

Abstract
The proinflammatory transcription factor nuclear factor-kappaB (NF-κB) plays a central role in host defence against pneumococcal disease. Both rare mutations and common polymorphisms in the NFKBIA gene encoding the NF-κB inhibitor IκB-α associate with susceptibility to bacterial disease, but the possible role of polymorphisms within the related IκB-ζ gene NFKBIZ in the development of invasive pneumococcal disease has not previously been reported. To investigate this further, we examined the frequencies of 22 single-nucleotide polymorphisms spanning NFKBIZ in two case-control studies, comprising UK Caucasian (n=1008) and Kenyan (n=723) individuals. Nine polymorphisms within a single UK linkage disequilibrium block and all four polymorphisms within the equivalent, shorter Kenyan linkage disequilibrium block displayed either significant association with invasive pneumococcal disease or a trend towards association. For each polymorphism, heterozygosity was associated with protection from invasive pneumococcal disease when compared to the combined homozygous states (e.g. for rs600718, Mantel-Haenszel 2×2 χ2=7.576, P=0.006, OR=0.67, 95% CI for OR: 0.51-0.88; for rs616597, Mantel-Haenszel 2×2 χ2=8.715, P=0.003, OR=0.65, 95% CI: 0.49-0.86). We conclude that multiple NFKBIZ polymorphisms associate with susceptibility to invasive pneumococcal disease in humans. The study of multiple populations may aid fine-mapping of associations within extensive regions of strong linkage disequilibrium (‘transethnic mapping’).
Keywords: genetic polymorphism, pneumococcal disease, nuclear factor-kappaB, IkappaB-zeta, NFKBIZ
INTRODUCTION
Streptococcus pneumoniae is a major global health problem and a leading cause of death in children world-wide (1). Invasive pneumococcal disease (IPD) occurs when Streptococcus pneumoniae invades a normally sterile site, most commonly blood (bacteraemia), and is associated with a mortality of at least 20% even in developed countries (1-3). Although asymptomatic nasopharyngeal carriage of the pneumococcus is common in the general population, invasive disease develops in only a minority of individuals and the factors that determine invasion remain poorly understood (4). Increasing evidence from mouse models and rare human primary immunodeficiency states supports a central role for the proinflammatory transcription factor nuclear factor-kappaB (NF-κB) in host defence against pneumococcal disease (5-10).
Activation of NF-κB occurs following stimulation of a variety of immune receptors, including toll-like receptors (TLRs), members of the interleukin-1 (IL-1) and tumour necrosis factor (TNF) receptor superfamilies, and the interleukin-17 (IL-17) receptor. Control of NF-κB is mediated through direct interactions with a family of IκB inhibitors. The best-studied IκB members are the proteins IκB-α, IκB-β and IκB-ε, which trap NF-κB in the cytoplasm of unstimulated cells (11). The most recently described member of the IκB family is the protein IκB-ζ, encoded by the gene NFKBIZ. IκB-ζ differs from the major IκBs -α, -β and -ε in that it is localised in the nucleus rather than the cytoplasm (12, 13). Unlike the cytoplasmic IκBs, very little IκB-ζ protein is detected in resting macrophages or lung primary cells; IκBζ mRNA is however rapidly induced following TLR/IL-1 signalling (12, 14-16). Two variants of IκB-ζ mRNA appear to be generated as a result of alternative splicing: long IκB-ζ(L) and short IκB-ζ(S). The short form lacks the exon 3 which contains the initiation codon of IκB-ζ(L), and as a result encodes a shorter protein from a downstream initiation site, lacking the amino-terminal 99 amino acids of IκB-ζ(L) (13, 14). The IκBζ promoter region contains NF-κB binding sites, and indeed NF-κB has been shown to be essential for the induction of IκBζ (14). IκB-ζ in turn acts as an inhibitor of NF-κB and is likely to be regulated by NF-κB in a fast negative feedback loop, as has been demonstrated for the inhibitor IκB-α (16).
A further difference between IκB-ζ and the cytoplasmic IκBs is that, in addition to exerting an inhibitory effect on NF-κB, IκB-ζ also demonstrates intrinsic transcriptional activity and appears to be essential for the activation of a subset of inflammatory genes including interleukin-6 (IL-6) (12, 13, 15, 17). IκB-ζ additionally appears to act as a negative regulator of TNF-α signalling in mice (15, 18), and may therefore have a role in specific signalling in response to subgroups of pro-inflammatory cytokines. The functional consequences of IκB-ζ activation remain incompletely understood, and are further complicated by apparent differences in IκB-ζ signalling between cell lines, mice and humans (16, 18).
