

Abstract
The maximum likelihood method provides a powerful approach for many problems in cryo-EM image processing. This contribution aims to provide an accessible introduction to the underlying theory and reviews existing applications in the field. In addition, current developments to reduce computational costs and to improve the statistical description of cryo-EM images are discussed. Combined with the increasing power of modern computers and yet unexplored possibilities provided by theory, these developments are expected to turn the statistical approach into an essential image processing tool for the electron microscopist.
Introduction
The cryo-EM single-particle reconstruction (SPR) problem is a very difficult one. Given a large number of very noisy electron-microscope images, each showing a macromolecular “particle” in a random position and orientation, the problem is to deduce the three-dimensional (3D) structure of the particles that were imaged. It is amazing that standard software packages now allow even casual users to perform such reconstructions, in a numerical process that seems little short of magical. The goal of this chapter is to describe the basis of a particularly powerful approach to the SPR problem and related tasks in 2D crystallography and electron tomography.
Maximum-likelihood estimates
When we use an SPR algorithm to obtain a 3D model from a set of images 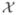 , how do we know that we have found the best model? Formally, we would want to know what quantity is optimized to yield the model. Traditional SPR refinement algorithms use an iterative process. The images are aligned and sorted to give the best match to a set of projections of the nth model. Given the angles of each projection, a synthesis of the 3D map from the images is performed by standard methods, to yield the (n + 1)st model. This process is repeated until the model does not change. Arguably this process yields a least-squares estimate, as the traditional cross-correlation based projection-matching step is equivalent to a least-squares optimization. In the optimization process a numerical density value is obtained for each voxel of the model, and as a byproduct the values of orientation angles for each particle are also found. There is however no theory that says that the model obtained in this way is the most reliable one.
, how do we know that we have found the best model? Formally, we would want to know what quantity is optimized to yield the model. Traditional SPR refinement algorithms use an iterative process. The images are aligned and sorted to give the best match to a set of projections of the nth model. Given the angles of each projection, a synthesis of the 3D map from the images is performed by standard methods, to yield the (n + 1)st model. This process is repeated until the model does not change. Arguably this process yields a least-squares estimate, as the traditional cross-correlation based projection-matching step is equivalent to a least-squares optimization. In the optimization process a numerical density value is obtained for each voxel of the model, and as a byproduct the values of orientation angles for each particle are also found. There is however no theory that says that the model obtained in this way is the most reliable one.
The statistical approaches discussed in this chapter seek to maximize a probability function. Suppose we group the desired 3D map along with any other quantities that we wish to estimate into a more generalized model Θ. In a sense, what we would like to maximize is the probability P(Θ|) that this model is the correct one, given the data. Unfortunately, there are both philosophical and practical problems surrounding this quantity. Some people would say that Θ is not a random variable in the first place, so how can one define a probability function. But if the philosophical problems are bypassed, perhaps by imagining an ensemble of possible true structures Θ that could have given us our dataset , there is still the problem of computing this quantity. An easy way around these problems is to compute the probability of observing given Θ. This is a valid and computable probability, and it is given a special name, the likelihood 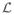 (Θ) = P(|Θ). What is unusual here is that the likelihood is expressed as a function of the model Θ rather than of the data .
(Θ) = P(|Θ). What is unusual here is that the likelihood is expressed as a function of the model Θ rather than of the data .
The choice of the likelihood as the quantity to be maximized can be understood by applying Bayes’ rule,
| (1) |
We know what P(|Θ) is, that is just the likelihood. P() is an imponderable: what is the probability of obtaining this dataset instead of some other? However, since it doesn’t depend on Θ we won’t have to worry about it, and for our present purposes we can replace it by a constant. The resulting quantity to be maximized is P(|Θ)P(Θ) and is called the posterior probability. The model Θ that maximizes it is called the maximum a posteriori (MAP) estimate, and we will discuss briefly the use of this estimation approach at the end of this chapter. The term P(Θ) is called the prior probability, as it reflects any knowledge we might have about the model in the absence of any data. For now, suppose we believe that all possible models are equally likely, so that P(Θ) is also a constant. Then maximizing P(Θ|) becomes the same as maximizing the likelihood.
Finding the model Θ that gives the maximum value of the likelihood—this is called the maximum-likelihood estimate or MLE—is a very good way to find the best model. The MLE is asymptotically unbiased and efficient; that is, in the limit of very large datasets, the MLE is as good or better than any other estimate of the true model.
Introduction to the EM-ML algorithm
Finding the model Θ that maximizes the likelihood is a daunting task. In principle, the density value of each voxel in the 3D map must be varied until the best set of densities is found—an optimization problem with on the order of 106 unknowns! Fortunately there is a straightforward way to find the ML estimate, called the expectation maximization algorithm (Dempster et al., 1977). We will abbreviate this as the EM-ML algorithm to avoid confusion with the terminology of EM for “electron microscopy”. The idea behind the EM-ML algorithm is illustrated here with a simple example based on the Gaussian mixture model (Redner & Walker, 1984), while a formal presentation of the theory is considered in the next section of this chapter. Additional examples designed to mimic cryo EM are worked out in Yin et al. (2003) and continued to consider resolution in Prust et al. (2009). More sophisticated miniature examples are considered in Yin et al. (2004).
Imagine a series of position measurements xi with i = 1, …, N coming from a single-molecule optical tracking experiment. Typically the observed position shows Gaussian-distributed errors from measurement noise. One can record a large number of position measurements and compute for example the MLE of the mean and variance directly from the measurements.
Suppose that the single molecule under observation undergoes a conformational change between two states, which we’ll call state 0 and state 1. With this change the reporter group changes its true x-coordinate between two discrete values. We would like to estimate these two values. One approach would be to make a histogram of the measured values, as in Figure 1, and do some sort of fit to the histogram. A more powerful way however is to find the MLE of the two mean values directly from the measured values.
Figure 1.

Histogram of values of a random variable x. The values were drawn from a distribution consisting of a mixture of two Gaussians with means of 0 and 2.5.
