

Abstract
Biomedical research has undergone a major shift in emphasis over the past decade from characterizing the genomes of organisms to characterizing their proteomes. The high-throughput approaches that were successfully applied to sequencing of genomes, such as miniaturization and automation, have been adapted for high-throughput cloning and protein production. High-throughput platforms allow for a multi-construct, multi-parallel approach to expression optimization and construct evaluation. We describe here a series of baculovirus transfer and expression vectors that contain ligation-independent cloning regions originally designed for use in high-throughput Escherichia coli expression evaluation. These new vectors allow for parallel cloning of the same gene construct into a variety of baculovirus or Escherichia coli expression vectors. A high-throughput platform for construct expression evaluation in baculovirus-infected insect cells was developed to utilize these vectors. Data from baculovirus infection expression trials for multiple constructs of two target protein systems relevant to the study of human diseases are presented. The target proteins exhibit a wide variation in behavior and illustrate the benefit of investigating multiple cell types, fusion partners and secretion signals in optimization of constructs and conditions for eukaryotic protein production.
Introduction
The current model for drug discovery includes multiple approaches for identification and validation of potential drug targets. These approaches include biochemical characterization of functions and pathways thought to be associated with specific disease states, high-throughput screening of chemical libraries and determination of three-dimensional structures of protein targets, alone and in the presence of ligands. These approaches have common need for large amounts of specific proteins obtained with a high level of purity. Beyond drug discovery efforts, the completion of the human genome has resulted in many more biomedical researchers cloning genes and purifying proteins to advance their work. The ability to produce sufficient amounts of a particular protein with the necessary properties and activities for a given application has become the primary bottleneck in many biomedical research projects.
Recombinant gene expression has almost completely displaced purification from natural sources as the method of choice for obtaining protein for research purposes. The availability of commercial expression systems has made this technology commonplace. As target complexity has increased, recombinant expression systems have become increasingly sophisticated, with a wider number of cell types being used as the heterologous host. The high-throughput approaches that were successfully applied to sequencing the genome, such as miniaturization into multi-well plate formats compatible with automated liquid handling, have been adapted for high-throughput cloning and protein production (1). These new high-throughput processes allow for a multi-construct, multi-parallel approach to expression optimization and construct evaluation, which in turn may accelerate a variety of downstream applications requiring abundant, soluble protein. And yet protein production is still frequently an obstacle that must be overcome.
Due to its low cost, compatibility with automation and ease of scale-up, E. coli remains the most widely used host for high-throughput protein production. The extensive use of E. coli as an expression host has been accompanied by considerable improvement in its ability to produce difficult targets. However, substantial limitations remain. It is estimated that only a little more than 10% of the proteins of higher eukaryotes can be expressed solubly in E. coli (2, 3). Problems are especially acute for membrane proteins or those requiring post-translational modifications for folding or function. Many kinases that are part of signaling pathways may be produced solubly in E. coli but are inactive because the protein or pathway responsible for activation is not present in the host cell. This reduces the protein’s utility for applications requiring activity such as high-throughput screening. These limitations necessitate the use of eukaryotic systems for producing many protein targets.
The most commonly used eukaryotic form of protein production in high-throughput expression platforms is the baculovirus-infected insect cell system (1, 4). The adaptation of insect cells to growth in suspension allows for culturing in shaker-flasks and small-volume deep-well blocks similar to bacterial culturing, and the introduction of serum-free medias has led to a large reduction in the costs associated with insect cell culture. Another advantage of using insect cells is the similarity of their post-translational modifications to those of mammalian cells.
The Midwest Center for Structural Genomics developed a high-throughput platform for E. coli-based expression evaluation and a series of ligation-independent cloning (LIC) vectors (5) that have been made widely available to academic researchers. The form of ligation-independent cloning described here relies on the addition to the ends of inserts of sequences complimentary to sequences in the vectors. These complementary sequences are rendered single-stranded through the exonuclease activity of T4 DNA polymerase and annealed with similarly treated vector to create stable, though not covalently closed, circular DNA, which may be used to transform bacteria. We have used these vectors and developed additional variants to establish a high-throughput platform for E. coli-based expression evaluation (6). We sought to develop additional vectors using this design that were compatible with producing recombinant baculovirus to extend the system into high-throughput eukaryotic expression analysis. A number of previous reports describing high-throughput cloning and expression analysis using baculovirus-infected insect cells emphasized the development of cloning methods (7), defined parameters for process steps such as transfection and cell growth in deep-well blocks (8, 9) and described alterations in the baculovirus genome to enhance stability in high-throughput formats (10). Here we present the construction and use of a series of plasmids for examining the effect of various fusion protein tags alone and in combination with different signal peptides on soluble protein expression in the cytosol and secreted into the media. We have drawn on the earlier descriptions of process steps and used this information, along with methods from our bacterial high-throughput system, to develop a process to test the plasmids and demonstrate their utility.
Methods and Materials
Construction of LIC baculovirus expression vectors
The plasmid pFastBac Dual from Invitrogen was used as the parent vector for construction of LIC-compatible baculovirus shuttle vectors. The first step in this construction was to remove by PCR mutagenesis two of the four SspI restriction sites found in this plasmid. These sites are in the region upstream of the ampicillin resistance gene (β-lactamase). One site begins at base 538 and the other at base 669. The site beginning at base 669 overlaps the −10 region of the β-lactamase gene promoter (11) so the sequence was mutated to the −10 consensus sequence TATAAT from TAATAT(T). The SspI sequence is underlined. The oligos used for the mutagenesis reaction had the following sequences: 5′ GCTGATTTAACAAAAATTTAACGCGAATTTTAACAAATAATTAACGTTTACAATTCGGGTGGC and 5′ CCCTGATAAATGCTTCAATATAATTGAAAAAGGAAGAGTATGAGTATTCAAC. Reactions were run using the QuikChange multi site-directed mutagenesis kit (Stratagene). Protocols found at www.westlab.org/protocols/protocols/quikchange.pdf were used with some modification. After transformation and colony selection, plasmid DNA was miniprepped using Qiagen minispin columns. The samples were screened for the presence of the mutations by digesting with SspI followed by electrophoresis using a 1% agarose E-gel 48 (Invitrogen).
The two remaining SspI sites in the dual promoter region of pFastBac Dual were removed by digesting the plasmid with restriction enzymes XhoI and BamHI that cut on either side of the dual promoter region. The large plasmid fragment was gel purified and ligated with a synthetic version of the pPolH promoter in which the SspI site has been mutated. This synthetic promoter was constructed from the following oligos: 5′AAAATGATAACCATCTCGCAAATAAATAAGTATTTTACTGTTTTCGTAACAGTTTTGTAATAAAAAA and 5′GTGGGACGGTATGAATAATCCGGAATTATTTATAGGTTTTTTTATTACAAAACTGTTACGAAAACAG. Restriction sites for XhoI and BamHI were added by PCR to the synthetic promoter using the following oligos: 5′GTCACTCGAGGGAGATAATTAAAATGATAACCATCTCG and 5′GATCGGATCCCGCGCCCGATGGTGGGACGGTATGAATAA. Positive clones were identified by PCR and confirmed by DNA sequencing. The resulting plasmid was used as the parent vector for construction of the LIC-region-containing plasmids.
The SspI-minus pFB vector constructed above was digested with BamHI and PstI. The large DNA fragment from this digestion was gel purified. The LIC regions from pMCSG7 (5), pMCSG9 (12), pMocr (6) and pGB1 (6) (encoding His6 tags, fusion proteins, and TEV cleavage sites) were PCR amplified using the following oligos: 5′GATCGGATCCACCATGCACCATCATCATCATCATTCT, 5′GTACATGCATGTCGACCGGAGCTCGAATTCGG. These oligos add a BamHI site followed by a Kozak sequence adjacent to the ATG translational start site of these regions and an NsiI site at the 3′ end of the LIC region. These fragments were digested with BamHI and NsiI and then ligated with the BamHI- and PstI-digested vector fragment. NsiI and PstI have complimentary overhang sequences. Transformations were carried out as described above, colonies were selected and positive clones were identified by PCR and confirmed by DNA sequencing.