Recent work has demonstrated that both rare mutations and common polymorphisms in the NFKBIA gene encoding the NF-κB inhibitor IκB-α are associated with susceptibility to bacterial disease in humans (5, 6, 19). The possible role of polymorphism within the related IκB-ζ gene NFKBIZ in the development of human disease has not previously been studied, however. To investigate this further we studied the frequencies of polymorphisms in the NFKBIZ gene in European and African individuals with IPD and controls.
SUBJECTS AND METHODS
Sample information
The UK Caucasian IPD sample collection has been previously described (20). Blood samples were collected on diagnosis from all hospitalised patients with microbiologically-proven IPD (defined by the isolation of Streptococcus pneumoniae from a normally sterile site, most commonly blood) as part of an enhanced active surveillance programme in three hospitals in Oxfordshire, UK (John Radcliffe, Horton General, and Wycombe General Hospitals). Consecutive cases of IPD were enrolled between June 1995 and May 2001. There were no exclusion criteria for the study. DNA samples were available from 275 patients. Clinical details, including age, gender, clinical presentation and the presence of underlying risk factors, were recorded on a standardized proforma. During the study, Oxfordshire was a region of very low HIV prevalence and HIV testing was not routinely performed. Frequencies of initial clinical presentation were as follows: pneumonia 69%, isolated bacteraemia 15%, meningitis 11%, and other presentations 5%. The mean age of the patients was 58 years, ranging from 0 to 94 years; 50% were male. Pneumococcal serotypes were identified using polyclonal rabbit antisera (Statens Seruminstitut, Copenhagen, Denmark). The distribution of serotypes was very similar to that of previous UK studies, with serotype 14 being the commonest.
The control group comprised a combination of 163 UK healthy adult blood donors and 570 cord blood samples. For the cord samples, blood was collected anonymously from the discarded umbilical cords of healthy neonates born at the John Radcliffe Hospital, Oxford, UK, as previously described (20). Examination of microsatellite markers excluded contamination with maternal DNA. The study of cord blood samples is intended to reveal background population allele frequencies; recent extensive use of a UK birth cohort control group for association studies of multiple disease phenotypes has confirmed the validity of such an approach (21). The mean age of the adult blood donors was 38 years, and 50% were male; 54% of the cord blood donors were male. Individuals of non-European ancestry were excluded from cases and controls. The study was approved by the research ethics committees of the participating hospitals.
The Kenyan bacteraemia case-control collection has also been previously described (22). Kenyan children (less than 13 years old) with bacteraemia were recruited from Kilifi District hospital between 1998 and 2002. The 687 bacteraemic cases comprised patients with isolated Gram-positive and Gram-negative infections, diagnosed using standard blood culture techniques. The most frequent organisms isolated were Streptococcus pneumoniae (25%), non-Typhi Salmonella species (16%), Haemophilus influenzae (14%), and Escherichia coli (8%). The 550 community controls were individually matched to a subset of the cases on the basis of time (recruited within 14 days), location of homestead, age, and sex. Only children with complete data for HIV, malnutrition, and malaria status were included in the analysis. Ethical approval for the study was given by the Kenya Medical Research Institute (KEMRI) National Scientific Steering and Research Committees.
Genotyping techniques
DNA extraction from blood was performed using Nucleon II kits (Scotlab Bioscience, Buckingham, UK). Polymorphisms within NFKBIZ and neighbouring chromosomal regions were selected from the dbSNP (www.ncbi.nlm.nih.gov) and ensembl (www.ensembl.org) databases. Polymorphisms were chosen on the basis of their likely functionality, as well as to provide an overview of the extent of linkage disequilibrium across the gene and flanking regions. Genotyping was performed using the Sequenom Mass-Array® MALDI-TOF primer extension assay (23). A touchdown PCR protocol was used, with cycling conditions as follows: 95°C for 15 minutes; 94°C for 20 seconds; 65°C for 30 seconds; 72°C for 30 seconds; steps 2 to 4 repeated for 5 cycles; 94°C for 20 seconds; 58°C for 30 seconds; 72°C for 30 seconds; steps 5 to 7 repeated for 5 cycles; 94°C for 20 seconds; 53°C for 30 seconds; 72°C for 30 seconds; steps 8 to 10 repeated for 38 cycles; final extension at 72°C for 3 minutes. Primer sequences are listed in table E1 (online data supplement). Each genotyping plate contained a mixture of case and control samples.