Computing the likelihood starts with the probability-density function (PDF) of the observed values, a mixture of two Gaussians,
| (2) |
In this example we are interested in determining the model parameters Θ = {μ0, μ1} assuming that the other parameters a0, a1 and σ are already known. The probability of a particular measured value would be vanishingly small were it not for the fact that any physical measurement has a finite resolution. Letting ε be the resolution of measurement, the probability of measuring the first value is
| (3) |
and the probability of the measurement of the entire data set is
| (4) |
Since ε does not vary when Θ is changed, it is irrelevant to the maximization process. Thus in the literature this factor is traditionally ignored; the likelihood is instead written simply as the probability-density function, which in this case, is a product of simpler PDFs:
| (5) |
Nevertheless maximizing this product of PDFs appears daunting.
In the case of a single Gaussian distribution, the maximization of the likelihood turns out to have a very simple form. The ML estimates of the mean and standard deviation are simply equal to the mean and standard deviation of all the measured values. In this special case maximum-likelihood and least-squares estimation give the same answer. Is there a way we can exploit this simple ML estimation approach for the present problem involving a mixture of two Gaussians?
We could just set a threshold, and divide the data set into two halves. We would then compute the average of all the xi values that fell below the threshold, and do the same for all those above the threshold, and call these our estimates of the means. However, taking the averages of these two “classes” of measurements will produce estimates that are biased, being spread more widely than the two correct means.
Estimating the two means would be very simple if we had independent information about the underlying state of the molecule. Let the yi be a set of “switch” variables such that when yi = 0 we know that the corresponding xi is obtained from the molecule in state 0, and when yi = 1 the molecule is in its state 1. Then it is very easy to obtain the means μ0 and μ1 which are the MLEs of the positions:
| (6) |
Unfortunately we do not have access to the yi values; they are so-called “hidden variables”. The EM-ML algorithm however provides a simple way to iteratively converge on the ML mean values. It starts with an initial guess of the parameters of the two underlying Gaussian distributions. For example, the initial guess could have means that are too far apart, as in the top panel of Figure 2A. The first step of the expectation-maximization algorithm is to provide an estimate of the hidden yi variable corresponding to each measurement xi. The estimate is computed as the expectation value ŷi, which in this case is simply equal to the probability of the molecule being in state 1. It is computed from each measurement xi, based on the current estimate of the Gaussian distributions.
Figure 2.

EM-ML estimation of the means and amplitudes in a mixture of two Gaussians as in Figure 1. A, top panel, a histogram of 1000 simulated measurements is shown, along with two Gaussian components computed on the basis of initial guesses for the mean and amplitude of the components. The middle panel shows how the expectation ŷ varies with the measured value x. Instead of there being a strict classification of x values into one or the other component, these ŷ values take on intermediate values in the range of x values where the components overlap. The bottom panel illustrates how a weighted average can give rise to the mean value for the left-hand component. The bins of the histogram have been scaled by the values of ŷ, while the initial guess of the PDF is plotted as a smooth curve. The center of mass of the weighted histogram yields the new estimate for μ1. B, the same plots are shown after the two Gaussian components have been updated with new means and amplitudes by one EM-ML iteration. C, the result after convergence with 24 iterations. The simulation was based on means of 0 and 2.5. The EM-ML estimated means were −0.06 and 2.46.
The second step is the maximization step, where we compute the MLEs of the means based on the inferred ŷ values. In this case the MLEs are simply computed as weighted averages of the xi values, weighted by either 1 − ŷi or ŷi to yield the two estimates μ0 and μ1. The calculation is exactly the same as in Eq. (6) except that ŷi replaces yi. This gives us two new mean values, which we can then use for another round of the EM-ML algorithm.
The remarkable property of the expectation-maximization iteration is that the new estimated values of the means are guaranteed to result in an increased likelihood of the model. Figure 2 provides an illustration of the EM-ML algorithm. In the figure, histograms are used as a visualization device, but the underlying computations do not involve the binning or sorting of events at all. The figure shows one EM-ML iteration, and also the result after 25 iterations, when the parameters have reached a fixed value.
This simple example demonstrates three important features of the EM-ML algorithm. Firstly, the algorithm “fills in” missing information through the computation of the expectation values of hidden variables. Secondly, it exploits the hidden variable values to make the process of computing the MLE much easier. Thirdly, perhaps the most interesting aspect of the EM-ML algorithm is that it makes use of “fuzzy” estimates of the hidden variables. Even though the underlying yi values, could we measure them, take only the values 0 and 1, the expectation values ŷi vary continuously over the range of 0 to 1. The maximization of the likelihood nevertheless converges correctly when these expectation values are used.
Relevance to SPR problems
The features of this simple example are mirrored in the much more complex computations involved in single-particle reconstruction. In EM-ML processing of particle images, “hard” values for the orientation angles of each particle are not assigned. Instead the orientation of each particle is described in a fuzzy way as a probability density function, giving the probability that the particle assumes each possible orientation. Similarly, when EM-ML is used to simultaneously reconstruct several different conformations (i.e. distinct 3D maps) from images of mixed populations of particles, the assignment of a given particle image to a given conformation is treated as a hidden variable, and is made in a “fuzzy” way.
This chapter reviews applications of the EM-ML algorithm to a range of image processing tasks across various cryo-EM modalities. First, a formal description of the theory is presented along with a typical example from cryo-EM image processing. This theoretical framework is then used as a common basis to describe existing EM-ML approaches for the analysis of single-particles (with and without symmetry), 2D crystals and sub-tomograms. In addition, the validity of the most common statistical data model and possible alternatives is discussed and an overview of approaches to accelerate the intensive EM-ML computations are presented. The chapter concludes with a discussion on the perspectives of the statistical approach for cryo-EM image processing.