An additional series of LIC vectors was then constructed in which a secretion signal sequence was fused upstream of the polyhistidine sequence that is common to the E. coli fusion vectors. This was accomplished using two rounds of PCR. The first round fused the signal sequence to each of the four LIC regions used above, and the second round introduced the BamHI restriction site with the Kozak sequence and ATG to the end of the signal sequence. The signal sequences used were from honeybee melittin (HBM) (13), Autographa californica multicapsid nuclear polyhedrosis virus (AcMNPV) gp64 (Ac64) protein (14), and the Orgyia psuedotsugata multicapsid nuclear polyhedrosis virus (OpMNPV) gp64 (Op64) protein (15). The gp64 proteins are major envelope glycoproteins of baculovirus. The following oligos were used to add the signal sequence to the end of the various fusion LIC regions: HBM 5′AACGTTGCCCTTGTTTTTATGGTCGTGTACATTTCTTACATCTATGCGGCCCACCATCATCATCATCATTCTTCTGGT, Ac64 5′GTTTTATATGTGCTTTTGGCGGCGGCGGCGCATTCTGCCTTTGCGGCGGAGCACCATCATCATCATCATTCTTCTGGT, and Op64 5′GTTGTATTTATTTTGCTTTGTGTTCGGTTCTCGGCGCCCGCCGAGCACCATCATCATCATCATTCTTCTGGT. These were used with the NsiI site oligo described above. The BamHI sites oligos used for these fragments were: HBM 5′GATCGGATCCACCATGAAATTCTTAGTCAACGTTGCCCTTG, Ac64 5′GATCGGATCCACCATGGTCAGCGCTATTGTTTTATATGTGC, and Op64 5′GATCGGATCCACCATGGTGAGAATTGTTGTATTTATTTTGCTTTG. These PCR reactions also used the NsiI site oligo described above and construction proceeded as previously described.
High-throughput construct design, cloning, expression and purification
Design
Protein sequences were analyzed for ordered and disordered regions using server based programs. Where available, a full-length native construct was included as a comparative standard for protein production level of the various constructs. Oligos for PCR construction were designed using Clone-manager (Scientific and Educational Software).
PCR construction and cloning
The constructs were produced in 50 μL PCR reactions. The reactions were cleaned up using MinElute 96 UF PCR purification kits by Qiagen. These were run on a Biomek FX liquid handler. The inserts and vectors were prepared and annealed as previously described (6). Positives clones were identified by PCR analysis of miniprep DNA from transformed bacterial colonies.
Transposition
A 1 μL aliquot of the DNA solution for each positive clone was used to transform 20 μL of Z-competent DH10 Bac cells (Invitrogen). Transformation, plating and purification of recombinant bacmid DNA were carried out as described online at tools.invitrogen.com/content/sfs/manuals/bac.pdf. M13 forward and reverse primers were used to confirm the presence of recombinant bacmid by PCR screening followed by electrophoresis using a 1% agarose E-gel 48.
Transfection
The following protocol was adapted from McCall et al., 2005 (8). For each bacmid transfected a 150 μL aliquot of serum-free media (Insect-Xpress, Lonza) was pipeted into a well of a 24-well block and mixed with 20 μL of Cellfectin (Invitrogen) and 7 μL of the bacmid. The block was incubated at room temperature for thirty minutes and then 850 μL of 2 ×106 cell/mL Spodoptera frugiperda 9 (Sf9) cells was added and the block was sealed with a gas-permeable film. The blocks were incubated at 27 °C with shaking at 120 RPM for four hrs. The film was removed and 3 mL of media with 10 % FBS was added to each sample well and the blocks were re-sealed. These were then incubated at 27 °C with shaking at 300 RPM for one week. The blocks were then spun at 3000 RPM and 4 °C for 15 min in a Beckman Coulter Allegra X-12R centrifuge. The media was drawn off and pipeted into a 5.0 mL cryovial with a separate 100 μL aliquot pipeted into a 1.5 mL eppendorf tube. Both samples were wrapped in foil and stored at 4 °C.
Titering by QPCR
The recombinant baculovirus DNA was purified using a MagNA Pure Compact instrument (Roche Applied Sciences) with nucleic acid isolation kit I. The MagNA Pure program for sample processing was Total NA plasma_100_400_V3_2. The sample input was 100 μL P0 virus and the elution volume in the program was set at 100 μl. The recombinant baculovirus DNA elutions obtained from the MagNA Pure Compact were used as templates for QPCR reactions.
Purified recombinant baculovirus DNA samples were analyzed by quantitative PCR using a Lightcycler 1.5 instrument (Roche Applied Sciences). Reactions were set up with Lightcycler TaqMan Master mix to a final volume of 20 μL with 0.5 μM outside primers. Primer sets were designed to target either the ie1 gene of AcNPV DNA or the SV40 polyA sequence adjacent to the cloned gene of interest. The internal probe oligo for each set of targets was labeled at the 5′ end with 6-FAM and the 3′ end with TAMRA. A 5 μL sample of each purified viral DNA was added to the reaction as template. Cycling conditions began with a 95 °C enzyme activation for 10 min followed by 40 cycles of 95 °C denaturation, 55 °C annealing and 60 °C extension for 20 sec. Viral titers were estimated as pfu/mL using a standard calibration curve of crossing point versus log concentration (Figure 2, inset) for a dilution series of DNA of known concentration. Standard DNAs were pFastBac plasmid (polyA primers) or purified viral genome (ie1 primers).
Figure 2.
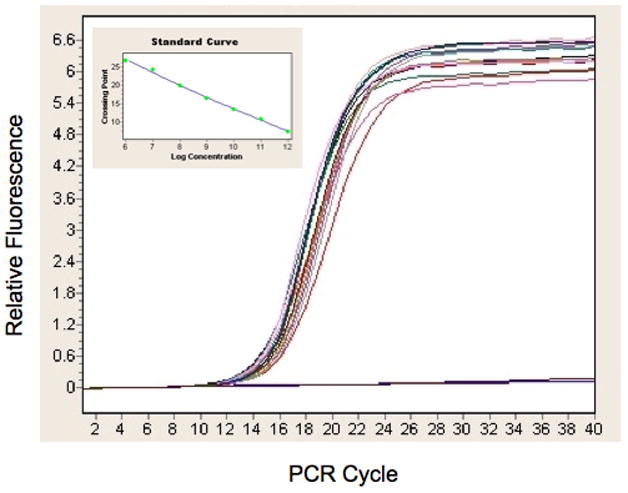
Quantitation of recombinant baculovirus. The evolution of fluorescent signal is plotted against the cycle number in a quantitative polymerase chain reaction assay using genomes purified from recombinant baculovirus as template. The inset is the standard curve based on DNAs of known concentration that was used for absolute quantification of this data to arrive at titers in genomes per mL for the virus samples. The data shown are for the P0 virus from the YFP validation constructs.
Small-scale expression trials in baculovirus-infected insect cells
Expression trials were carried out in 24-well blocks. Wells were seeded with 2 × 106 insect cells/mL, High-five or Sf9 as indicated in the figure legends, and then infected at a multiplicity of infection of 2 or 5 as indicated. The blocks were sealed with gas permeable film and incubated at 27 °C with shaking at 300 RPM for the indicated time. The cells and/or media were harvested by centrifugation at 1000 × g for 10 min at 4 °C. To analyze for secreted protein, the media was decanted into a 15 mL conical tube and stored at 4 °C until further handling. For analysis of intracellular protein, the media was removed from the cell pellet and the block was frozen at −80 °C until purification.
High-throughput purification from baculovirus-infected insect cells or conditioned media
Frozen culture blocks containing insect cell pellets were thawed at 25 °C. A 1.25 mL aliquot of lysis buffer (50 mM NaPO4 (pH 7.5), 300 mM NaCl, 2 mM MgCl2, 8 U/mL Benzonase (Novagen), Complete protease inhibitor, EDTA-free (Roche Diagnostics)) was added to each insect cell pellet in the block and these were shaken at 25 °C for 30 min. Contents of each well were transferred to 1.7 mL microfuge tubes and centrifuged for 10 min at 20,000 × g.
The soluble fractions were transferred to a 96-well deep well block containing a 120 μL slurry of 75% PBS and 25% Ni-NTA Agarose (Qiagen) and incubated 1 hour at 4 °C for batch binding of protein to the resin. Purification was performed on a Biomek FX. The resin and soluble fraction were transferred to a Whatman 96-well 2 mL glass filled 10 μm polypropylene filter. The resin was washed six times by 400 μL of 20 mM imidazole in PBS. The protein was eluted by 250 mM imidazole in PBS.