General PCR conditions for amplifying products prior to sequencing were as follows: 95°C for 15 minutes, and then 40 cycles of 95°C for 30 seconds, 55-65°C for 30 seconds, and 72°C for 60 seconds, followed by 72°C for five minutes. Direct sequencing was performed using BigDye v3.1 terminator mix (ABI) followed by ethanol precipitation. Plates were run on an ABI 3700 capillary sequencer and sequence analysis was performed with the Lasergene DNAstar package (Lasergene), using SeqMan software. Primer sequences are listed in table E2 (online data supplement).
Statistical analysis
Statistical analysis of genotype associations and logistic regression was performed using the program SPSS v16.0. Analysis of linkage disequilibrium (LD) was performed using the Haploview v4.1 program (24). Haplotype blocks were defined as regions demonstrating strong evidence of historical recombination between less than 5% of SNP-pair comparisons (25). All control genotype distributions were in Hardy-Weinberg equilibrium.
RESULTS
A total of twenty-two single-nucleotide polymorphisms (SNPs) were genotyped in the UK Caucasian IPD cases and controls, of which seven SNPs were non-polymorphic or very rare (table 1). Of the remaining fifteen SNPs, seven appeared to be associated with susceptibility to IPD at the 0.05 significance level (table 1). The seven associated SNPs span a distance of 20kb across the gene, from intron 2 (rs685666) to the 3′ untranslated region (rs601225). Flanking polymorphisms on either side of NFKBIZ were also genotyped to determine the extent of LD and to exclude the possibility that the associated SNPs were simply markers linked to a true association in a neighbouring gene. The disease-associated SNPs were all found to be located within a single block of strong LD which involves NFKBIZ and extends in a 3′ direction (figure 1a). No other genes are predicted to lie within this region, and the lack of associated SNPs outside this LD block suggests that the observed association is indeed localised to NFKBIZ.
Table 1.
NFKBIZ and flanking regions polymorphism allele frequencies and P values in the UK Caucasian IPD and Kenyan pneumococcal bacteraemia case-control studies.
| Polymorphisme/ Locationf |
UK Caucasian study | Kenyan study | ||
|---|---|---|---|---|
| Minor allele frequency (%) |
P valuea | Minor allele frequency (%) |
P valuea | |
| rs9841857 −20918 |
23.2 | 0.517 | 1.0 | 0.272 b |
| rs11718446 −18387 |
25.6 | 0.782 | 20.5 | 0.055 |
| rs685666 −10717 |
23.9 | 0.036 | 8.2 | 0.135 |
| rs6441627 c −7960 |
24.2 | 0.011 | 7.8 | 0.171 b |
| rs616597 −1214 |
22.1 | 0.001 | 5.7 | 0.159b |
| rs600718 −565 |
22.4 | 0.010 | 5.8 | 0.022 b |
| rs587555 +611 |
33.7 | 0.050 | 35.8 | 0.969 |
| rs677011 +3564 |
34.0 | 0.042 | 46.8 | 0.169 |
| rs622122 +5465 |
37.4 | 0.077 | 34.5 | 0.813 |
| rs601225 +9711 |
35.7 | 0.049 | 32.4 | 0.715 |
| rs9856113 d +16854 |
43.9 | 0.086 | 20.7 | 0.158 |
| rs771574 +49743 |
31.6 | 0.941 | 16.2 | 0.545 |
| rs771785 +134870 |
47.3 | 0.237 | 39.7 | 0.989 |
| rs771757 +164708 |
19.6 | 0.858 | 31.3 | 0.389 |
| rs9813042 +239780 |
37.1 | 0.247 | 14.1 | 0.936 |
P values are derived from 3×2 Chi-squared comparisons of genotypes, except where indicated. P values are uncorrected for multiple comparisons. Significant P values at 0.05 level are highlighted in bold italics. Derived from sample sizes: UK study 275 IPD cases and 733 controls; Kenyan study 173 pneumococcal bacteraemia cases with 550 controls.
Two-sided Fisher’s exact test.
Duplicate nomenclature, also named rs645781.
Duplicate nomenclature, also named rs694936.
The following NFKBIZ SNPs were either non-polymorphic or very rare (minor allele frequency <0.01) in the UK population studied: rs3821727; rs1043339; rs7644329; rs11710599; rs13077110; rs7645725; rs7628891.
SNP positions are relative to start of translation in exon 3 of IκB-ζ(S).
Figure 1. Location of SNPs and linkage disequilibrium (LD) map for NFKBIZ in the (a) UK and (b) Kenyan populations studied.