Theoretical basis
The Maximum Likelihood Estimator
At the heart of the maximum likelihood method lies a parameterized, statistical model that is used to describe the underlying physics of the data formation process. Again, we will denote the set of model parameters by Θ, and our data set of N independent measurements by = (X1, X2, …, XN). Then, the statistical model is expressed in the form of the probability density function P(|Θ), the probability of observing the data given Θ. For a given set of model parameters, the PDF will show that some data are more probable than others. In the practical situation however, we are interested in the opposite. We have already observed the data and are looking for those model parameters that best fit the data. In particular, we want to find those model parameters that make the data “more likely” than any other parameter set would make them.
To that purpose, and as explained in more detail in the introduction, we define the likelihood function (Θ) = P(|Θ) as a function of Θ. Whereas the PDF is defined as a function of the data given a particular set of model parameters, the likelihood function is a function of the model parameters for a given data set. The method of maximum likelihood aims at finding the set of parameters that maximizes (Θ). This is the maximum likelihood estimator (MLE) of Θ:
| (7) |
Assuming that all observations are statistically independent, the likelihood can be written as a product of the PDFs of the individual observations:
| (8) |
and since maxima are unaffected by monotone transformations, for computational convenience one often takes the logarithm of this expression:
| (9) |
Depending on the form of P(Xi|Θ), finding the maximum of L(Θ) may be straightforward or extremely difficult. As we will describe in the example below, analytical expressions to obtain the MLE may be obtained directly for simple problems. For many other problems direct optimization of the likelihood function is analytically intractable and more elaborate techniques, like the EM-ML algorithm must be employed.
An example of direct MLE calculation in cryo-EM
One example of straightforward likelihood optimization in cryo-EM is the estimation of the underlying signal from a series of noisy, structurally homogeneous and aligned 2D images. Let’s assume the following data model:
| (10) |
where Xi are the observed images of J pixels each and with pixel values Xij; A is the underlying 2D image (with pixel values Aj) that is common to all images; and Gi are images of independent noise with pixel values Gij taken from a Gaussian distribution with zero mean and unity standard deviation.
Note that in this case our set of model parameter set Θ consists of image A and parameter σ. The PDF of observing pixel value Xij is then given by a Gaussian distribution centered at Aj and with standard deviation σ (see Figure 3). Furthermore, because we assume independence between all pixels, the PDF of observing the entire image Xi may be expressed as the multiplication over the PDFs of all individual pixels:
| (11) |
where ||Xi − A||2 denotes the sum of the squared residuals over all pixels .
Figure 3.

An example of direct MLE calculation. A, an image A with a zoom window centered on one individual pixel value with value Aj is shown. B, three copies of image A with different instances of white Gaussian noise are shown together with zoom windows on the same pixel j with pixel values X1j, X2j and X3j. C, the PDF of the jth pixel in the noisy images is shown as a Gaussian curve centered at Aj and with standard deviation σ. Direct calculation of is performed by averaging over X1j, X2j and X3j. D, the MLE of the entire image A is shown together with a zoom window, centered on . Note that the MLE of the image will approach the image in A if larger numbers of noisy images are available.
Thereby, the log-likelihood function, as defined in Eq. (9) reduces to:
| (12) |
The MLE for the underlying signal may then be obtained directly by setting the partial derivatives of Eq. (12) with respect to Aj equal to zero, and solving for Aj. This yields the well-known equation to calculate the average image:
| (13) |
Similarly, the MLE for σ would yield the formula for calculating the root mean square deviation between the average and the observed images.
Equation (12) illustrates that under the assumption of independent, Gaussian noise with equal standard deviation, the MLE is equal to the least squares estimator (LSE), which aims at minimizing . This equality has been used erroneously to argue against the application of maximum likelihood methods in cryo-EM. However, as we will see below, for more complicated problems the method of maximum-likelihood will actually yield very different results from conventional approaches based on least-squares estimation.
Incomplete data problems
For many problems the likelihood function cannot be optimized directly, but it can be simplified by assuming the existence of additional, “hidden” variables. Without the hidden variables, the data is considered to be incomplete. The complete data would comprise both the observed and the hidden variables, and finding the MLE for the complete data problem would be a trivial task. In some cases the hidden variables actually correspond to incompleteness in the data vectors themselves, due to problems in the data collection process. More often however, the hidden variables correspond to aspects of physical reality that could in principle be measured but are not observed for practical reasons.
Let be the incomplete, observed data and assume that a complete data set (, 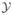 ) exists. Then, the MLE is determined by the so-called marginal likelihood of the observed data:
) exists. Then, the MLE is determined by the so-called marginal likelihood of the observed data:
| (14) |
where P(|Θ) is the unconditional probability of , regardless of the values of , and this probability is obtained by integrating the joint probabilities over all possible values of . This is called marginalization. P(|, Θ) is the probability of given that has happened, and P(|Θ) is the prior probability of happening.
It is important to note that because of the marginalization Eq. (14) is only a function of Θ and not of the hidden variables . Thereby, the problem at hand is only to find those parameter values ΘMLE that maximize the marginal likelihood function.
An example of an incomplete data problem in cryo-EM
The first description of a maximum-likelihood approach for an incomplete data problem in cryo-EM was described by Sigworth (1998). He considered the problem of finding AMLE, cf Eq. (13), for a set of noisy 2D images with unknown in-plane rotations and translations. Or in other words, he presented a ML approach for the 2D alignment of a structurally homogeneous set of images. In this case, the data are modelled as:
| (15) |
where Rφi denotes the in-plane transformation that brings the common underlying signal in register with the ith image. This transformation comprises a single rotation αi and two translations in perpendicular directions xi and yi.
The incompleteness of the observed data lies in the fact that the relative orientations of all images have remained unobserved in the experiment. The complete data set would be (, ), with = (φ1, φ2, …, φN), and finding the MLE for the complete data set would be as trivial as described in the simple example above.
For the incomplete case, the marginal log-likelihood function, cf. Eq. 14, is given by:
| (16) |
For any given transformation φ and parameter set Θ, the conditional probability of observing image Xi is again expressed as a multiplication over J Gaussian distributions, this time centered at the correspondingly oriented reference image RφA:
| (17) |
Note that, apart from the model parameters Θ, this probability is also a function of the hidden variable φ. This concept is further illustrated in Figure 4.