For secreted proteins the media that had been pipeted off after centrifugation of the block was transferred to Amicon Ultra-4 centrifugal filter devices (Millipore) with a 10 kDa molecular weight cut-off. These were spun at 3000 × g at 15 °C for 15 min intervals. The samples were concentrated from 5 mL to 1 mL and then diluted to 5 mL with phosphate buffered saline (PBS) (50 mM sodium phosphate pH 7.5, 300 mM NaCl). The centrifugation and dilution were repeated and then the sample was centrifuged to concentrate the sample once more. The final 1 mL samples were transferred to a 96-well deep well block and the purification procedure was identical to that of the intracellular protein as described above.
Amylose purification and size-exclusion chromatography (SEC) characterization of NS1 fusions
Purification of the HBM-MBP-NS1(JEV) fusion from conditioned media by amylose affinity chromatography. Media from an 800 mL infection cultured for 72 hours was passed over a 0.5 mL amylose column at 100 mL per hour in a 4 °C room. The column was washed with 20 mL of 50 mM phosphate (pH 7.5) with 150 mM NaCl. The MBP-NS1 fusion protein was then eluted from the column with four 0.5 mL aliquots of 50 mM phosphate (pH 7.5) with 150 mM NaCl and 20 mM maltose. A 350 μL sample from the amylose affinity purification was loaded onto a Superose 300 column and run at a flow rate of 0.5 mL/minute with 50 mM phosphate (pH 7.5) with 150 mM NaCl. Fractions of 0.5 mL were collected.
Sample analysis
All analysis of PCR reaction products was done using a Labchip 90 instrument (Caliper) and analysis of high-throughput protein expression samples was done using the Labchip 90 or by SDS-PAGE using Criterion 12 lane 4–20% Tris-Gly gels (BioRad) followed by staining with Bio-Safe Coomassie stain (BioRad).
Results
Construction of new LIC compatible baculovirus vectors
A series of ligation-independent cloning (LIC) compatible transfer and expression vectors for use in a high-throughput baculovirus expression platform were constructed. These vectors are derived from the commercially available pFastBac Dual vector (Invitrogen). This vector was chosen because the steps of the Bac (bacteria) to Bac (baculovirus) system are compatible with and easily adapted to similar steps in the high-throughput bacterial process currently in use in our lab. Using this system, the recombinant baculovirus DNA is produced by transposition in bacteria (16,17), which reduces the probability of a non-recombinant baculovirus background. The efficiency of transposition can be monitored using common techniques such as PCR.
Our cloning process relies on the LIC sequence designed at the Midwest Center for Structural Genomics (MCSG) which utilizes restriction digestion at a unique Ssp I site to open the vector as the first step in creating the single-strands that anneal with insert DNA. Of four Ssp I sites in commercially available pFastBac Dual, two are distant from the cloning region of the plasmid, just upstream of the beta-lactamase gene that confers ampicillin resistance. These two sites were removed by PCR mutagenesis. Two other Ssp I sites are found in the dual promoter region, one in the AT-rich region of the p10 promoter and the other between the pPolh promoter and the multiple cloning sequence. The dual promoter was replaced with a synthetic version of the pPolh promoter in which the downstream Ssp I site was mutated. The cloning regions from bacterial expression vectors developed by the MCSG (5, 12) were then inserted adjacent to this promoter. The plain polyhistidine tag (pMCSG7) and the polyhistidine-maltose binding protein (MBP) (pMCSG9) fusion versions of these plasmids as well as polyhistidine-Mocr (6) and polyhistidine-GB1 (6) fusion versions were all inserted into the pFastBac Dual derived vectors (Fig. 1A).
Figure 1.
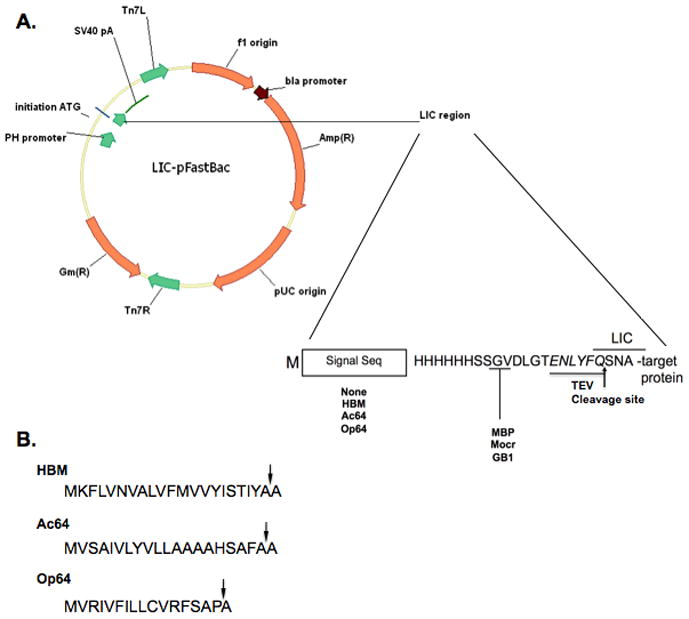
A. Plasmid map of the LIC compatible baculovirus expression vectors that have been constructed. The circle diagram shows the major features of the pFastBac plasmids used as the parent for vector construction. The inset shows the features of the new vectors and their placement. Each of the sixteen variants contains a His6 tag followed by a TEV cleavage site, shown in italics. A LIC sequence in the DNA overlaps a portion of the TEV cleavage site coding sequence. In some variants a sequence encoding a signal peptide precedes the His6 tag and some variants have a fusion protein sequence between the His6 tag and the TEV recognition sequence. B. Amino acid sequences of the signal peptides used in vector construction. The sequence of the signal peptide from: honeybee melittin (HBM), Autographa californica multicapsid nuclear polyhedrosis virus gp64 protein (Ac64), and Orgyia psuedotsugata multicapsid nuclear polyhedrosis virus gp64 protein (Op64). The site of cleavage by signal peptidase for each signal peptide is indicated by an arrow.
Variants of each fusion were also constructed with three different secretion signal sequences inserted between the initiating codon and the fusion tag sequence (Fig. 1A) for use in producing secreted proteins. The signal sequences used in these constructs were from the honeybee melittin gene (HBM) (13) and the major envelope protein, gp64, of the baculoviruses Autographa californica (Ac64) (14) and Orgyia psuedotsugata (Op64) (15) (Fig. 1B). Previous studies showed that the use of insect derived processing sequences may lead to enhanced expression and secretion as compared to use of the native signal sequence (18, 19), although this strategy has not been effective with every target protein (20). We created three signal sequence variants because previous work has indicated that different signal peptides may display variable effectiveness in combination with different target proteins (21).
Vector and process validation using yellow fluorescent protein as a model
Prior reports from industrial laboratories identified ranges and optimal conditions for use of 24-well blocks for transfection of recombinant baculovirus DNA (8), growth of various insect cell lines (9) and recombinant gene expression by baculovirus infection of various insect cell lines (8, 9). We adapted these protocols and merged them with elements of our existing bacterial process to establish a high-throughput process for generation of recombinant baculovirus and small-scale expression evaluation.
To validate both the new vectors and the high-throughput process steps, we chose the gene for yellow fluorescent protein (YFP) as a model target. The gene was constructed with the complimentary sequences to the LIC region in the vectors, incubated with T4 DNA polymerase to generate single-stranded termini in preparation for annealing, and then annealed into all sixteen variants of the new baculovirus vectors. Four colonies from the transformation of bacteria with each vector annealing were screened by PCR for the presence of insert. The positive clones were moved to the transposition step to make recombinant baculovirus DNA. Recombinant bacmid DNA was purified and used to transfect Sf9 cells to produce the recombinant baculovirus.
Transfections were carried out using suspension cultures in 24-well blocks. The final cell concentration of 0.5 × 106 cells/ml and seven day incubation identified as optimal by McCall et al., 2005 (8) were used but the final volume was limited to 4 ml/well to reduce the potential for cross-contamination. A humidified incubator was not available so over the course of the incubation some volume was lost by evaporation through the gas permeable cover. Upon harvest, we observed an average reduction in volume from 4 mL to 2.5 mL. The viral genome DNA was purified and used as template for quantitative polymerase chain reaction (QPCR). Although a variety of methods for titering virus are available (22, 23) we chose QPCR for its speed and accuracy when freshly prepared virus with high infectivity is used (24, 25). The titers for the virus from this validation trial ranged from 7.6 × 108 to 2.1 × 109 genomes/mL (Fig. 2). This titer range is somewhat higher than the 4 – 6.5 × 108 pfu/mL reported by McCall et al., 2005 (8), which is probably due, at least in part, to the concentration of the samples by evaporation.