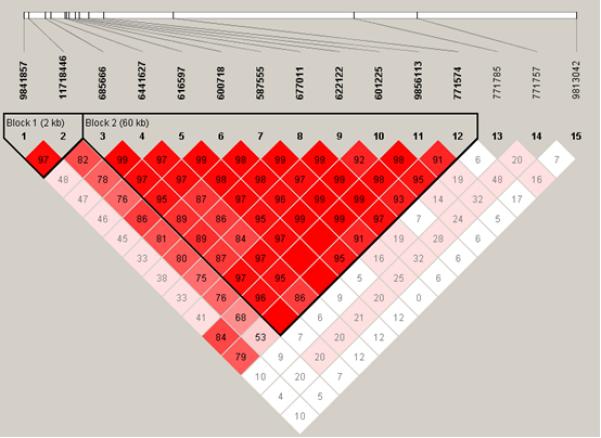
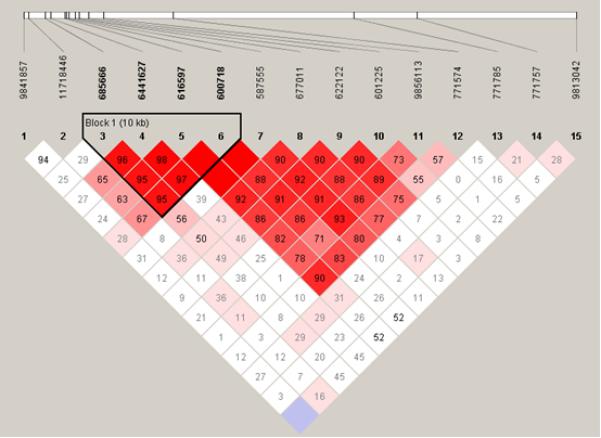
Polymorphisms are identified by their dbSNP rs numbers, and their relative positions are marked by vertical lines. Unfilled squares indicate a high degree of LD (LD coefficient D’=1) between pairs of markers. Numbers indicate the D’ value expressed as a percentile. Red squares indicate pairs in strong LD with LOD scores for LD≥2, pink squares D’<1 with LOD≥2, blue squares D’=1 with LOD<2, white squares D’<1.0 and LOD<2.
In an attempt to fine map the pneumococcal disease association within such a high-LD region, the NFKBIZ polymorphisms were next genotyped in the Kenyan bacteraemia case and control study (table 1). As expected, the extent of LD appeared to be shorter in this African population, with evidence of prior recombination breaking up the extensive 60kb LD block which was observed in Europeans (figure 1b). A smaller LD block was predicted in the Kenyan population, containing only the SNPs rs685666, rs6441627, rs616597 and rs600718 (figure 1b). Each of these four linked SNPs displayed either a trend towards susceptibility or a significant association with pneumococcal bacteraemia in the Kenyan study (table 1), although no association with overall bacteraemia was observed. Of these SNPs, rs600718 and rs616597 displayed the most statistically significant associations with IPD in the Kenyan and UK Caucasian populations; their genotypic distributions are presented in table 2. Inclusion of patient covariates (age, co-morbid conditions, and gender) within a conditional logistic regression framework demonstrated no effect of these factors on genotype in the UK or Kenyan studies, and additionally no effect of HIV infection and protein-energy malnutrition in the Kenyan study. Neither of the SNPs was associated with outcome from IPD in these groups (data not shown).
Table 2.
NFKBIZ polymorphism rs616597 and rs600718 genotype frequencies in UK Caucasian individuals with IPD and controls and in Kenyan individuals with bacteraemia (overall, Gram-positive, and pneumococcal) and controls.
| Polymorphism /location [major/minor allele] |
Population | Status | Genotype distributiona | Total | Chi- squareb (P value) |
||
|---|---|---|---|---|---|---|---|
| AA | Aa | aa | |||||
| rs616597 Intron 2 -1214 [C/A] |
UK | Control | 423 (60.0%) |
254 (36.0%) |
28 (4.0%) |
705 | 12.677 (0.001) |
| IPD | 169 (63.8%) |
73 (27.5%) |
23 (8.7%) |
265 | |||
| Kenyan | Control | 468 (89.3%) |
55 (10.5%) |
1 (0.2%) |
524 | 0.908c | |
| Bacteraemia | 587 (88.7%) |
73 (11.0%) |
2 (0.3%) |
662 | |||
| Gram-positive bacteraemia |
287 (89.7%) |
32 (10.0%) |
1 (0.3%) |
320 | 0.956c | ||
| Pneumococcal bacteraemia (IPD) |
159 (94.1%) |
10 (5.9%) |
0 (0%) |
169 | 0.159c | ||
| rs600718 Intron 2 -565 [T/A] |
UK | Control | 430 (59.6%) |
262 (36.3%) |
30 (4.2%) |
722 | 9.138 (0.010) |
| IPD | 164 (63.1%) |
75 (28.8%) |
21 (8.1%) |
260 | |||
| Kenyan | Control | 487 (89.2%) |
59 (10.8%) |
0 (0%) |
546 | 0.147 c | |
| Bacteraemia | 603 (88.3%) |
75 (11.0%) |
5 (0.7%) |
683 | |||
| Gram-positive bacteraemia |
297 (88.9%) |
33 (9.9%) |
4 (1.2%) |
334 | 0.040 c | ||
| Pneumococcal bacteraemia (IPD) |
162 (93.6%) |
10 (5.8%) |
1 (0.6%) |
173 | 0.022 c | ||
Number of individuals (%); AA, wild-type homozygote; Aa, heterozygote; aa, mutant homozygote.
