determined by the ANOVA test. (* p<0.05).
GNP dose dependency
In order to check for GNP dose dependent biodistribution we performed an additional study using a 20-fold mass dose of 1.4nm GNP. The entire biodistribution 24 h later did not significantly differ for each organ, tissue, and body fluid from the data reported here (data of the 20-fold mass dose are not shown).
Effect of Surface charge of 2.8nm carboxylated versus aminated GNP
After 24 h most of the applied GNP, either negatively or positively charged, were found in the liver. These were significantly higher for negative 2.8nm GNP (81%) than for positive 2.8nm GNP (72%) (Fig. 5A). In contrast, in the spleen the positively charged GNP showed a significantly higher accumulation of 11.4% than the negative GNP of 8.6%, Fig. 5A. Both are factors of 6 and 5, respectively, and higher than for the TPPMS coated GNP, Fig. 1A. For the lungs, an increased accumulation of carboxyl- and amino-coated GNP by a factor of 10 was observed compared to the TPPMS coated GNP.
For the heart, kidneys, and remaining carcass, the negative GNP accumulated significantly less than the positive GNP (Fig. 5B). But in the urine a significantly higher amount of negative GNP (0.71%) than positive GNP (0.51%) was detected.
Significantly more positively than negatively charged GNP were cleared through the hepato-biliary pathway (Fig. 5B).
Discussion
Physiological GNP dose
This study aims to determine quantitative, but macroscopic NP distribution at a GNP dose of no acute toxicity to any of the organs and tissues of the rats. The usage of precisely determined doses for measuring the biodistribution of GNP is challenging. In fact, we observed variable GNP retentions in the syringe and cannula with which the GNP suspensions were injected intravenously into the tail vein. This attributed systematically to the dead space of the syringe and cannula of about 50–70μl, but also to additional incidental wall losses which we carefully determined by radio-analysis at each application and was used for the proper correction of GNP dose delivery to the rat. In our recent study we intentionally aimed for low GNP doses and did not apply more than 0.17 mg/kg body weight. Whereas in other studies found in open literature, doses between 0.1 and 2700 mg/kg body weight were administered (see Table 1 in [18]), our low GNP dose assures that it is very unlikely to expect any acute toxic effects or responses of the GNP surrounding tissues. These low mass doses became possible, including their detection, in the various organs, tissues, and excreta over a dynamic range of five orders of magnitude by using a stable-confined, radioactive 198Au label of the GNP. The independence of GNP dose at this low end was demonstrated by an additional 24 h biodistribution study using a 20-fold higher mass dose of 1.4nm GNP; the entire biodistribution in each organ, tissue, and the blood did not significantly differ from the lower dose data reported here.
Furthermore, we radio-labelled the GNP by neutron activation only to the extent required to detect the GNP in the various samples. As a result, the delivered radio-dose was far below the level required to cause any acute radio-toxic effect – even on a nanoscopic level, with the small GNP from 1.4nm to 18nm consisting of maximally one radio-isotope per GNP and the 80nm and 200nm GNP consisting, on average, 20 and 300 radio-isotopes per GNP. Such low numbers of radio-atoms clearly excludes any radio-dose effect in the direct vicinity of these NP and, at the same time, also warrants no instabilities of the gold core lattice. Using 198Au as a radio-isotope of the same chemical element as the GNP provides the same chemistry of both radio-isotope and core NP matrix, thereby, preventing leaching of the radio-isotope out of the insoluble matrix of the gold NP. Yet, the selection of such rigid experimental parameters has some implications on experimental outcomes. Admittedly, these are the morphologic localisations within the tissues which are impossible to detect for neither the very small nor the very large GNP. In fact, the localization of 1.4nm and 2.8nm GNP in biological tissue matrix are currently impossible with any electron microscopy technology (high resolution TEM imaging of these small NP requires a very clean substrate surface). Additionally, the silver enhancement method does not work for such small gold NP below 3nm either, because of the lack of silver binding [23]. On the other hand, for 80nm and 200nm GNP, the number of GNP and the required magnification of the micrographs requires complete screening of thousands of 2–5μm × 2.5μm grid fields for electron microscopic examination making it virtually impossible to detect these GNP in organs and tissues which have accumulated from less than 1% of the administered dose. Here, we show 18nm GNP in Kupffer cells, as well as endothelial cells, and hepatocytes. Particularly the single 18nm GNP found in hepatocytes and endothelial cells indicate little agglomeration in the blood Fig. 3A–C. Increasing agglomeration of GNP in Kupffer cells over retention time has recently been reported [24] and may indicate agglomeration within the Kupffer cell as a result of endosomal fusion.