Expression trials using Sf9 cells or High-five cells were also set up in 24-well blocks. These were infected with P0 virus at a multiplicity of infection of 5. The cultures were harvested after 48 or 72 hours and the fusion protein targets were purified by metal affinity chromatography either from the cell pellet or from the media after a buffer exchange. The eluates from the metal affinity chromatography step were analyzed either on the Labchip 90 (Fig. 3) or by SDS-PAGE (Fig. 4). All the fusion tags yielded soluble cytosolic protein in each cell line (Fig. 3). Protein secreted into the media was detected for every combination of signal sequence and fusion tag tested but with a wide variation in yield (Fig. 4). The pattern of performance for combinations of secretion signal and fusion partner was similar between High-five cells and Sf9 cells though there are some significant differences between the cell lines. High-five cells have slightly higher yields at the 48-hour timepoint but for most clones displays lower yield then Sf9 cells at the 72-hour timepoint. There may be some proteolysis occurring with some of the proteins leading to the lower yields in High-five cells relative to Sf9 at 72 hours as well as relative to the High-five 48-hour samples. This is most apparent in the HBM-MBP clone at 72 hours in High-five cells where a lower band running at a molecular weight close to MBP alone is detected. Several other clones are no longer detected in the media at 72 hours but can still be found in the cell pellet (supplementary Fig. 1). These trials confirm that both the process we developed and the vectors we constructed are capable of being used for parallel construct evaluation at small-scale.
Figure 3.
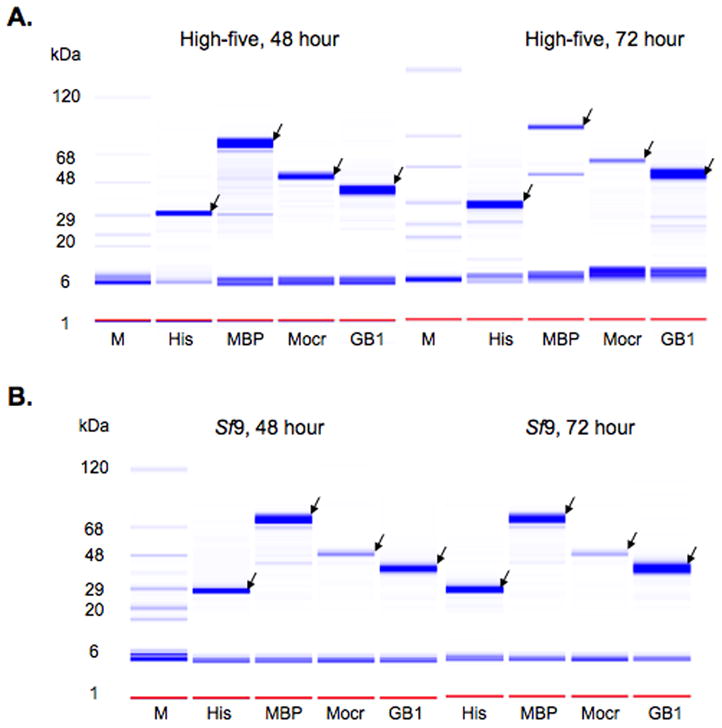
Intracellular production of YFP as a function of cell type, time and fusion partner. YFP fusions produced in High-five cells (A) and Sf9 cells (B) at were analyzed at 48 and 72 hour timepoints using the Labchip 90. The data are shown as a gel representation similar to SDS-PAGE stained with Coomassie blue. Lane M is the molecular weight marker, the fusion partners are indicated at the bottom of each lane. The arrows indicate proteins at the expected molecular weights.
Figure 4.
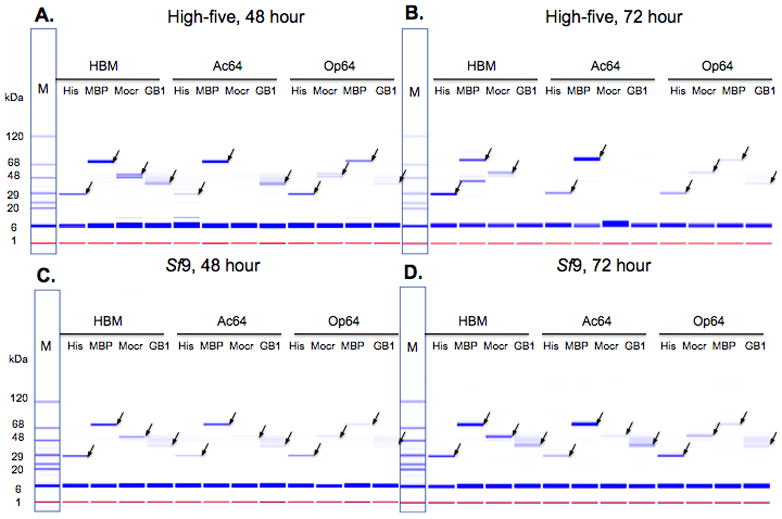
Production of YFP secreted into the media. The media of recombinant baculovirus-infected cells was buffer exchanged by spin-filtration into phosphate-buffered saline, subjected to nickel chelate chromatography and analyzed using the Labchip 90. The data are shown as a gel representation similar to SDS-PAGE stained with Coomassie blue. Lane M is the molecular weight marker. Protein is from the media of infected High-five (A, B) or Sf9 (C, D) cell cultures at the indicated length of time of incubation. The arrows indicate proteins at the expected molecular weights.
Parallel cloning and expression trials of human IKKε using E. coli and baculovirus
The IκB kinases, or IKKs, are essential components of NF-κB signaling pathways (26). These kinases phosphorylate the IκB proteins, which leads to release of NF-κB to activate gene transcription. Three IKKs directly phosphorylate the IκBs: IKKα, IKKβ and IKKε. In humans, IKKε is predominantly expressed in immune cells (27), which may indicate a special role for this protein in inflammatory response. A recent study of the effects of a high-fat diet on IKKε knockout mice indicated that IKKε might be an important target for development of therapies for metabolic disorders such as obesity, diabetes, insulin resistance and associated complications (28).
To facilitate both assay development and structural biology for IKKε, several different gene constructs were designed and produced. These included a full-length construct and four truncations that removed the leucine zipper and helix-loop-helix domains from the serine/threonine kinase catalytic domain (27). These five constructs were each cloned into five different E. coli expression vectors that varied by the fusion tag they encoded. The cloning efficiency was noticeably lower than average for these constructs. The data from extensive growth and expression experimentation suggested that the wild-type IKKε protein was toxic to the E. coli host cells and no wild-type protein was obtained. We also made the same five constructs as two inactive mutants of IKKε. These constructs expressed well and the projected yields from E. coli are sufficient to support an effort to determine the three-dimensional structure of this protein in the presence and absence of ligands. However, these constructs are incompatible with assay development and inhibitor screening because the proteins are inactive.
Our inability to produce active IKKε in E. coli necessitated the use of an alternative protein production system. The five wild-type construct fragments used for LIC insertion into the E. coli vectors were used for LIC insertion into three of our new baculovirus vectors. The validated high-throughput baculovirus process was used to produce recombinant virus for these fifteen clones in parallel and to carry out expression testing with P0 virus. The MBP fusion of the active form displayed the best production of soluble protein. The full-length version of IKKε had the best yield and the truncations were undetected or displayed significant proteolysis over the course of the expression trial (Fig. 5A). The initial expression trials were carried out in High-five cells. We next compared production of soluble protein between High-five cells and Sf9 (29) using the MBP fusion. Additionally, a GST -IKKε fusion was also tested for production alongside the MBP fusion in Sf9 cells. GST fusions are often used for heterologous expression of kinases. Both of the fusions also had an N-terminal His6 tag that was used for partial purification. The High-five cells yielded slightly more soluble protein than Sf9 cells for the MBP fusion (Fig. 5A versus 5B). The MBP fusion displayed a significant advantage in yield compared with the GST fusion (Fig. 5B). When scaled up, the MBP-full-length IKKε fusion yields approximately 2 mg purified protein per 800 mL of infected culture, in line with the yield suggested by our 5 mL trials. This protein is fully active and was used successfully for assay development (Dr. Stuart Decker, per. comm.).
Figure 5.