Genotypic 3×2 Chi-squared comparison, 2 degrees of freedom.
Genotypic two-sided Fisher’s exact test P value.
For the UK study, the direction of association best fits a model of heterozygote protection, with an excess of both wild-type and mutant homozygotes in the IPD groups (analysing heterozygotes against combined homozygotes for rs600718, 2×2 χ2=4.70, P=0.03, odds ratio (OR)=0.71, 95% confidence interval (CI) for OR: 0.52-0.97; for rs616597, 2×2 χ2=6.20, P=0.01, OR=0.68, 95% CI: 0.50-0.92). The Kenyan results are also consistent with a heterozygote protective effect against pneumococcal bacteraemia (for rs600718, 2×2 χ2=3.82, P=0.05, OR=0.51, 95% CI for OR: 0.25-1.01; for rs616597, 2×2 χ2=3.15, P=0.07, OR=0.54, 95% CI: 0.27-1.08), although this cannot be concluded with complete confidence owing to the rarity of the mutant allele for each SNP in this population. Comparison of odds ratios for rs600718 and rs616597 did not demonstrate any evidence of heterogeneity between the UK and Kenyan IPD-control groups for either SNP; after combining and stratifying the two study groups, heterozygosity was associated with protection from IPD when compared to the combined homozygous states (for rs600718, Mantel-Haenszel 2×2 χ2=7.576, P=0.006, OR=0.67, 95% CI for OR: 0.51-0.88; for rs616597, Mantel-Haenszel 2×2 χ2=8.715, P=0.003, OR=0.65, 95% CI: 0.49-0.86).
In the context of the shorter LD observed in the Kenyan population, these results localise the IPD disease association to a limited region within NFKBIZ. No further polymorphisms in the vicinity of rs600718 were listed on databases. In an attempt to identify novel polymorphisms, a 2.2 kb region surrounding SNPs rs600718 and rs616597 was sequenced directly in 96 UK Caucasian individuals. The sequenced region comprised the exon downstream of the two SNPs (exon 3 of IκB-ζ(S)) and the entire intron within which the SNPs were located. Only a single new variant was identified, located 21bp 3′ of exon 3 of IκB-ζ(S): one individual was found to be CT heterozygous at this position, the remaining individuals sequenced were TT homozygotes. The rarity of this intronic variant suggests that it is unlikely to be of any significance to the present study, however. The sequencing also confirmed the genotyping accuracy of SNPs rs600718 and rs616597 (100% concordance between sequencing and Sequenom genotyping).
DISCUSSION
This study has identified associations between multiple NFKBIZ polymorphisms and susceptibility to IPD. The finding that the same SNP (rs600718) independently associates with susceptibility to IPD in both UK and Kenyan populations suggests that this is likely to be a real effect. Although the LD architecture differs between these European and African populations, it is noteworthy that nine SNPs within a single UK LD block and all four SNPs within the Kenyan LD block displayed either significant association with IPD or a trend towards association. In this context, we believe that the results are unlikely to represent an artefact from multiple testing or population stratification. The absence of an association between NFKBIZ polymorphisms and susceptibility to bacteraemia overall suggests that the effect is likely to be specific to IPD.
Although the finding of multiple associated SNPs within a large LD block provides reasonably compelling evidence of an association with this genomic region, it also renders the localisation of the functional variant within this block extremely difficult (26). A theoretical approach to this problem is through the study of the same disease phenotype in another ethnic group. African populations are ‘older’ than European populations, and as a result have undergone a greater number of recombination events, leading to less extensive LD (25, 27). The study of a second population may therefore aid fine-mapping of associations within extensive regions of strong LD (‘transethnic mapping’). Furthermore, it has been argued that the demonstration of genetic loci that impact on disease susceptibility across different populations is perhaps of even more value than the identification of population-specific effects (28). Genetic studies incorporating more than one ethnic group remain relatively unusual however, reflecting the challenge in assembling adequate sample collections from individuals with the same phenotype in different populations.