Protein coating
Changes in protein coatings of GNP are likely to cause a different biokinetic behavior of these GNP. It is well known, and it was recently confirmed, that PEGylation of 5nm GNP induced prolonged blood circulation times; so different bioaccumulations occur for PEGylated versus phosphinated GNP [25]. There is growing evidence that numerous serum proteins bind to negatively and positively charged NP [26, 27]; for example, binding patterns were reported for quantum dots, gold, silica, polystyrene-latex, and copolymeric NP [14, 15, 28–32]. Additionally, in a recent study (ACS Nano 2009) the biochemistry of the binding of several important serum proteins to gold nanoparticles between 5–100 nm was thoroughly investigated in a size-dependent manner. Thereby the hydrodynamic diameter can increase by as much as a factor of two [31]. It was shown that the phosphor binding site of TPPMS coated GNP can be substituted or detached in biologic systems while a disulfide bridge remains stable [33]. In our experiment we used phosphinated GNP and therefore hypothesize that TPPMS coating is rapidly being replaced by proteins. In the case of the carboxylated or aminated GNP protein, binding may occur to the surface molecules and/or after replacement of them. In any case, the GNP-protein-conjugate grows considerably in its hydrodynamic size and the original charge may change, and even alternate, according to the protein pattern on the GNP surface.
Charge dependency of 2.8nm sized GNP
The distribution of GNP with the same 2.8nm size depends on their charge. The significantly lower accumulation of positively charged 2.8nm GNP in the liver may explain the mostly significantly higher accumulation in the other organs and in the remainder than for the negatively charged GNP. Yet, the binding of different serum proteins as a result of the different GNP charges and surface molecules is another option for the different accumulation pattern (Fig. 5A, B) which requires further investigation.
It was shown that the surface charge influences the toxicity of GNP. While anionic GNP appear nontoxic the cationic GNP were found to be moderately toxic, which resulted from their interaction with the cell membrane [34]. Moreover, a microscopic evaluation demonstrated the uptake of neutral and negative GNP by red blood cells while the positively charged GNP were attached to the cell surface [35]. This could also have influenced the biodistribution of the differently charged GNP.
In this study we also used GNP smaller than 10nm, and to our knowledge, there are almost no other papers available on the biodistribution so far. There is one study which focuses on the biodistribution of differently charged gold-dendrimers with a gold core diameter of 5nm, but these NP are not comparable to our GNP since they exhibit a large dendrimeric shell and contain several 5nm GNP in one dendrimer NP [15].