Figure 4.

The PDF of a noisy image as a function of its relative orientation with respect to a reference image. A, Xi, a rotated and noisy version of reference image A from Figure 3A is shown. B, top panel, the probability P(Xi|φ, Θ) of observing Xi given a model Θ that comprises image A is shown (on an arbitrary scale) as a function of the relative orientation φ. The bottom panel shows RφA, i.e. image A rotated according to φ. Note that P (Xi|φ, Θ) is highest when φ corresponds to the correct orientation of Xi.
An additional advantage of the maximum likelihood approach is the natural way in which prior knowledge about the hidden variables may be handled. The term P(φ|Θ) provides a statistical description of the distribution of the hidden variables. If we assume that the in-plane rotations are evenly distributed and that particle picking has left residual origin offsets with Gaussian distributions in both directions, the probability density of φ may be calculated as:
| (18) |
where ξx and ξy are the expected values for the in-plane translations in directions x and y, and σxy is the standard deviation in the translations in either direction.
Thereby, prior knowledge that large origin offsets are less likely than small offsets is translated in an effective downweighting of larger offsets in the probability calculations of all possible orientations. Although a similar term may also be incorporated into maximum cross-correlation approaches (Sigworth, 1998), this is not common practice in conventional approaches for the alignment of cryo-EM images. Instead, one often expresses this prior knowledge in a less powerful way: by limiting the searches for the optimal offsets to a user defined maximum value in all directions.
From Eqs. (17) and (18), we can see that Θ = (A, σ, ξx, ξy, σxy) and the task at hand is to find those parameters ΘMLE that maximize the marginal likelihood function as defined in Eq. (16).
The EM-ML algorithm
The EM-ML algorithm is a general tool to find MLEs for incomplete data problems (Dempster et al., 1977). The intuitive idea behind this algorithm is an old one. Because one does not know parameter estimates Θ nor the hidden variables , one iteratively alternates between estimating both. For a given set of model parameters Θ one estimates the hidden variables, for the resulting hidden variables one finds the best model parameters, and one repeats this process until the model parameters no longer change.
However, rather than finding the best given an estimate Θ(n) at the nth iteration, the EM-ML algorithm computes a distribution over all possible values of . To this purpose, in the so-called expectation (E) step one first calculates the expected value of the complete-data log-likelihood function with respect to the missing data , given the observed data and the current parameter estimates Θ(n):
| (19) |
Here, P(|, Θ(n)) is the conditional probability of the missing variables in terms of the observed measurements and the current model parameters, which is calculated using Bayes’ rule:
| (20) |
The integration of P(|, Θ(n)) over all possible values of represents the above-mentioned distribution of the hidden variables. Given this distribution, in the subsequent maximization (M) step one computes new estimates for the model parameters by maximizing the corresponding expectation:
| (21) |
The new model parameters are then used for the next E-step, and the process is repeated as necessary. It can be shown that each iteration is guaranteed to increase the log-likelihood and the algorithm is guaranteed to converge to a local maximum of the likelihood function (Dempster et al., 1977).
An example of EM-ML in cryo-EM
For the example of aligning a set of structurally homogeneous images described above, the E-step of the EM-ML algorithm yields:
| (22) |
which can be rewritten by substitution of Eqs. (17) and (18), and by separating terms C that are not related to A:
| (23) |
In the subsequent M-step one maximizes Q(Θ, Θ(n)) with respect to the model parameters. From Eq. (23) it can be readily seen that obtaining new estimates for A corresponds to solving a weighted least-squares problem with weights P(φ|Xi, Θ(n)). The result is a weighted average comprising contributions from all possible values of φ for every image Xi:
| (24) |
The concept of calculating A(n+1) as a probability weighted average is further illustrated in Figure 5. All other model parameters are updated using similar probability-weighted average calculations.
Figure 5.

Reference image calculation by probability-weighted averaging. A, three noisy versions of reference image A from Figure 3A in different orientations (Xi, with i = 1, 2, 3). B, images for all sampled φ. The opacity of the images is used to illustrate the weight P(φ|Xi, Θ(n)) in the weighted average calculation that produces the updated estimate of the reference A(n+1), see Eq. (24). Note that X1 corresponds to the same image that was shown in Figure 4A and that the columns correspond to the same orientations as in Figure 4B.
Comparison with conventional methods
It is interesting to compare the EM-ML approach with conventionally more popular methods in cryo-EM. In particular, it has been common practice to include the hidden variables as part of the unknown model parameters. For the example of single-reference 2D-alignment described above, the model thereby comprises the estimate for the underlying signal (A) and the optimal orientations for each of the individual observations ( for i = 1, …, N). One typically minimizes the following least squares target:
| (25) |
by iteratively alternating between estimating the underlying signal and estimating the orientations. For a given estimate A(n), one calculates the optimal orientation for each image Xi as the one that minimizes the squared difference ||Xi − RφiA||2. Then, one calculates a new estimate for the underlying signal by:
| (26) |
The orientation that minimizes the squared difference term mentioned above in turn maximizes the inner product between images Xi and RφiA. This inner product is also called the cross-correlation, and hence the term “maximum cross-correlation” approach.
Comparison of Eqs. (25) and (16) illustrates that the maximum cross-correlation and maximum likelihood approaches pursue different objectives. The principal difference lies in the marginalization over the hidden variables in the latter. Consequently, in the maximum likelihood approach model parameters are calculated as probability-weighted averages over all possible orientations, while only the “best” orientation is considered for each image in the maximum cross-correlation approach, cf. Eqs. (26) and (24).
The advantages of the maximum likelihood approach appear at low signal to noise ratios. The higher the level of noise in the data, the higher the number of false peaks that occur in the cross-correlation function. Thereby, the orientations that maximize the cross-correlation function are less likely to reflect the optimal ones. The statistical description of the noise in the maximum likelihood approach allows to deal with these ambiguities in the cross-correlation function. Correspondingly, the maximum likelihood approach will yield better results than the maximum cross-correlation approach for data with high levels of noise. Interestingly, the two approaches become equivalent in the absence of noise: when σ approaches zero, probability distributions P(φ|Xi, Θ(n)) become delta functions centered at . In other words, the maximum cross-correlation approach may be considered as a special case of the maximum likelihood method that ignores the presence of noise in the data.