The differing frequency of the rs600718 and rs616597 mutant alleles between European and African populations is noteworthy: frequencies of approximately 22% in the UK Caucasians and nearly 6% in the Kenyans were observed. The UK genotype distributions suggest that mutant homozygotes are particularly vulnerable to IPD, and in the setting of a developing country with a very high burden of pneumococcal disease mortality, such genotypes are likely to be subject to significant negative selective pressure. The mutant allele frequency noted in the Kenyan population may therefore reflect a balance between a deleterious effect of the mutant homozygous state versus a possible evolutionary benefit from heterozygosity. Heterozygote protection against major infectious disease phenotypes is well-described (29-32), and may also result from additional selective pressures acting at this locus – investigation of NFKBIZ in the setting of other major infectious and inflammatory disease phenotypes may therefore be of interest.
To our knowledge this is the first reported disease association with NFKBIZ, and the identity of the IPD-associated polymorphism which exerts a functional effect on NFKBIZ remains unclear. Study of the Kenyan population suggests that the true functional variant is likely to be either rs600718 itself, or a polymorphism within this LD block tagged by rs600718. The SNP rs600718 is located in the intron between exon 1 of IκB-ζ(L) and exon3 of IκB-ζ(S); it does not itself appear to disrupt a splice site and its possible effects on IκB-ζ gene function are unknown. Stabilisation of IκBζ mRNA has recently been shown to be required for stimulus-specific IκBζ induction (14), and it is possible that a NFKBIZ polymorphism may exert a functional effect through modulation of the rate of mRNA degradation.
IκB-ζ is known to be highly expressed in the lungs and on mucosal surfaces (16, 33), consistent with a role in host defence against the pneumococcus. The mechanism by which IκB-ζ variation influences the development of IPD is likely to be complex, given the apparent dual actions of IκB-ζ on NF-κB signalling. The requirement for IκB-ζ in the expression of a subset of inflammatory genes, in particular IL-6, may account for its role in susceptibility to IPD. A further possibility is that IκB-ζ variation modulates IL-17A signaling in response to pneumococcal infection. Interleukin-17A has been shown to mediate immunity to pneumococcal colonisation, perhaps in part reflecting the induction of human β-defensin 2 (hBD-2), which exerts a potent inhibitory effect on pneumococcal growth (34-36). Indeed, IκB-ζ has recently been reported to play a key role in the induction of hBD-2 expression in airway epithelium in response to IL-17A (36). There is increasing evidence that IL-17A is central to the control of inflammatory airway responses (37), and the possible role of NFKBIZ polymorphisms in the regulation of pneumococcal clearance following IL-17A-induced hBD-2 expression is worthy of further investigation. IκB-ζ is itself the subject of considerable interest and ongoing research is likely to clarify its precise signalling roles. This study adds to the increasing evidence that control of NF-κB is central to host defence against pneumococcal disease in humans, and for the first time highlights a role for IκB-ζ in this process.
Supplementary Material
Acknowledgments
SJC is a Wellcome Trust Clinical Research Fellow; CCK is a scholar of the Agency for Science, Technology and Research (A-STAR), Singapore and member of the MBBS-PhD programme, Faculty of Medicine, National University of Singapore; AR is supported by the EU FP6 GRACE grant and the Academy of Finland; DWC is supported by the NIHR Biomedical Research Centre, Oxford; JAS is funded by the Wellcome Trust; TNW is funded by the Wellcome Trust, European Network 6 BioMalpar consortium Project and the MalariaGen Network funded by Bill and Melinda Gates; AVSH is a Wellcome Trust Principal Fellow. This paper is published with the permission of the director of the Kenya Medical Research Institute (KEMRI).
Supported by the Wellcome Trust, United Kingdom.
Footnotes
Conflict of interest statement: The authors declare no conflict of interest.
This article has an online supplementary information section, which is accessible from the Genes and Immunity website.