Dispersion in blood and kidney clearance towards urine
In physiology it is well described that molecular structures smaller than 50 kDa including NP below a hydrodynamic diameter of 5.5nm are almost quantitatively cleared by the kidneys into the urine. This is shown for 1.9nm Gold NP of undisclosed surface modification to be used as an X-ray contrast agent [13], and for quantum dots with zwitterionic surface coating [14, 30]. Pan et al. [33] just recently suggested that the TPPMS coating is rapidly removed in an in vitro cell culture system so that the naked GNP surface is likely to bind proteins. If this change in surface modification occurs in the blood it is likely that the most abundant protein, albumin, will be the first to bind to these GNP which we have shown in our ongoing work (data not shown). Roecker and co-workers [36] showed that iron-platinum NP of 11nm original size, with a negatively charged carboxyl surface coating, show a dense package of albumin binding to these NP which gain size of up to 17nm. Since both the 1.4nm GNP and the 2.8nm GNP are smaller than a 3.5nm sized albumin molecule, they may bind to one or a few albumin molecules according to their negative surface charge. Hence, the GNP may be hidden within the conjugate and its diameter is likely to increase > 5.5nm. At this size a reduced clearance of the kidneys is expected [30], which is consistent with our observation. The fact that our TPPMS coated 1.4nm GNP and 2.8nm GNP of both positive and negative charges were not quantitatively cleared into the urine, but only percentages of up to 5%, clearly indicated that protein binding depends on the surface modification and GNP size. This is supported by the decreased renal clearance of those anionically or cationically coated QD which bound serum proteins in vivo resulting in an increased QD-conjugate size [14]. The NP retention in the blood rose with an increase of the VSSA from 5nm to 1.4nm GNP; it ran in parallel with GNP retention in the kidneys, Fig. 1G+H; i.e. a higher GNP concentration in the blood led to a higher accumulation in the tissue of the kidneys. At the same time, the higher GNP concentration in the blood led to more clearance into the urine, Fig. 1I. As a result, the GNP percentage in the urine also ran in parallel with GNP percentage in the blood and kidneys and increased with VSSA, as indicated in Fig. 1G–I and the slope of the linear fit of the blood, kidneys, and urine in Table 3. All slopes were in a rather small range compared to the other organs and tissues. In comparison to another study, our findings of 5nm GNP in urine were much lower [15].
The high fraction of 18nm GNP associated with blood cells and, likewise, those of the 80nm and 200nm GNP are a plausible mechanism for explaining the prolonged 24 h circulation time in the blood. Further studies are needed in order to resolve these kinds of mechanisms including endocytosis by monocytes, as well as the incorporation in red blood cells as demonstrated recently [35, 37]. In contrast, the high GNP percentages in blood serum of 1.4nm to 5nm GNP points towards different mechanisms such as conjugation with serum proteins [28, 29] which are able to increase GNP hydrophilicity and dispersion in blood. In fact, Pan and co-workers [11] just recently reported rapid replacement of the TPPMS surface coating in an in vitro study allowing for avid binding of serum proteins to the highly negatively charged naked GNP surface area.
Accumulation in organs and tissues
Our findings of the highest percentages found in the liver for all sizes, and surface modifications agree with studies of other groups [18, 38, 39]. Especially the early distribution study by Säterborg [40] shows comparable data; they also used radio-labelled GNP (25nm) which are comparable to the 18nm GNP used in our study. Their results are confirmation of our investigations as the highest accumulation is detected in the liver [40]. Another study investigated the biodistribution of GNP of different sizes [41]; however, it is difficult to compare it with our results because it is not clear where the injected GNP ended up. In fact, less than 1% of the given dose is detected for all examined organs together and it is unclear where the remaining 99% is retained. Also, their application of a non-physiological dose of 1g/kg is much too high [41]. The size dependent GNP accumulation appears to be biphasic with almost no size dependency of the 18nm to 200nm GNP, but with a linear relation to VSSA of the 1.4nm to 5nm GNP. This is surprising as the increasing GNP surface area per volume was expected to lead to an increased receptor mediated GNP binding and endocytosis by the Kupffer cells in the liver. In fact, the contrary is the case. It appears that GNP ≤ 5nm are too small to be trapped by membrane receptors of Kupffer cells. Indeed, we expect that GNP uptake in the liver is known to start immediately after GNP injection (first path kinetics). At this early stage, only abundantly available proteins, such as albumin, may have bound to GNP, but the entire dynamics of in vivo protein binding and exchange on the surface of NP is not yet understood. Regarding increasing Brownian motion of GNP-protein-conjugates which are decreasing in size appears to be plausible to counteract the binding process to membrane receptors of Kupffer cells. The diffusion coefficiency of 5nm GNP-protein-conjugates in water is 1.3 10−10 m2 s−1, respectively, while the diffusion coefficiency of a 50nm conjugate is 1.3 10−11 m2 s−1. The equation for the diffusional displacement distance s is:
with time t and the diffusion coefficient D ~ 1/d′; with d′ the diameter of the GNP-protein conjugate
According to this equation, the displacement distance is 0.51 and 0.16 μm, respectively, within 1 msec. The displacement distance of 510nm/msec of a 5nm NP is 100-fold of the NP diameter and appears to be sufficiently fast enough to hinder receptor binding of the Kupffer cells, while the rather low movement of 160nm/msec of a 50 nm NP is just threefold of the NP diameter and may therefore less affect the receptor binding on the membrane.