EM-ML approaches in cryo-EM
The EM-ML algorithm has been applied to a variety of tasks in cryo-EM image processing. Although the actual applications differ widely, the theoretical framework described in the previous section may be employed to put all these approaches on a common basis. Most approaches share the assumption of independent Gaussian noise in the data. Thereby, the optimization strategy typically remains the same and the differences between the existing approaches lie in the unknown model parameters (Θ) and the hidden variables ().
Single particle analysis
As described in the example above, it was Sigworth (1998) who introduced the concept of optimizing a marginal likelihood function to the field. His approach to align a structurally homogeneous set of 2D images was tested on simulated data that were in accordance with the assumption of white and Gaussian noise. These simulations showed that the ML approach has a reduced sensitivity towards the choice of the starting model and allows recovering the underlying signal from much noisier data than the maximum cross-correlation method. This paper was followed by a series of contributions applying similar ideas to single-particle analysis (see Table 1).
Table 1.
An overview of EM-ML approaches in single-particle analysis.
| Approach | Data model | Model Parameters Θ | Hidden variables
|
|---|---|---|---|
| Sigworth 1998 | Xi = Rφi A + σGi | A, σ, ξx, ξy, σxy | αi, xi, yi |
| kerdenSOM | Xi = Aki+ σGi | Ak, σ, πk | ki |
| ML2D | Xi = Rφi Aki + σGi | Ak, σ, πk, σxy | ki, αi, xi, yi |
| ML3D | Xi = Pφi Vki + σGi | Vk, σ, πk, σxy | ki, αi, βi, γi, xi, yi |
| MLn3D | Xi = siPφi Vki + σGi + ni | Vk, σ, πk, σxy, si, ni | ki, αi, βi, γi, xi, yi |
Symbols Xi, Rφi, A, σ, αi, xi, yi, ξx, ξy and σxy were defined in the section that provided the theoretical basis. Subscripts k, with k = 1, …, K are used to indicate the kth model in multi-reference refinement schemes. Then πk are prior probabilities of that model and ki indicates that model k is the correct one for the ith observed image. Vk indicates a three-dimensional model and Pφi is a projection operation, with φi = (αi, βi, γi, xi, yi) comprising three Euler angles and two in-plane translations. si and ni are multiplicative and additive parameters for each ith image.
Firstly, Pascual-Montano et al. (2001) described a neural network for the classification of a set of pre-aligned 2D images called kerdenSOM, for kernel-density self-organizing map. In this work, the data model comprises multiple 2D models (Ak with k = 1, …, K) and the assignments ki of the experimental images to these models are treated as hidden variables. This results in a ML variant of the conventional k-means classifier [see Frank (2006)], where experimental images are not assigned to a single model but contribute to all models with varying weights. In addition, this contribution considered the K models to be arranged in a 2D map and defined a regularization term to the log-likelihood in order to impose similarity between neighbouring models in the map. Thereby, the behaviour of a neural network, or self-organizing map, was achieved. This has the additional advantage that K does not necessarily needs to reflect the number of different classes in the data. The efficiency of this approach was demonstrated for cryo-EM images of large T-antigen and for rotational spectra of negatively stained particles of hexameric helicase G40P.
Secondly, Scheres et al. (2005a) proposed a 2D multi-reference alignment scheme, called ML2D. As for the kerdenSOM algorithm, in this algorithm Θ comprises multiple 2D images Ak with k = 1, …, K. In this case however, the problems of alignment and classification were tackled simultaneously by treating both the in-plane transformations φi = (αi, xi, yi) and the class assignments ki as hidden variables. The ML method was again shown to yield much better model estimates than the maximum cross-correlation approach for simulated data with white Gaussian noise. It was also shown that these advantages were strongly reduced for simulated data with dependent noise. Nevertheless, for experimental cryo-EM images the ML2D algorithm was shown to be robust to the choice of the initial starting models and reference-free alignments and classifications could be obtained by starting the multi-reference alignments from averages of random subsets of the unaligned images. Application of this approach to cryo-EM images of large-T antigen particles led to the first-time visualization of an overhanging dsDNA probe in this complex.
The next EM-ML approach to single particle analysis concerned a 3D multi-reference refinement scheme (Scheres et al., 2007a). In this case K 3D reference maps Vk are refined simultaneously against a structurally heterogeneous set of projections. The hidden variables of this problem comprise six parameters for every image: its class assignment ki and its 3D orientation as described by three Euler angles and two in-plane translations φi = (αi, βi, γi, xi, yi).
For 3D refinements, the maximization step of the EM-ML algorithm is more complicated than in the 2D case. By expressing the 3D electron densities of the K models as weighted sums of smooth radial basis functions called blobs (Marabini et al., 1998) the reconstruction problem was expressed as a system of linear equations. Optimization of the log-likelihood function was shown to correspond to finding a weighted least squares solution to the reconstruction problem, for which purpose a modified version of the algebraic reconstruction technique (ART) (Eggermont et al., 1981) was developed.
Again, the ML approach was shown to be robust to high levels of noise and relatively insensitive to the starting model. Most significantly, it allowed separation of projections from distinct 3D structures by starting multi-reference refinements from random variations of a single, strongly low-pass filtered initial model. Thereby, the classification protocol, termed ML3D, is unsupervised as it does not depend on any prior knowledge about the structural variability in the data. Its efficiency and potentially wide applicability was demonstrated for two challenging cryo-EM data sets. ML3D classification separated projections of 70S E. coli ribosomes in a ratcheted conformation and in complex with elongation factor G (EF-G) from unratcheted ribosomes without EF-G. Projections of large-T antigen were classified according to various degrees of bending along the central axis of the dodecameric complex.