References
- 1.World Health Organisation Pneumococcal vaccines. Wkly epidemiol record. 2003;14:110–19. [Google Scholar]
- 2.Balakrishnan I, Crook P, Morris R, Gillespie SH. Early predictors of mortality in pneumococcal bacteraemia. Journal of Infection. 2000;40:256–61. doi: 10.1053/jinf.2000.0653. [DOI] [PubMed] [Google Scholar]
- 3.Parsons HK, Dockrell DH. The burden of invasive pneumococcal disease and the potential for reduction by immunisation. International Journal of Antimicrobial Agents. 2002;19:85–93. doi: 10.1016/s0924-8579(01)00491-5. [DOI] [PubMed] [Google Scholar]
- 4.Bogaert D, de Groot R, Hermans PWM. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect Dis. 2004;4:144–54. doi: 10.1016/S1473-3099(04)00938-7. [DOI] [PubMed] [Google Scholar]
- 5.Courtois G, Smahi A, Reichenbach J, Doffinger R, Cancrini C, Bonnet M, et al. A hypermorphic IκBα mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J Clin Invest. 2003;112:1108–1115. doi: 10.1172/JCI18714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janssen R, van Wengen A, Hoeve MA, ten Dam M, van der Burg M, van Dongen J, et al. The same IκBα mutation in two related individuals leads to completely different clinical syndromes. J Exp Med. 2004;200:559–568. doi: 10.1084/jem.20040773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmeck B, Zahlten J, Moog K, van Laak V, Huber S, Hocke AC, et al. Streptococcus pneumoniae-induced p38 MAPK-dependent phosphorylation of RelA at the interleukin-8 promoter. J Biol Chem. 2004;279:53241–53247. doi: 10.1074/jbc.M313702200. [DOI] [PubMed] [Google Scholar]
- 8.Amory-Rivier CF, Mohler J, Bedos JP, Azoulay-Dupuis E, Henin D, Muffat-Joly M, et al. Nuclear factor-kappaB activation in mouse lung lavage cells in response to Streptococcus pneumoniae pulmonary infection. Crit Care Med. 2000;28:3249–56. doi: 10.1097/00003246-200009000-00021. [DOI] [PubMed] [Google Scholar]
- 9.Jones MR, Simms BT, Lupa MM, Kogan MS, Mizgerd JP. Lung NF-κB activation and neutrophils recruitment require IL-1 and TNF receptor signaling during pneumococcal pneumonia. Journal of Immunology. 2005;175:7530–7535. doi: 10.4049/jimmunol.175.11.7530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 11.Baldwin AS. The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–81. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 12.Yamazaki S, Muta T, Takeshige K. A novel IκB protein, IκB-ζ, induced by proinflammatory stimuli, negatively regulates nuclear factor-κB in the nuclei. J Biol Chem. 2001;276:27657–62. doi: 10.1074/jbc.M103426200. [DOI] [PubMed] [Google Scholar]
- 13.Motoyama M, Yamazaki S, Eto-Kimura A, Takeshige K, Muta T. Positive and negative regulation of nuclear factor-κB-mediated transcription by IκB-ζ, an inducible nuclear protein. J Biol Chem. 2005;280:7444–51. doi: 10.1074/jbc.M412738200. [DOI] [PubMed] [Google Scholar]
- 14.Yamazaki S, Muta T, Matsuo S, Takeshige K. Stimulus-specific induction of a novel nuclear factor-κB regulator, IκB-ζ, via Toll/Interleukin-1 receptor is mediated by mRNA stabilization. J Biol Chem. 2005;280:1678–1687. doi: 10.1074/jbc.M409983200. [DOI] [PubMed] [Google Scholar]
- 15.Yamamoto M, Yamazaki S, Uematsu S, Sato S, Hemmi H, Hoshino K, et al. Regulation of Toll/Il-1-receptor-mediated gene expression by the inducible nuclear protein IκBζ. Nature. 2004;430:218–222. doi: 10.1038/nature02738. [DOI] [PubMed] [Google Scholar]
- 16.Totzke G, Essmann F, Pohlmann S, Lindenblatt C, Jänicke RU, Schulze-Osthoff K. A novel member of the IkappaB family, human IkappaB-zeta, inhibits transactivation of p65 and its DNA binding. J Biol Chem. 2006;281(18):12645–54. doi: 10.1074/jbc.M511956200. [DOI] [PubMed] [Google Scholar]
- 17.Trinh DV, Zhu N, Farhang G, Kim BJ, Huxford T. The nuclear I kappaB protein I kappaB zeta specifically binds NF-kappaB p50 homodimers and forms a ternary complex on kappaB DNA. J Mol Biol. 2008;379(1):122–35. doi: 10.1016/j.jmb.2008.03.060. [DOI] [PubMed] [Google Scholar]
- 18.Yamamoto M, Takeda K. Role of nuclear IkappaB proteins in the regulation of host immune responses. J Infect Chemother. 2008;14(4):265–9. doi: 10.1007/s10156-008-0619-y. [DOI] [PubMed] [Google Scholar]