However, in other organs there is comparably little binding of 18nm to 200 GNP and from 5nm down to 1.4nm binding and accumulation increases with increasing VSSA. This surface area proportional increase was expected in general, but the microscopic mechanisms leading to this increase are still not understood. However, an important determinant is the GNP concentration in blood which also increased with increasing VSSA. The increasing presence of GNP is merely likely to increase the probability of retention. It is noteworthy that this biphasic size-dependent pattern applies to organs as diverse as the brain, uterus, heart and kidneys. The relatively high levels and the same pattern of size dependency in the remaining carcass underlines the role of blood circulation leading to a wide GNP distribution and accumulation throughout the entire body. Note that these considerations apply to the GNP retained in the organs and tissues, but not to the GNP amount suspended in the blood volume of each organ or tissue; the latter was estimated and subtracted from the measured GNP – see Methods.
At later time points, abundant proteins are likely to be replaced by higher affinity proteins in the blood which will change the accumulation patterns, not only in the liver, but also in other organs. The fact that we still find GNP in the blood 24 h after the i.v. injection sheds light on the dynamics of interactions of GNP with serum proteins and blood cells, as well as macrophages in the liver and spleen, and additionally with endothelial cells. The larger percentages of GNP with ≥ 18nm diameter found in blood cells may be one mechanism, among others, in preserving GNP retention in blood. Additionally, clearance processes within 24 h of the previously accumulated GNP in various organs and tissues cannot be excluded either.
Although GNP percentages in the brain are low, we consistently found accumulation of all GNP and this accumulation pattern fits into those of the blood, and several other organs being size-independent of between 18nm and 200nm while accumulation increased with increasing VSSA from 5nm to 1.4nm. This emphasizes once more that the blood-brain-barrier is not completely tight sealed for NP circulating in blood as we have previously shown for iridium NP and elemental carbon NP [42–44]. This may be a concern in the case of chronic exposure of biopersistent NP reaching blood circulation.
Interestingly enough, no size dependent accumulation of about 2% of TPPMS-coated GNP of the initial GNP dose was observed in the spleen over the entire size range. This was unexpected since the spleen belongs to the reticulo-endothelial-system (RES) well known for its size dependent particle accumulation; for instance, submicron-sized polystyrene particles ranging from 60 to 250 nm were accumulated in the spleen of rats from 0.3 to 6.5% of the ID [45]. However, the carboxylated, as well as the aminated 2.8nm GNP, were accumulated by factors of 4 and 5, respectively, when compared to all TPPMS coated GNP. The different behaviour between our GNP of different coatings and the submicron-sized polystyrene particles suggests different protein binding patterns. It is quite surprising that the much higher blood concentration of the 1.4nm and 5nm GNP compared to the > 18nm GNP did not lead to an increased uptake in the spleen as was seen in several other organs – see above.
Hepato-biliary clearance
In the liver, NP – not immediately trapped by Kupffer cells - are translocated through the fenestrated vascular endothelium into the Dissé spaces to be taken up by hepatocytes and processed into biliary canaliculi [46]. From there they are drained via the biliary duct to the beginning of the small intestine where they become included in the fecal excretion. Concordingly, we found the 18nm GNP, not only in Kupffer cells, but also in endothelial cells and hepatocytes, highlighting the hepato-biliary pathway. Hepato-biliary clearance of the TPPMS coated GNP showed an inverse linear relationship to the GNP diameter over the entire size range of 2.8nm to 200nm; i.e. the smaller the GNP the higher the penetration through the various physiological structures of the liver into the intestine as outlined above. The clearance of the carboxylated 2.8nm GNP fitted very well into this trend of size dependency. The hepato-biliary clearance of the carboxylated 2.8nm GNP occurred to be about half for negatively charged GNP compared to positively charged GNP which is in line with another study using quantum dots of opposite charges [15]. Only the smallest GNP of 1.4nm did not follow this linear size relationship, but were cleared with an even higher percentage of 4.6 ± 0.4%. According to this complex pathway, the rate-determining processes are yet to be identified.