More recently, ML3D classification was observed to yield suboptimal results in certain cases. For structurally heterogeneous cryo-EM data sets on E. coli 70S ribosomes and on human RNA polymerase II, rather than separating different conformations reconstructions at distinct intensities were obtained. The origin of the problem was found in a typical cryo-EM preprocessing step: image normalization.
In the normalization process one aims to minimize additive and multiplicative variations in the signal among all images. Since the abundant noise makes it impossible to normalize the signal itself, it is common practice to normalize the noise instead. However, variations in signal-to-noise ratios or artifacts in the images often lead to small, remaining variations in the signal. These variations can often be ignored in conventional refinement schemes because normalized cross-correlation coefficients are invariant to additive or multiplicative factors. In ML refinements however, the squared distance metric inside the PDF calculation, see Eq. (15), is highly sensitive to these variations.
To reduce the corresponding sensitivity of the ML approach to normalization errors, the model parameter set Θ was extended with a multiplicative and an additive factor for the signal in each experimental image (si, respectively ni in Table 1). For both the 70S ribosome and the RNA polymerase II data sets, the corresponding approach, which was termed MLn3D, successfully classified distinct structural states (Scheres et al., 2009b). For the 70S ribosome, this resulted in a previously unobserved conformation with spontaneous ratcheting and two tRNAs in hybrid states (Julian et al., 2008).
Icosahedral viruses
The computation of structures having icosahedral symmetry from cryo-EM images is a particular case of single particle analysis. However, because of the high-order rotational symmetry of the icosahedral group, several special approaches have been developed which are described in this section.
Incorporation of the icosahedral symmetry is often done by representing the electron scattering intensity of the particle as a weighted sum of basis functions. Because all of the operations in the icosahedral group are rotational operations, it is most natural to use the spherical coordinate system (r,θ, φ) in which case the symmetry constrains the angular behavior of the basis functions (i.e., θ and φ) but not the radial behavior of the basis functions (i.e., r). The usual choice is to use basis functions that are a product of angular and radial functions where the angular part is a linear combination of spherical harmonics as introduced by Laporte (1948) and developed in subsequent years by many authors including Zheng & Doerschuk (1996). The experimental images are roughly linear transformations of the unknown weights in the weighted sum of basis functions, which is important for practical computation of the 3D reconstruction. However, as in the general single-particle case described above, this 3D reconstruction is dependent on unknown parameters, e.g., the projection orientations φi and class assignments ki. Different types of maximum likelihood estimators result depending on the treatment of these parameters.
In the original work of Vogel et al. (1986); Provencher & Vogel (1988); Vogel & Provencher (1988), which resulted in the “ROSE” algorithm, a Gaussian maximum likelihood estimator is used which estimates both the weights in the weighted sum of basis functions and the parameters that determine the linear transformation. That is, rather than treating the unkown projection orientations φi and class assignments ki as hidden variables as described for the general single-particle case, this approach includes these parameters in the model Θ. The advantage of this approach is that no probabilistic information on the behavior of the parameters is required with the disadvantage of having to solve a difficult optimization problem, especially as the number of images grows.
This type of approach was further developed by Doerschuk & Johnson (2000) and Yin et al. (2001, 2003) who proposed to treat the projection orientations and class assignments as hidden variables () and use an EM-ML algorithm to optimize the corresponding marginal likelihood function of Eq. 14. In addition, these authors use alternative radial basis functions that are linear combinations of spherical Bessel functions. These functions have two advantages over the Laguerre polynomials used in the ROSE algorithm. The first advantage is that they are non-zero only for a range of r, which allows straightforward masking of the 2D images and 3D reconstruction. The second advantage is that the 3D Fourier transform of the product of the angular and radial basis functions can be computed symbolically so the projection slice theorem can be used to compute the projection of the electron scattering intensity in an arbitrary direction. Kam & Gafni (1985) describe an alternative approach using much of the same mathematics for the description of the electron scattering intensity. However, the optimization problem that is solved does not appear to be a maximum likelihood problem.
The approach of locking symmetry into the basis functions is general although, depending on the number of operators in the symmetry group, it is more or less valuable in terms of reducing computation. Zheng & Doerschuk (1996) describe the necessary basis functions for all of the platonic symmetries. Prust et al. (2009) describes application of the approach to the rotational symmetry of the tail of infectious bacteriophage P22. Chen (2008) describes application of the approach to objects with helical symmetry using standard basis functions (Moody, 1990), Lee et al. (2009) describes new basis functions focused on helical symmetry and Lee (2009) uses these basis functions to solve 3D reconstruction problems for objects with helical symmetry. Even in the case where there is no symmetry, describing the electron scattering intensity of the object by a weighted sum of basis functions is possible. Approaches based on weighted sums of basis functions for this case appear to be essentially the same as the single-particle methods described above.
Besides providing numerical values for each of the unknown parameters, maximum likelihood theory also provides some information on the size of the errors. The key result (Efron & Hinkley, 1978) is that the error between the MLE and the true values of the model parameters is approximately Gaussian distributed with mean vector 0 and a covariance matrix that is the negative inverse of the matrix of mixed second-order partial derivatives, i.e. the Hessian, of the log likelihood function with respect to the model parameter vector. However, there are at least two important challenges in using this result in cryo-EM. First, it may be more or less difficult to connect these error estimates with the most common method for measuring the performance of a cryo EM 3D reconstruction, which is the Fourier Shell Correlation (FSC) (Harauz & van Heel, 1986). Second, in the case where the connection can be made, it is quite likely that there are computational complexity issues since the number of model parameters is typically so large that computation of the Hessian matrix is impractical. Still, these ideas have been used to create a maximum likelihood variant of the FSC for a virus problem with symmetry, specifically, in the ab initio 3D reconstruction of the tail of the infectious bacteriophage P22 (Prust et al., 2009). In this situation the first challenge was solved by a Monte Carlo procedure and the second challenge was not present because the resolution was low so the number of parameters was relatively small. In a higher resolution problem, it is probably necessary to make a diagonal approximation to the Hessian matrix.