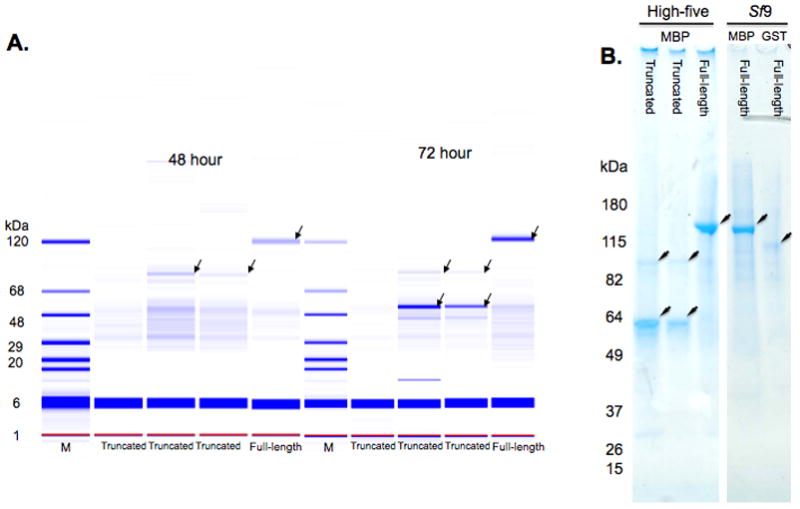
Expression analysis of human wild-type IKKε constructs. A. Labchip 90 analysis of soluble expression in High-five cells. The IKKε constructs were produced as fusion proteins with MBP. The samples are the eluates from the nickel chelate chromatography step of high-throughput expression and purification. M is a protein molecular weight ladder. Each construct is identified at the bottom of the lane. The arrow identifies the protein migrating at the expected molecular weight for each construct. The lower arrows in the truncated (2–352 and 2–356) samples identify major proteolysis products of IKKε. These bands increase from 48 hours to 72 hours of incubation while the band at the expected molecular weight becomes weaker. B. SDS-PAGE analysis of MBP or GST fusions of IKKε from infection of High-five or Sf9 cells. In the 4–20% gradient gel stained with Coomassie blue, the first three lanes are MBP fusion proteins from a 72-hour infection of High-five cells. The remaining two lanes are full-length IKKε fusions with MBP (first lane) and GST (second lane) from a 72-hour infection of Sf9 cells.
High-throughput construct expression evaluation of the NS1 protein from four different strains of Flavivirus
Flavivirus are spread by tick and mosquito vectors and represent a serious global threat to human health (30, 31). The flavivirus nonstructural protein 1 (NS1) is a highly conserved glycoprotein of ~350 amino acids ranging from 43–48 kDa in mature form. The protein is monomeric when excised from polyprotein but has been observed as a homodimer when translocated to the lumen of the endoplasmic reticulum (32) and is thought to form a soluble hexamer when secreted from infected mammalian cells (33). The function of the NS1 protein is not fully understood but properly glycosylated NS1 is required for viral RNA replication (34). This makes NS1 a particularly interesting target for structure determination and mechanistic study.
Several rounds of evaluation of constructs and fusion partners with NS1 cDNAs from yellow fever virus (YFV), west Nile virus (WNV), dengue type 2 (Den2) and Japanese encephalitis virus (JEV) for soluble expression in E. coli failed to identify combinations compatible with structural studies. We selected several of the available constructs for each cDNA to evaluate with our baculovirus process. Two constructs for each virus were chosen to clone into each of four different vectors for intracellular expression. The constructs chosen were the longest one available for each gene and one of the C-terminal truncations that removes a cysteine-rich region. Although the constructs for each virus were not identical they were similar in length. The longest construct for each virus was also cloned into the four secretion vectors that displayed the best yield with YFP (Fig. 4). This gave a total of 48 clones, 32 for intracellular expression and 16 to be secreted. The 48 clones were moved through our process in parallel, and recombinant virus obtained from the transfections (P0) was used without amplification for expression trials in Sf9 and High-five cells.
We detected no soluble protein production from any of the intracellular expression clones (supplementary Fig. 2 and 3). For the secreted clones, the YFV protein construct (2–350) yielded soluble product with every fusion partner in both cell lines. The truncated JEV protein construct (2–290) did not yield any detectable product. The Den2 protein construct (2–352) and the truncated WNV protein construct (17–306) were detectable only when fused with MBP. Gel representations of the partially purified MBP-NS1 fusions purified from media or cell pellets from each cell line are shown in Fig 6. The levels of protein purified from the media were similar for Ac64-MBP-YFV and -Den2 from each cell line while the yield for HBM-MBP-Den2 was somewhat higher from media from High-five cells than Sf9 cells. Ac64-MBP-WNV NS1 was significantly more abundant in both media and cell pellets from Sf9 cells then from High-five cells, whereas HBM-MBP-WNV NS1 was detected only in the media from High-five cells (Figure 6A). Lanes corresponding to HBM-MBP-WNV NS1 in Fig 6 panels B, C and D display a strong band at the size of MBP alone, ~46 kDa, and a band at this size is also detected in panel A but at lower intensity. This may be indicative of a sensitivity of the HBM-MBP-WNV NS1 clone to proteolysis. The level of YFV and Den2 NS1 proteins purified from Sf9 cell pellets was 2–3 fold higher than for High-five cells (Fig. 6C and D, Supplementary Table 1). The molecular weights assigned to the proteins by the Labchip were higher than the predicted molecular weights, consistent with NS1 glycosylation. Additionally, the molecular weights of the protein purified from cell pellets is approximately 2 kDa higher than for protein purified from the media, which corresponds to the difference expected from removal of the signal sequence by signal peptidase. The WNV clone showed the greatest difference between the predicted molecular weights (78.5 kDa unprocessed, 76.5 kDa processed) and the observed molecular weights (91 kDa from the cell pellet, 89 kDa from the media). Additional glycosylation may account for this difference because WNV NS1 has three potential glycosylation sites whereas there are only two sites in the other three NS1 proteins.
Figure 6.
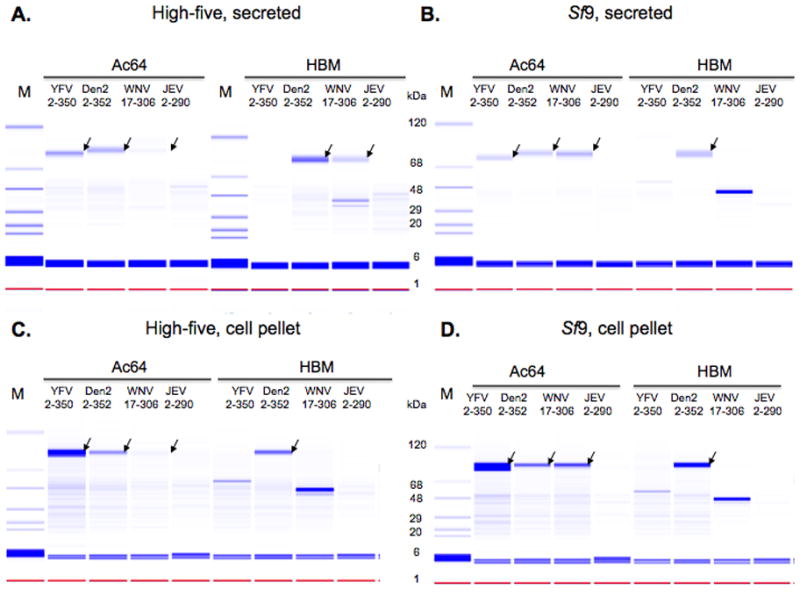
Expression analysis of flavivirus NS1 protein varying the secretion signal. Various NS1-MBP fusions were purified from the media or cell pellets of recombinant baculovirus-infected insect cell cultures and analyzed with the Labchip 90. Protein from the media of infected High-five (A) or Sf9 (B) cell cultures was buffered exchanged by spin-filtration into phosphate-buffered saline and then subjected to nickel chelate chromatography. Cell pellets from High-five (C) or Sf9 (D) cultures were resuspended in lysis buffer, the soluble fraction was separated from the insoluble fraction and then subjected to nickel chelate chromatography. Samples from the eluates were then run on the Labchip 90. Molecular weight markers are in the M lanes. Signal peptide sequences, viral origins and construct amino acid compositions are indicated. The WNV constructs have N- and C-terminal truncations and the JEV constructs have a C-terminal truncation as described in the text. The arrows indicate protein bands consistent with the expected molecular weight of mature, processed or unprocessed NS1 fusion protein.