Conclusion
Size and surface charges of GNP are strong determinants of the biokinetic fate in the organism. The most GNP accumulation was in the liver and the amount increased with the size of GNP. In contrast, there was little size dependency of accumulation of 18nm to 200nm GNP in most other organs and in the carcass as well as the blood. But for GNP between 1.4nm and 5nm the accumulation increased sharply with decreasing size; the increase was linearly proportional to the volumetric specific surface area of the GNP, i.e. proportional to the inverse of the GNP diameter. The different charges of the 2.8nm GNP led to significantly different accumulations in most organs, tissues, and body fluids.
From the different accumulation patterns observed as a result of different sized and surface-charged GNP we conclude that the alterations of the GNP surface area, due to dynamic protein binding and exchange, are major mechanisms determining GNP accumulation in the various organs and tissues. A better understanding of this complex GNP biokinetics is essential for the development of improved drug delivery, as well as for dose estimates of secondary organs and tissues used in risk assessment.
Acknowledgments
We thank Dr. Carsten Rudolph from Dr. von Hauner Children’s Hospital, Ludwig-Maximillians-University of Munich, for his support of the zeta-potential measurements. We also thank Dr. Dorothea Alber and Gregor Bukalis from the Helmholtz Zentrum in Berlin for performing the neutron activation of our GNP at the research reactor BER II. This work was partially supported by the German Research Foundation FOR-627, SPP-1313 and PAK 56, the EU-FP6 project Particle-Risk (012912 (NEST)), and the EU FP7 projects NeuroNano (NMP4-SL- 2008-214547), ENPRA (NMP4-SL-2009-228789) as well as InLiveTox (NMP-2008-1.3-2 CP-FP 228625-2) and US-NIH grant HL074022.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature
- 1.Boisselier E, Astruc D. Gold nanoparticles in nanomedicine: preparations, imaging, diagnostics, therapies and toxicity. Chemical Society Reviews. 2009;38:1759–82. doi: 10.1039/b806051g. [DOI] [PubMed] [Google Scholar]
- 2.Giljohann D, Seferos D, Daniel W, Massich M, Patel P, Mirkin C. Gold Nanoparticles for Biology and Medicine. Angewandte Chemie International Edition. 2010;49:3280–3294. doi: 10.1002/anie.200904359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sperling RA, Rivera Gil P, Zhang F, Zanella M, Parak WJ. Biological applications of gold nanoparticles. Chemical Society Reviews. 2008;37:1896–908. doi: 10.1039/b712170a. [DOI] [PubMed] [Google Scholar]
- 4.Medley CD, Smith JE, Tang Z, Wu Y, Bamrungsap S, Tan WH. Gold nanoparticle-based colorimetric assay for the direct detection of cancerous cells. Anal Chem. 2008;80:1067–1072. doi: 10.1021/ac702037y. [DOI] [PubMed] [Google Scholar]
- 5.Bhattacharya R, Mukherjee P. Biological properties of “naked” metal nanoparticles. Advanced Drug Delivery Reviews. 2008;60:1289–1306. doi: 10.1016/j.addr.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 6.Bhattacharya R, Patra CR, Verma R, Kumar S, Greipp PR, Mukherjee P. Gold nanoparticles inhibit the proliferation of multiple myeloma cells. Advanced Materials. 2007;19:711–716. [Google Scholar]
- 7.Ghosh P, Han G, De M, Kim CK, Rotello VM. Gold nanoparticles in delivery applications. Advanced Drug Delivery Reviews. 2008;60:1307–1315. doi: 10.1016/j.addr.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 8.Lasagna-Reeves C, Gonzalez-Romero D, Barria MA, Olmedo I, Clos A, Ramanujam VMS, Urayama A, Vergara L, Kogan MJ, Soto C. Bioaccumulation and toxicity of gold nanoparticles after repeated administration in mice. Biochemical and Biophysical Research Communications. 2010;393:649–655. doi: 10.1016/j.bbrc.2010.02.046. [DOI] [PubMed] [Google Scholar]