- 19.Chapman SJ, Khor CC, Vannberg FO, Frodsham A, Walley A, Maskell NA, et al. IκB genetic polymorphisms and invasive pneumococcal disease. American Journal of Respiratory and Critical Care Medicine. 2007;176:181–187. doi: 10.1164/rccm.200702-169OC. [DOI] [PubMed] [Google Scholar]
- 20.Roy S, Knox K, Segal S, Griffiths D, Moore CE, Welsh KI, et al. MBL genotype and risk of invasive pneumococcal disease: a case-control study. Lancet. 2002;359:1569–73. doi: 10.1016/S0140-6736(02)08516-1. [DOI] [PubMed] [Google Scholar]
- 21.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berkley JA, Lowe BS, Mwangi I, Williams T, Bauni E, Mwarumba S, et al. Bacteremia among children admitted to a rural hospital in Kenya. N Engl J Med. 2005;352(1):39–47. doi: 10.1056/NEJMoa040275. [DOI] [PubMed] [Google Scholar]
- 23.Jurinke C, van den Boom D, Cantor CR, Koster H. The use of MassARRAY technology for high throughput genotyping. Adv Biochem Eng Biotechnol. 2002;77:57–74. doi: 10.1007/3-540-45713-5_4. [DOI] [PubMed] [Google Scholar]
- 24.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 25.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 26.Todd JA. Statistical false positive or true disease pathway? Nature Genetics. 2006;38:731–33. doi: 10.1038/ng0706-731. [DOI] [PubMed] [Google Scholar]
- 27.Wall JD, Pritchard JK. Haplotype blocks and linkage disequilibrium in the human genome. Nature Reviews Genetics. 2003;4:587–97. doi: 10.1038/nrg1123. [DOI] [PubMed] [Google Scholar]
- 28.NCI-NHGRI Working Group on Replication in Association Studies Replicating genotype-phenotype associations. Nature. 2007;447:655–60. doi: 10.1038/447655a. [DOI] [PubMed] [Google Scholar]
- 29.Dean M, Carrington M, O’Brien SJ. Balanced polymorphism selected by genetic versus infectious human disease. Annu Rev Genomics Hum Genet. 2002;3:263–92. doi: 10.1146/annurev.genom.3.022502.103149. [DOI] [PubMed] [Google Scholar]
- 30.Mead S, Stumpf MP, Whitfield J, Beck JA, Poulter M, Campbell T, et al. Balancing selection at the prion protein gene consistent with prehistoric kurulike epidemics. Science. 2003;300:640–3. doi: 10.1126/science.1083320. [DOI] [PubMed] [Google Scholar]
- 31.Carrington M, Nelson GW, Martin MP, Kissner T, Goedert JJ, Kaslow R, et al. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science. 1999;283:1748–52. doi: 10.1126/science.283.5408.1748. [DOI] [PubMed] [Google Scholar]
- 32.Khor CC, Chapman SJ, Vannberg FO, Murphy C, Dunne A, Ling EY, et al. A functional variant in MAL/TIRAP and protection against invasive pneumococcal disease, bacteraemia, malaria and tuberculosis. Nature Genetics. 2007;39(4):523–528. doi: 10.1038/ng1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ueta M, Hamuro J, Yamamoto M, Kaseda K, Akira S, Kinoshita S. Spontaneous ocular surface inflammation and goblet cell disappearance in IκBζ gene-disrupted mice. Invest Ophthalmol Vis Sci. 2005;46:579–88. doi: 10.1167/iovs.04-1055. [DOI] [PubMed] [Google Scholar]
- 34.Lu YJ, Gross J, Bogaert D, Finn A, Bagrade L, Zhang Q, et al. Interleukin-17A mediates acquired immunity to pneumococcal colonization. PLoS Pathog. 2008;4(9):e1000159. doi: 10.1371/journal.ppat.1000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee HY, Andalibi A, Webster P, Moon SK, Teufert K, Kang SH, et al. Antimicrobial activity of innate immune molecules against Streptococcus pneumoniae, Moraxella catarrhalis and nontypeable Haemophilus influenzae. BMC Infect Dis. 2004;4:12. doi: 10.1186/1471-2334-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kao CY, Kim C, Huang F, Wu R. Requirements for two proximal NF-kappaB binding sites and IkappaB-zeta in IL-17A-induced human beta-defensin 2 expression by conducting airway epithelium. J Biol Chem. 2008;283(22):15309–18. doi: 10.1074/jbc.M708289200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aujla SJ, Dubin PJ, Kolls JK. Interleukin-17 in pulmonary host defense. Exp Lung Res. 2007;33(10):507–18. doi: 10.1080/01902140701756604. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.