We later obtained full-length clones of the WNV and JEV NS1 cDNA and used these to produce longer constructs similar to those for YFV and Den2. The WNV and JEV 2–352 constructs were cloned into vectors with each of the three signal sequences fused to MBP. Recombinant virus was made and expression trials were run in 24-well blocks. Protein was purified from both the media and cell pellets using metal affinity chromatography and analyzed by SDS-PAGE (Fig. 7A). For each cDNA, the levels of protein production and secretion varied considerable among the different signal sequences. Clones with the Op64 sequence produced the most protein overall for each construct (Fig. 7A) but the highest level of secreted protein was observed with the JEV clone with the HBM signal sequence. Based on the concentration of protein in this 5 mL sample we projected the clone would yield approximately 6.5 mg purified protein per 800 mL of infected culture. To determine the accuracy of this prediction, the clone was then used in an 800 mL infection and the protein was purified directly from the media using an amylose column (Fig. 7B). The total yield of purified protein was 5.8 mg. We next used size-exclusion chromatography to determine the aggregation state of the purified protein (Fig. 8A). Fractions from the two major peaks were analyzed by SDS-PAGE (Fig. 8B). Based on the size-exclusion chromatography, the purified NS1 protein was mostly monomeric with a lesser dimeric component.
Figure 7.
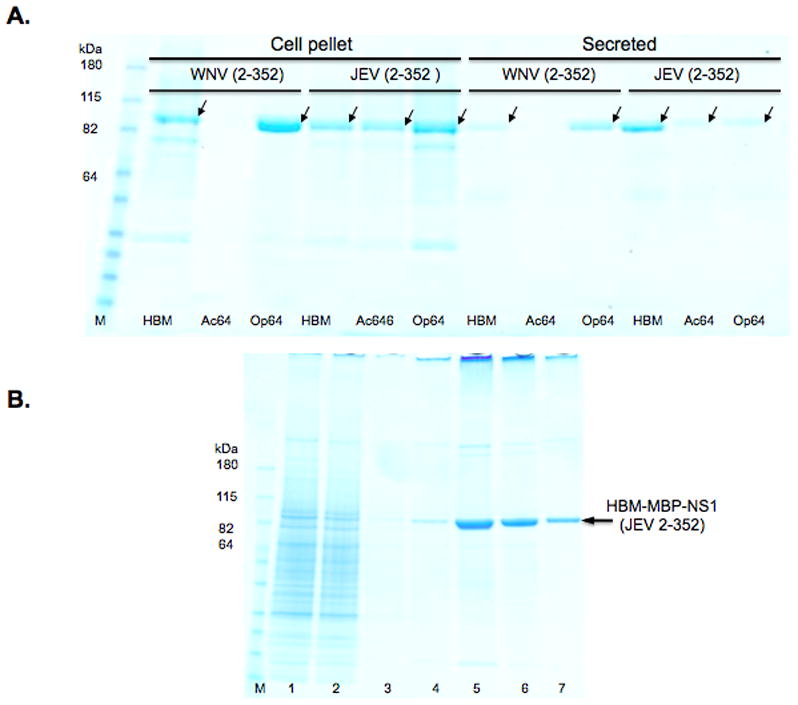
Expression analysis of full-length WNV and JEV NS1 protein varying the secretion signal. A. SDS-PAGE of MBP-NS1 fusion proteins purified from cell pellets or the media of recombinant baculovirus-infected Sf9 cells. In the 4–20% gradient gel stained with Coomassie blue, lane M is the molecular weight markers. Signal peptide, viral origins and construct amino acid compositions are indicated. The arrows indicate protein bands consistent with the expected molecular weight of mature NS1 protein. B. SDS-PAGE analysis of amylose affinity chromatography of HBM-MBP-NS1 (JEV 2–352). Column fractions on a 4–20 % gradient gel that was stained with Coomassie blue. Molecular weight marker (M), conditioned media (1), amylose column flow-through (2), wash (3), elution fractions (4–7). The MBP-NS1 protein fusion is indicated by an arrow.
Figure 8.
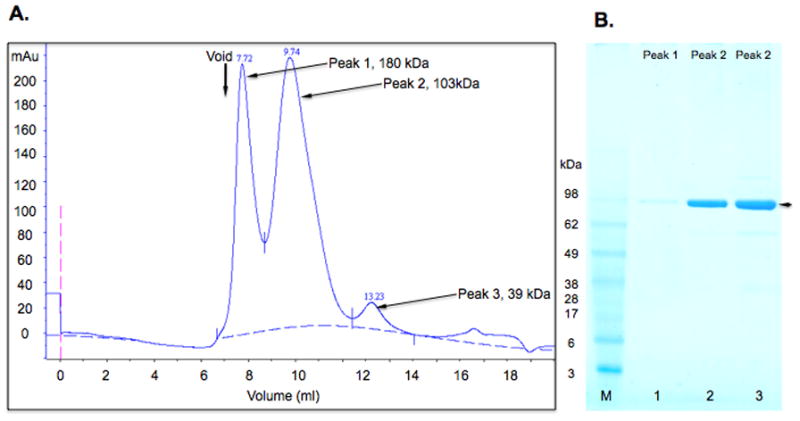
Characterization of the aggregation state of purified JEV MBP-NS1. A. Size exclusion chromatography profile for MBP-NS1 (JEV) fusion protein purified from conditioned media. The column void volume is indicated. Molecular weights of peaks were assigned by fitting elution volumes to a standard curve. B. SDS-PAGE analysis. 30 μL aliquots of fractions from Peak 1 (180 kDa ), lane 1, and Peak 2 (103 kDa), lanes 2 and 3, were analyzed on a 4–20 % gradient gel that was stained with Coomassie blue. The MBP-NS1 fusion protein is indicated with an arrow.
Discussion
The first, and in many cases most difficult, hurdle in many biomedical research efforts is obtaining sufficient amounts of target protein with the properties required for the method or techniques being applied. One approach to addressing this challenge has been development of high-throughput processes for multi-construct multi-parallel evaluation of protein production. Although E. coli is still the most common system employed for heterologous protein production the limitations of bacterial expression necessitate the use of eukaryotic systems for production of some targets. The baculovirus-infected insect cell system is the most commonly used of the eukaryotic systems and high-throughput processes for this system have previously been described (7–10). We adapted steps from these earlier processes for use with a new matrix of transfer and expression plasmids (Fig. 1A). We fused elements of E. coli expression vectors designed for high-throughput applications with a commercially available baculovirus transfer and expression vector to develop a high-throughput baculovirus-based process to allow for evaluation of protein production in parallel with an existing E. coli high-throughput process. A major feature of this matrix is the combination of various fusion protein tags with three signal peptide sequences for expression analysis of secreted proteins (Fig. 1B).
The development of high-throughput expression testing processes has been enabled by the development of several ligation-independent cloning systems. The Gateway system (Invitrogen) relies on enzymatic recombination to create the final expression clone. The large number of Gateway-adapted vectors makes it a very flexible system but the requirement for proprietary enzymes is a disincentive to generating large numbers of clones. This system also introduces up to 8 amino acids into the target protein through the translation of the recombination sites that must be added to the gene fragment. The In-Fusion system (Clontech) relies on the use of complimentary sequences in the vector and insert, and employs a proprietary enzyme to render these sequences single stranded and join them. This system has the advantage of not adding any amino acids and may be used with any vector (7). Our new vectors contain the ligation-independent cloning region developed at the Midwest Center for Structural Genomics (5). The LIC protocol we employ also relies on complimentary sequences in the vector and insert, which are rendered single-stranded through the exonuclease activity of T4 DNA polymerase. The use of T4 DNA polymerase to generate the single-strands is less expensive than the cost per reaction of In-Fusion reagents. The cloning efficiency of this method and In-Fusion should be comparable since the length of the single-stranded tails (15 bases) is the same. One disadvantage of the MCSG-based method is the extra alanine codon at the beginning of the insert to provide a guanine to act as a stop to the T4 exonuclease activity. We chose the MCSG-based method because its LIC sequence allows gene fragments constructed for annealing with existing pMCSG E. coli vectors (5, 12) also to be annealed into the new baculovirus vectors for parallel expression trials in E. coli and baculovirus-infected insect cells.