The theory in the previous paragraph describes performance once the data is recorded. An analogous theory may be used to “predict” performance before any data is recorded. Given a PDF P(|Θ), the Cramer-Rao bound (CRB) [e.g., Marzetta (1993)] will give the minimal achievable variance for any unbiased estimator, including the unbiased MLE. Thereby, one could design the optimal experiment by computing this bound for different experimental strategies and choosing the experiment that makes the bound as small as possible. The required calculations are similar to the calculations described in the previous paragraph and have been demonstrated in a tentative way in Doerschuk & Johnson (2000).
2D crystallography
Electron crystallography is a cryo-EM method that has provided the highest-resolution structures of protein assemblies. Because the penetration distance of electrons is limited to approximately 100 nm, the acquisition of data from 3D protein crystals—as are used for X-ray crystallography—is not practical. However, planar 2D crystals, composed of a single layer or double-layer of proteins, can be imaged in the electron microscope. Of particular interest are 2D crystals formed of membrane proteins embedded in a lipid bilayer membrane, as analysis of these crystals provides the most reliable structural information for these proteins. For further details on 2D crystallization and the subsequent structure determination process the reader is referred to the chapters by Stahlberg and by Walz in this issue.
In a 2D crystal there are fewer lattice contacts than in 3D crystals, and the crystal lattice often shows substantial disorder. Fortunately, in the electron microscope an image can be formed of a 2D crystal, and the lattice disorder can be removed by computational ”unbending” of the crystal image. In this process the micrograph is analyzed to locate the center of each unit cell. To the extent that its center deviates from an ideal lattice, each unit cell is then shifted to bring it into the proper position.
In the end, the goal of analysis of a crystal image is to estimate the 2D density of a unit cell of the lattice. The “unbending” method is equivalent to a conventional alignment and averaging of 2D images, and a superior alternative is ML estimation of the unit-cell image as in Eq. (24). Zeng et al. (2007) implemented the EM-ML algorithm, where each data image Xi is a unit cell in the micrograph, and the transformations φ are constrained to small translations from lattice points and to a small angular deviations from the lattice directions. That is, the hidden variables φi are modeled as Gaussian-distributed with small standard deviations which are estimated as part of the model. The result is improved resolution in structures obtained from 2D crystals having substantial disorder. In the ML estimation it was assumed that the disorder in the crystals was confined to in-plane translations and rotations; however, a further extension can be imagined in which a 3D algorithm similar to ML3D could be employed to account for small out-of-plane deviations of the unit cells as well. In addition, one might envision improvements by relating the disorder parameters of neighbouring unit cells to each other, as crystal disorder is often to some extent a continuous phenomenon.
Tomography
In electron tomography a reconstruction of a unique 3D object, for example an entire cell, is obtained by combining a series of projections that are recorded at different tilt angles. The process of tomographic reconstruction is described in more detail in the chapter by Amat in this issue, and its application to HIV-1 is described in the chapter by Liu. Electron tomograms are typically extremely noisy because the electron dose over the whole tilt series needs to be limited in order to prevent radiation damage. Still, averaging over multiple copies of the same macromolecular complex may improve the signal-to-noise ratios provided that the individual sub-tomograms may be aligned (and classified in the case of structural heterogeneity). Apart from the increased dimensionality of the data vectors, sub-tomogram averaging is conceptually very similar to 2D averaging approaches in single-particle analysis. Again, the unkown orientations φi = (αi, βi, γi, xi, yi, zi) and/or the class assignments ki of the individual sub-tomograms may be treated as hidden variables and the EM-ML algorithm may be employed to obtain MLEs of the underlying signals.
The first reported ML approach to sub-tomogram averaging was the application of the kerdenSOM algorithm to aligned sub-tomograms of insect flight muscles (Pascual-Montano et al., 2002). In this case, as for the classification of 2D images described above, the class assignments ki are treated as hidden variables and an additional regularization term to the marginal log-likelihood function results in a neural network-like behaviour of the K output classes. However, compared to 2D averaging an additional complication arises in sub-tomogram averaging that was not taken into account in the kerdenSOM approach. Due to experimental limitations on the tilt angle, electron tomography data cannot be measured in its totality. In the case of single-axis tilting, a wedge-shaped region in Fourier space remains experimentally inaccessible. This region is commonly referred to as the missing wedge and sub-tomogram averaging procedures that do not take the missing wedges into account have been observed to yield suboptimal results (Walz et al., 1997). A variant of the kerdenSOM algorithm that takes missing wedges into account was mentioned in a structural study on cadherins (Al-Amoudi et al., 2007), but details concerning this algorithm were never described.
Conventional alignment and classification approaches for sub-tomograms have typically restricted the cross-correlation measure to the observed regions in Fourier space. Subtomogram averaging may then be performed by weighted averaging, i.e. by dividing the sum of all sub-tomograms by the times that each point in Fourier space has been measured. However, the EM-ML algorithm itself may provide a more elegant solution to the missing wedge problem, since this algorithm was ultimately designed to deal with incomplete data problems. Apart from the unknown orientations φi and the class assignments ki, one may also treat the data points inside the missing wedges as hidden variables. An EM-ML algorithm that optimizes the corresponding marginal log-likelihood function was recently introduced (Scheres et al., 2009a). This algorithm “estimates” the missing data points by calculating probability-weighted averages over all possible values, just like it does for the orientation and class assignments. However, the possible values for each of the missing data points range from minus infinity to infinity, which obviously prohibits the use of numerical integrations. Fortunately, an analytical solution exists where the missing data points for each individual subtomogram are replaced by the corresponding values in the (oriented) reference to which the subtomogram is compared. In this way, the incomplete experimental data are complemented with the currently available information from the model. Therefore, this algorithm does not need to divide the sum of all sub-tomograms by the number of times each point in Fourier space was observed, which prevents numerical instabilities for data points that were (almost) never observed.