- 9.Schmid G. The relevance of shape and size of Au55 clusters. Chemical Society Reviews. 2008;37:1909–1930. doi: 10.1039/b713631p. [DOI] [PubMed] [Google Scholar]
- 10.Bar-Ilan O, Albrecht RM, Fako VE, Furgeson DY. Toxicity assessments of multisized gold and silver nanoparticles in zebrafish embryos. Small. 2009;5:1897–910. doi: 10.1002/smll.200801716. [DOI] [PubMed] [Google Scholar]
- 11.Pan Y, Neuss S, Leifert A, Fischler M, Wen F, Simon U, Schmid G, Brandau W, Jahnen-Dechent W. Size-dependent cytotoxicity of gold nanoparticles. Small. 2007;3:1941–9. doi: 10.1002/smll.200700378. [DOI] [PubMed] [Google Scholar]
- 12.Chithrani BD, Ghazani AA, Chan WC. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Letters. 2006;6:662–8. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 13.Hainfeld JF, Slatkin DN, Focella TM, Smilowitz HM. Gold nanoparticles: a new X-ray contrast agent. British Journal of Radiology. 2006;79:248–253. doi: 10.1259/bjr/13169882. [DOI] [PubMed] [Google Scholar]
- 14.Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Ipe BI, Bawendi MG, Frangioni JV. Renal clearance of quantum dots. Nature Biotechnology. 2007;25:1165–1170. doi: 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balogh L, Nigavekar SS, Nair BM, Lesniak W, Zhang C, Sung LY, Kariapper MST, El-Jawahri A, Llanes M, Bolton B, Mamou F, Tan W, Hutson A, Minc L, Khan MK. Significant effect of size on the in vivo biodistribution of gold composite nanodevices in mouse tumor models. Nanomedicine-Nanotechnology Biology and Medicine. 2007;3:281–296. doi: 10.1016/j.nano.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 16.Renaud G, Hamilton RL, Havel RJ. Hepatic metabolism of colloidal gold-low-density lipoprotein complexes in the rat: evidence for bulk excretion of lysosomal contents into bile. Hepatology. 1989;9:380–92. doi: 10.1002/hep.1840090307. [DOI] [PubMed] [Google Scholar]
- 17.Souris JS, Lee CH, Cheng SH, Chen CT, Yang CS, Ho JAA, Mou CY, Lo LW. Surface charge-mediated rapid hepatobiliary excretion of mesoporous silica nanoparticles. Biomaterials. 2010;31:5564–5574. doi: 10.1016/j.biomaterials.2010.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balasubramanian SK, Jittiwat J, Manikandan J, Ong CN, Yu LE, Ong WY. Biodistribution of gold nanoparticles and gene expression changes in the liver and spleen after intravenous administration in rats. Biomaterials. 2010;31:2034–2042. doi: 10.1016/j.biomaterials.2009.11.079. [DOI] [PubMed] [Google Scholar]
- 19.Kuo CT, Yu JY, Huang MJ, Chen CH. On the Size Evolution of Gold-Monolayer-Protected Clusters by Ligand Place-Exchange Reactions: The Effect of Headgroup-Gold Interactions. Langmuir. 2010;26:6149–6153. doi: 10.1021/la100810z. [DOI] [PubMed] [Google Scholar]
- 20.Oeff K, Konig A. Blood volume of rat organs and residual amount of blood after blood letting or irrigation; determination with radiophosphorus-labeled erythrocytes. Naunyn-Schmiedebergs Archiv für Experimentelle Pathologie und Pharmakologie. 1955;226:98–102. [PubMed] [Google Scholar]
- 21.Semmler-Behnke M, Kreyling W, Lipka J, Fertsch S, Wenk A, Takenaka S, Schmid G, Brandau W. Biodistribution of 1.4- and 18-nm Gold Particles in Rats. Small. 2008;4:2108–2111. doi: 10.1002/smll.200800922. [DOI] [PubMed] [Google Scholar]
- 22.Kreyling WG, Semmler-Behnke M, Chaudhry Q. A complementary definition of nanomaterial. Nano Today. 2010;5:165–168. [Google Scholar]