Using YFP as a model gene and protein, we demonstrated the ability to produce recombinant baculovirus with these vectors and to produce protein in baculovirus-infected insect cells (Fig. 3 and 4). The high-throughput process we adapted from elements of our bacterial process and previous reports of high-throughput baculovirus protocols from other labs has proven to be robust and reproducible. The volume and titer of P0 virus produced is sufficient for multiple small-scale expression trials. In each of the examples shown the titer for P0 virus averaged in the high 108 genomes/mL range, similar to the results reported by McCall et al., 2005 (8). The initial expression trials for each example were executed using P0 virus. Since these trials were run with cells at 2 × 106 cells/mL in 5 mL volumes, only 20–100 μL per well from the 2.5 mL obtained in the initial transfection were required to achieve multiplicities of infection of 2 or 5. We have also found the amount of protein produced in these 5 mL trials is sufficient for detection and quantitation using Labchip 90 or SDS-PAGE analysis followed by staining with Coomassie blue. The process reproducibly yields high-titer virus from the transfection step and we have also observed that the small-scale protein production can be reproduced at larger scale. We have scaled up full-length IKKε and several of the NS1 clones and found our 5 mL expression cultures were fairly accurate predictors of protein yield from 800 mL cultures.
In general, we chose to include steps that emphasized robustness over speed, such as following McCall et al., 2005 (8) in extending incubation of transfections to seven days rather than the more commonly used three days. One choice that emphasized speed was the use of QPCR for viral titer determination. This technique can take as little as two hours for DNA preparation and quantitation as compared to a week or more for plaque titering. However, because it counts the copies of a target DNA sequence present in the sample QPCR data may not reflect the actual number of infection competent virus present (23, 25). By restricting our use of this technique to the titering of freshly made virus at low passage number the quantitation of DNA target sequences should stay tightly correlated with the number of infectious particles. One advantage of QPCR is the ability to monitor for the loss of sequences from the recombinant virus during passaging. We designed one set of oligos that target a sequence linked to the inserted gene, the SV40 polyA sequence, and another that target a sequence in the viral genome retained in defective interfering particles, the ie1 gene promoter (35, 36). By comparing the titers from these two sets of oligos, the percentage of recombinant genomes in the viral preparation can be determined. This allows for a distinction between loss of the recombinant gene in a high-titer virus preparation and overall low viral titer. Our use of the Bac to Bac system also influenced our decision to use QPCR for titering because it can play an important role at an earlier step in the process. When using the Bac to Bac system, the gene of interest is recombined into baculoviral DNA in bacteria and purified from there for transfection. The presence of two other plasmids in these cells complicates the quantitation of specific DNAs yielded by the purification using standard spectrophotometric techniques. QPCR supplies a solution through its ability to quantitate specific DNAs in complex mixtures, in this case by using only the ie1-specific primers. This allows for data-driven decision-making at the transfection step of baculovirus generation. Using the QPCR data, the volume of the bacmid preparation used in the transfection step may be adjusted to compensate for variations in bacmid concentrations between individual preparations. This may be particularly important in a high-throughput setting where the number of samples is large and the DNA preparation method is automated. In this situation QPCR may provide an additional quality control step for monitoring the efficiency of the process.
We have presented data for two projects that illustrate the benefits of having the capability to run multi-construct, multi-vector expression trials in both E. coli and baculovirus-infected insect cells. For IKKε, the goals of the project were to screen a chemical library for novel ligands and solve three-dimensional structures of both the protein alone and bound with any novel ligands identified by the screen. These goals necessitated the use of both expression systems to obtain forms of the protein appropriate to each application. We determined that truncated versions of inactive mutants of the protein expressed in E. coli are produced at levels that will easily support a structural biology effort but we were unable to obtain active protein through bacterial production. The full-length protein produced in baculovirus-infected insect cells was active and produced in sufficient quantity to support assay development and high-throughput screening (Fig. 5). Consequently, each system yielded a form of the protein that meets the requirements of one of the major project objectives.
The NS1 protein of flavivirus is a relatively complex target for structure determination. Although the mature form of the protein is known to be glycosylated and there is evidence the glycosylation is required for NS1’s role in viral replication (34) it isn’t clear that the glycosylation is necessary for proper folding. Expression evaluation in E. coli of multiple constructs of NS1 cDNA from four different flaviviruses identified a number of truncations that were soluble when fused with MBP. Unfortunately these turned out to be false positives, as they were found to be high-molecular weight aggregates when subjected to size-exclusion chromatography. We moved a subset of the constructs into our baculovirus process using both intracellular expression vectors and secretion expression vectors. Only the secretion vectors yielded soluble protein, not only in the media but also in the cell (Fig 6, supplementary Fig 2, 3). The apparent molecular weights of the proteins obtained suggest that they were glycosylated as each form appears at a higher molecular weight than predicted from the amino acid sequence. The material found as soluble protein in the cell pellets had a slightly higher molecular weight than the material purified from the media. The difference may be due to removal of the signal peptide by signal peptidase for protein isolated from the media.
Previous reports indicated that the NS1 protein forms a dimer in infected mammalian cells (32) and a hexamer when secreted from these cells (33). We subjected the protein we purified from the conditioned media to size exclusion chromatography (SEC) to determine if the protein produced by insect cells also formed multimers. Although there was apparently some dimeric protein in SEC, most of the protein appeared to be monomeric (Fig. 8). This may be due to the presence of the MBP fusion tag or could indicate a difference between protein produced by insect cells and that produced by flavivirus-infected mammalian cells. We observed concentration and time-dependent precipitation in the fractions from the amylose purification of this protein. Thus the dimeric form of the protein we observed in SEC may represent an early stage of the aggregation that leads to precipitation. It has previously been shown that the addition of the mild detergent, n-octylglucoside, can disrupt the hexameric form of NS1 protein (33). The amylose affinity purification was done in the absence of any stabilizing additives such as glycerol, and the addition of glycerol or a mild detergent to the chromatography buffers may prevent or slow the aggregation of the protein. The aggregation state of the soluble protein found in the cell pellets remains to be determined. Although additional characterization will be required before proceeding to crystallization trials, these preliminary experiments suggest that our expression evaluation trials may have identified a viable source of protein for NS1 structure determination.
The results presented here illustrate the benefits of using a high-throughput parallel approach for expression evaluation. The ability to carry out parallel trials in both E. coli and baculovirus-infected insect cells proved beneficial for IKKε. Having a variety of combinations of fusion partners and signal peptides has also proven useful. The behavior observed for the NS1 proteins is particularly instructive. The four versions of the protein, although highly conserved, display distinctly different behaviors when fused to the various signal peptides (Fig. 6 and 7). The vectors described here provide broad flexibility in developing an expression strategy.
Supplementary Material
Acknowledgments
The HTP lab has been supported in part by a grant from the Office of the Vice-President of Research at the University of Michigan. W.C.B. is supported by NIH grant AI055672-1 to J.L.S.. Additional support for W.C.B. and J.R.R. was provided through the LSI Innovation Partnership. We are also grateful to our collaborators, Alan Saltiel and Stuart Decker of the University of Michigan and Richard Kuhn and Shilpa Parakh of Purdue University whose projects contributed to the work described here.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hunt I. From gene to protein: a review of new and enabling technologies for multi-parallel protein expression. Pro Expr Purif. 2005;40:1–22. doi: 10.1016/j.pep.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 2.Carpenter EP, Beis K, Cameron AD, Iwata S. Overcoming the challenges of membrane protein crystallography. Curr Opin Struct Biol. 2008;18:581–586. doi: 10.1016/j.sbi.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braun P, LaBaer J. High-throughput protein production for functional proteomics. TRENDS in Biotech. 2003;21:383–388. doi: 10.1016/S0167-7799(03)00189-6. [DOI] [PubMed] [Google Scholar]
- 4.Leder L, Freuler F, Forstner M, Mayr LM. New methods for efficient protein production in drug discovery. Curr Opin Drug Discov Dev. 2007;10:193–202. [PubMed] [Google Scholar]
- 5.Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. A new vector for high-throughput, ligation-independent cloning encoding a Tobacco Etch Virus protease cleavage site. Pro Expr Purif. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 6.Delproposto J, Majmudar CY, Smith JL, Brown WC. MOCR: A novel fusion tag for enhancing solubility that is compatible with structural biology applications. Pro Expr Purif. 2009;63:40–49. doi: 10.1016/j.pep.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berrow NS, Alderton D, Sainsbury S, Nettleship J, Assenberg R, Rahman N, Stuart DI, Owens RJ. A versatile ligation-independent cloning method suitable for high-throughput expression screening applications. Nuc Acids Res. 2007;35:e45. doi: 10.1093/nar/gkm047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCall EJ, Danielsson A, Hardern IM, Dartsch C, Hicks R, Wahlberg JM, Abbott WM. Improvements to the throughput of recombinant protein expression in the baculovirus/insect cell system. Pro Expr Purif. 2005;42:29–36. doi: 10.1016/j.pep.2005.03.021. [DOI] [PubMed] [Google Scholar]
- 9.Bahia D, Cheung R, Buchs M, Geisse S, Hunt I. Optimisation of insect cell growth in deep-well blocks: development of a high-throughput insect cell expression screen. Pro Expr Purif. 2005;39:61–70. doi: 10.1016/j.pep.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 10.Possee RD, Hitchman RB, Richards KS, Mann SG, Siaterli E, Nixon CP, Irving H, Assenberg R, Alderton D, Owens RJ, King LA. Generation of baculovirus vectors for the high-throughput production of proteins in insect cells. Biotech and Bioeng. 2008;101:1115–1122. doi: 10.1002/bit.22002. [DOI] [PubMed] [Google Scholar]
- 11.Tomalsky ME. Sequencing and expression of aadA, bla, and tnpR from the multi-resistance transposon Tn1331. Plaasmid. 1990;24:218–226. doi: 10.1016/0147-619x(90)90005-w. [DOI] [PubMed] [Google Scholar]
- 12.Donnelly MI, Zhou M, Sanville Millard C, Calncy S, Stols L, Eschenfeldt WH, Collart FR, Joachimiak A. An expression vector tailored for large-scale, high-throughput purification of recombinant proteins. Protein Expr Purif. 2006;47:446–454. doi: 10.1016/j.pep.2005.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tessiet DC, Thomas DY, Khouri HE, Laliberte F, Vernet T. Enhanced secretion from insect cells of a foreign protein fused to the honeybee melittin signal peptide. Gene. 1991;98:177–183. doi: 10.1016/0378-1119(91)90171-7. [DOI] [PubMed] [Google Scholar]
- 14.Jarvis DL, Garcia A., Jr Biosynthesis of the Autogrphica californica nuclear polyhedrosis virus gp64 protein. Virology. 1994;206:300–313. doi: 10.1006/viro.1994.1646. [DOI] [PubMed] [Google Scholar]
- 15.Blissard GW, Rohrmann GF. Location, sequence. Transcriptional mapping, and temporal expression of the gp64 envelope glycoprotein gene of the Orgyia psuedotsugata multicapsid polyhedrosis virus. Virology. 1989;170:537–555. doi: 10.1016/0042-6822(89)90445-5. [DOI] [PubMed] [Google Scholar]
- 16.Luckow VA, Lee SC, Barry GF, Olins PO. Efficient generation of infectious recombinant baculoviruses by site-specific transposon-mediated insertion of foreign genes into a baculovirus genome propagated in E. coli. J Virol. 1993;67:4566–4579. doi: 10.1128/jvi.67.8.4566-4579.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leusch MS, Lee SC, Olins PO. A novel host-vector system for direct selection of recombinant baculoviruses (bacmids) in Escherichia coli. Gene. 1995;160:191–194. doi: 10.1016/0378-1119(95)00233-v. [DOI] [PubMed] [Google Scholar]
- 18.Hillar A, Otulakowski G, Brodovich H. Purification and characterization of a recombinant rat prohaptoglobin expressed in baculovirus-infected Sf9 insect cells. Pro Expr Purif. 2007;55:246–256. doi: 10.1016/j.pep.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y-B, Wang Z-Y, Chen H-Y, Cui B-A, Wang Y-B, Zhang H-Y, Wang R. Secretory expression of porcine interferon-gamma in baculovirus using HBM signal peptide and its inhibition activity on the replication of porcine reproductive and respiratory syndrome virus. Veterinary Immunology and Immunopathology. 2009;1332:314–317. doi: 10.1016/j.vetimm.2009.05.017. [DOI] [PubMed] [Google Scholar]
- 20.Jarvis DL, Summers MD, Garcia A, Jr, Bohlmeyer DA. Influence of different signal peptides and prosequences on expression and secretion of human tissue plasminogen activator in the baculovirus system. J Biol Chem. 1993;268:16754–16762. [PubMed] [Google Scholar]
- 21.Futatsumri-Sugai M, Tsumoto K. Signal peptide design for improving recombinant protein secretion in the baculovirus expression vector system. Biochem Biophys Res Comm. 2010;391:931–935. doi: 10.1016/j.bbrc.2009.11.167. [DOI] [PubMed] [Google Scholar]
- 22.Janakiraman V, Forrest WF, Seshagiri S. Estimation of baculovirus titer based on viable cell size. Nat Protocols. 2006;15:2271–2276. doi: 10.1038/nprot.2006.387. [DOI] [PubMed] [Google Scholar]
- 23.Roldao A, Oliveira R, Carrondo MJT, Alves PM. Error assessment in recombinant baculovirus titration: Evaluation of different methods. J Virol Meth. 2009;159:69–80. doi: 10.1016/j.jviromet.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 24.Lo H-R, Chao Y-C. Rapid titer determination of baculovirus by quantitative real-time polymerase chain reaction. Biotechnol Prog. 2004;20:354–360. doi: 10.1021/bp034132i. [DOI] [PubMed] [Google Scholar]
- 25.Hitchman RB, Siaterli EA, Nixon CP, King LA. Quantitative real-time PCR for rapid and accurate titration of recombinant baculovirus particles. Biotechnol and Bioeng. 2007;99:1–5. doi: 10.1002/bit.21177. [DOI] [PubMed] [Google Scholar]
- 26.Perkins ND. Integrating cell-signalling pathways with NF-κB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- 27.Shimada T, Kawai T, Takeda K, Matsumoto M, Inoue J-I, Tatsumi Y, Kanamaru A, Akira S, K-i IK. a novel lipopolysaccharide-inducible kinase that is related to IκB kinases. Int Immunol. 1999;11:1357–1362. doi: 10.1093/intimm/11.8.1357. [DOI] [PubMed] [Google Scholar]
- 28.Chiang S-H, Bazuine M, Lumeng CN, Geletka LM, Mowers J, White NM, Ma J-T, Zhou J, Qi N, Westcott D, Delproposto JB, Blackwell TS, Yull FE, Saltiel AR. The protein kinase IKKε regulates energy balance in obese mice. Cell. 2009;138:961–975. doi: 10.1016/j.cell.2009.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kishore N, Huynh QK, Mathialagan S, Hall T, Rouw S, Creely D, Lange G, Caroll J, Reitz B, Donnelly A, Boddupalli H, Combs RG, Kretzmer K, Tripp CS. IKK-I and TBK-1 are enzymatically distinct from homologous enzyme IKK-2. J Biol Chem. 2002;277:13840–13847. doi: 10.1074/jbc.M110474200. [DOI] [PubMed] [Google Scholar]
- 30.Kramer LD, Styer LM, Ebel GD. A global perspective on the epidemiology of West Nile Virus. Annu Rev Entomol. 2008;53:61–81. doi: 10.1146/annurev.ento.53.103106.093258. [DOI] [PubMed] [Google Scholar]
- 31.Kyle JL, Harris E. Global spread and persistence of Dengue. Annu Rev Microbiol. 2008;62:71–92. doi: 10.1146/annurev.micro.62.081307.163005. [DOI] [PubMed] [Google Scholar]
- 32.Winkler G, Randolph VB, Cleaves GR, Ryan TE, Stollar V. Evidence that the mature form of the flavivirus nonstructural protein NS1 is a dimer. Virol. 1988;162:187–196. doi: 10.1016/0042-6822(88)90408-4. [DOI] [PubMed] [Google Scholar]
- 33.Flamand M, Megret F, Mathieu M, Lepault J, Rey FA, Deubel V. Dengue virus type 1 nonstructural glycoprotein NS1 is secreted from mammalian cells as a soluble hexamer in a glycosylation-dependent fashion. J Virol. 1999;73:6104–6110. doi: 10.1128/jvi.73.7.6104-6110.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tajima S, Takasaki T, Kurane I. Characterization of Asn130-to-Ala mutant of dengue type 1 virus NS1 protein. Virus Genes. 2008;38:323–329. doi: 10.1007/s11262-008-0211-7. [DOI] [PubMed] [Google Scholar]
- 35.Blissard GW, Rohrman GF. Baculovirus diversity and molecular biology. Ann Rev Entomol. 1990;35:1277–155. doi: 10.1146/annurev.en.35.010190.001015. [DOI] [PubMed] [Google Scholar]
- 36.Kool M, Voncken JW, Van Lieer FLJ, Tramper J, Vlak JM. Detection and analysis of Autographa californica nuclear polyhedrosis virus mutants with defective interfering properties. Virology. 1991;183:739–746. doi: 10.1016/0042-6822(91)91003-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.