- 23.Sadauskas E, Jacobsen NR, Danscher G, Stoltenberg M, Vogel U, Larsen A, Kreyling W, Wallin H. Biodistribution of gold nanoparticles in mouse lung following intratracheal instillation. Chemistry Central Journal. 2009;3 doi: 10.1186/1752-153X-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takenaka S, Karg E, Kreyling WG, Lentner B, Moller W, Behnke-Semmler M, Jennen L, Walch A, Michalke B, Schramel P, Heyder J, Schulz H. Distribution pattern of inhaled ultrafine gold particles in the rat lung. Inhalation Toxicology. 2006;18:733–40. doi: 10.1080/08958370600748281. [DOI] [PubMed] [Google Scholar]
- 25.Lipka J, Semmler-Behnke M, Sperling RA, Wenk A, Takenaka S, Schleh C, Kissel T, Parak WJ, Kreyling WG. Biodistribution of PEG-modified gold nanoparticles following intratracheal instillation and intravenous injection. Biomaterials. 2010;31:6574–6581. doi: 10.1016/j.biomaterials.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 26.Lynch I, Dawson KA. Protein-nanoparticle interactions. Nano Today. 2008;3:40–47. [Google Scholar]
- 27.Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson KA. Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:14265–70. doi: 10.1073/pnas.0805135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cedervall T, Lynch I, Foy M, Berggard T, Donnelly SC, Cagney G, Linse S, Dawson KA. Detailed identification of plasma proteins adsorbed on copolymer nanoparticles. Angewandte Chemie International Edition England. 2007;46:5754–6. doi: 10.1002/anie.200700465. [DOI] [PubMed] [Google Scholar]
- 29.Cedervall T, Lynch I, Lindman S, Berggard T, Thulin E, Nilsson H, Dawson KA, Linse S. Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:2050–5. doi: 10.1073/pnas.0608582104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi HS, Liu WH, Liu FB, Nasr K, Misra P, Bawendi MG, Frangioni JV. Design considerations for tumour-targeted nanoparticles. Nature Nanotechnology. 2010;5:42–47. doi: 10.1038/nnano.2009.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dobrovolskaia MA, Patri AK, Zheng JW, Clogston JD, Ayub N, Aggarwal P, Neun BW, Hall JB, McNeil SE. Interaction of colloidal gold nanoparticles with human blood: effects on particle size and analysis of plasma protein binding profiles. Nanomedicine-Nanotechnology Biology and Medicine. 2009;5:106–117. doi: 10.1016/j.nano.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gessner A, Lieske A, Paulke BR, Muller RH. Influence of surface charge density on protein adsorption on polymeric nanoparticles: analysis by two-dimensional electrophoresis. European Journal of Pharmaceutics and Biopharmaceutics. 2002;54:165–170. doi: 10.1016/s0939-6411(02)00081-4. [DOI] [PubMed] [Google Scholar]
- 33.Pan Y, Leifert A, Ruau D, Neuss S, Bornemann J, Schmid G, Brandau W, Simon U, Jahnen-Dechent W. Gold Nanoparticles of Diameter 1.4 nm Trigger Necrosis by Oxidative Stress and Mitochondrial Damage. Small. 2009;5:2067–2076. doi: 10.1002/smll.200900466. [DOI] [PubMed] [Google Scholar]
- 34.Goodman CM, McCusker CD, Yilmaz T, Rotello VM. Toxicity of gold nanoparticles functionalized with cationic and anionic side chains. Bioconjugate Chemistry. 2004;15:897–900. doi: 10.1021/bc049951i. [DOI] [PubMed] [Google Scholar]
- 35.Rothen-Rutishauser BM, Schurch S, Haenni B, Kapp N, Gehr P. Interaction of Fine Particles and Nanoparticles with Red Blood Cells Visualized with Advanced Microscopic Techniques. Environmental Science & Technology. 2006;40:4353–4359. doi: 10.1021/es0522635. [DOI] [PubMed] [Google Scholar]
- 36.Rocker C, Potzl M, Zhang F, Parak WJ, Nienhaus GU. A quantitative fluorescence study of protein monolayer formation on colloidal nanoparticles. Nature Nanotechnology. 2009;4:577–580. doi: 10.1038/nnano.2009.195. [DOI] [PubMed] [Google Scholar]
- 37.Geiser M, Rothen-Rutishauser B, Kapp N, Schurch S, Kreyling W, Schulz H, Semmler M, Im Hof V, Heyder J, Gehr P. Ultrafine particles cross cellular membranes by nonphagocytic mechanisms in lungs and in cultured cells. Environmental Health Perspectives. 2005;113:1555–60. doi: 10.1289/ehp.8006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De Jong WH, Hagens WI, Krystek P, Burger MC, Sips AJ, Geertsma RE. Particle size-dependent organ distribution of gold nanoparticles after intravenous administration. Biomaterials. 2008;29:1912–1919. doi: 10.1016/j.biomaterials.2007.12.037. [DOI] [PubMed] [Google Scholar]
- 39.Terentyuk GS, Maslyakova GN, Suleymanova LV, Khlebtsov BN, Kogan BY, Akchurin GG, Shantrocha AV, Maksimova IL, Khlebtsov NG, Tuchin VV. Circulation and distribution of gold nanoparticles and induced alterations of tissue morphology at intravenous particle delivery. Journal of Biophotonics. 2009;2:292–302. doi: 10.1002/jbio.200910005. [DOI] [PubMed] [Google Scholar]
- 40.Säterborg N-E. The Distribution of 198Au Injected Intravenously as a Colloid and in Solution. Acta Oncologica. 1973;12:509–528. doi: 10.3109/02841867309130417. [DOI] [PubMed] [Google Scholar]
- 41.Sonavane G, Tomoda K, Makino K. Biodistribution of colloidal gold nanoparticles after intravenous administration: Effect of particle size. Colloids and Surfaces B-Biointerfaces. 2008;66:274–280. doi: 10.1016/j.colsurfb.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 42.Kreyling WG, Semmler M, Erbe F, Mayer P, Takenaka S, Oberdörster G, Ziesenis A. Minute Translocation of Inhaled Ultrafine Insoluble Iridium Particles from Lung Epithelium to Extrapulmonary Tissues. Annals of Occupational Hygiene. 2002;46:223–226. doi: 10.1080/00984100290071649. [DOI] [PubMed] [Google Scholar]
- 43.Semmler M, Seitz J, Erbe F, Mayer P, Heyder J, Oberdorster G, Kreyling WG. Long-term clearance kinetics of inhaled ultrafine insoluble iridium particles from the rat lung, including transient translocation into secondary organs. Inhalation Toxicology. 2004;16:453–9. doi: 10.1080/08958370490439650. [DOI] [PubMed] [Google Scholar]
- 44.Kreyling WG, Semmler-Behnke M, Seitz J, Scymczak W, Wenk A, Mayer P, Takenaka S, Oberdorster G. Size dependence of the translocation of inhaled iridium and carbon nanoparticle aggregates from the lung of rats to the blood and secondary target organs. Inhalation Toxicology. 2009;21:55–60. doi: 10.1080/08958370902942517. [DOI] [PubMed] [Google Scholar]
- 45.Moghimi SM, Porter CJH, Muir IS, Illum L, Davis SS. Non-Phagocytic Uptake of Intravenously Injected Microspheres in Rat Spleen - Influence of Particle-Size and Hydrophilic Coating. Biochemical and Biophysical Research Communications. 1991;177:861–866. doi: 10.1016/0006-291x(91)91869-e. [DOI] [PubMed] [Google Scholar]
- 46.Johnston HJ, Hutchison G, Christensen FM, Peters S, Hankin S, Stone V. A review of the in vivo and in vitro toxicity of silver and gold particulates: particle attributes and biological mechanisms responsible for the observed toxicity. Critical Reviews in Toxicology. 2010;40:328–46. doi: 10.3109/10408440903453074. [DOI] [PubMed] [Google Scholar]
- 47.Tominaga T, Tenma S, Watanabe H, Giebel U, Schmid G. Tracer diffusion of a ligand-stabilized two-shell gold cluster. Chemistry Letters. 1996:1033–1034. [Google Scholar]