Similar to the ML2D approach, the EM-ML algorithm for sub-tomogram averaging tackles the problems of alignment and classification simultaneously through a multi-reference refinement scheme (with hidden variables φi and ki). Moreover, the algorithm may be run in a completely unsupervised manner by starting the multi-reference refinements from averages of random subsets of sub-tomograms in random orientations. The advantages of this approach were illustrated using simulated data and reference-free class averages were obtained for experimental sub-tomograms of groEL and groEL/groES complexes. In addition, the same approach was shown to be effective for the reference-free alignment of random conical tilt (RCT) reconstructions from single particle experiments on p53. Note that just like sub-tomograms, RCT reconstructions are 3D data vectors with missing regions in Fourier space, although RCT reconstructions have missing cones rather than missing wedges. An alternative maximum likelihood approach for the alignment of RCT reconstructions was mentioned by Sander et al. (2006), although also for this algorithm the details have not yet been presented.
The statistical data model
The data model that underlies the PDF calculations plays a crucial role in the EM-ML approach. The PDF defines the log-likelihood target function and is used to calculate the probability distributions over the hidden variables in the model updates. If the model does not describe the data accurately, the benefits of the statistical approach may be lost altogether. All approaches described above share similar assumptions about the signal and they all assume that the noise is independent, additive and Gaussian. As outlined above this model results in a computationally attractive algorithm, but how accurately does this model describe cryo-EM images?
Thin samples of biological molecules fulfil the weak phase approximation, the theory of image formation which is used to describe the phase-contrast images of weakly scattering specimens. Under this approximation, the contrast in cryo-EM images is linearly related to the projected object potential. The latter justifies the use of standard X-ray integrals in the projection operators of the 3D approaches described above. The same theory is also used to derive imaging effects of the electron microscope in the form of the contrast transfer function (CTF). The accuracy of this model, at least for thin specimens, is reflected in the (near-)atomic resolutions that have been obtained with it for single particles and 2D crystals (Cheng & Walz, 2009). However, of all the ML approaches described above only those for icosahedral viruses [e.g. Yin et al. (2003)] include an explicit description of the CTF. All the other approaches simply ignore the CTF or employ suboptimal CTF correction strategies [e.g. Zeng et al. (2007)]. Consequently, these approaches may fail to provide good model estimates in cases where the CTF plays an important role, as is often the case in medium-high resolution refinements. For more details on image restoration and the importance of CTF correction, the reader is referred to the chapter by Penczek in this issue.
The noise term in the data model is used to represent all features in the observed images that are not explained by the description of the signal. In cryo-EM images the major source of noise is shot noise. This type of noise follows a Poisson distribution and arises from statistical fluctuations in the small number of imaging electrons (typically 10–20 electrons per squared Angstrom). EM-ML algorithms for Poisson distributions are computationally more complicated than those for Gaussians. But as the Poisson distribution approaches the Gaussian for larger numbers of imaging electrons, the latter provides a good approximation for pixels that span many squared Angstroms (Sigworth, 2004). Apart from the abundant shot noise, several other sources of noise exist. Structural noise arises from the irreproducible density of the ice that surrounds the particle or the carbon layer that is used to support it. Also structural variability in the particles that is not described by the data model may be considered as a source of structural noise. Detector noise arises from the stochastic nature of the interactions of electrons with the detector—the digital camera or photographic film which is used to acquire the image. The result is a random variation in both the point-spread function and amplitude of the single-electron response of the detector. Baxter et al. (2009) experimentally measured the relative contributions of the different types of noise. For typical cryo-EM images on a well-behaved ribosome sample they found the combination of shot noise and detector noise to be one order of magnitude larger than the structural noise. The latter was found to be approximately of the same power as the underlying signal in the images. Taken together one may assume that, at least for pixels that span multiple squared Angstroms, Gaussian distributions describe the combination of the various independent sources of noise in cryo-EM images reasonably well.
The assumption of independent noise may be more problematic. The independence between all pixels was used in Eq. (11) to relate the PDF of an entire image with the PDFs of the individual pixels, and ultimately allows for fast calculations of the probabilities. Independent noise is uncorrelated from pixel to pixel and has a flat power spectrum. Therefore, it is also called white noise. Although shot noise is intrinsically white, the background noise in a cryo-EM micrographs is typically not white for several reasons. Most importantly, there is a fall-off with resolution of both the signal and the noise due to the point spread functions of the microscope and the electron detector. In addition, various sources of structural noise, such as density for the ice or carbon support or structural variations in the particles are expected to have strong correlations among nearby pixels. Ignoring these correlations in the PDF calculations will lead to a suboptimal behaviour of the EM-ML approach.
Two approaches to deal with non-white noise in cryo-EM images have been proposed. In the most straightforward approach, the data are adapted to the white-noise model by so-called pre-whitening of the observed images (Zeng et al., 2007; Sigworth, 2004). In this preprocessing step the higher frequencies in the images are boosted based on an estimate for the power spectrum of the noise. The resulting images have a flat power spectrum and are processed using the conventional ML approach. Alternatively, the model itself may be adapted to describe non-white data. The latter was achieved by reformulation of the PDF in the Fourier domain. Assuming independent, Gaussian noise on all Fourier components, the PDF may be calculated in a way that is highly similar to Eq. (11) but involves Fourier components rather than image pixels. The uniqueness of the expression in the Fourier domain lies in the possibility to estimate the standard deviation of the noise (σ) as a function of spatial frequency, thereby explicitly modeling non-white noise. In addition, the formulation in the Fourier domain allows for a convenient incorporation of the CTF into the data model. This approach was called MLF, for Maximum Likelihood in the Fourier domain, and it was implemented for the problems of 2D and 3D multi-reference refinements in single particle analysis (Scheres et al., 2007b). For simulated CTF-filtered images, the MLF approach was shown to be superior to the ML approach for white noise and its usefulness in the experimental situation was illustrated for structurally heterogeneous cryo-EM data sets on 70S E. coli ribosomes and large-T antigen complexes.
Perhaps the most basic of all assumptions in the data models discussed above is the one that is violated most often. By including an image in the data set one assumes it contains signal. However, especially for smaller particles (with molecular weights well under 1 MDa) it is often extremely difficult to distinguish genuine particles of interest from artifacts in the micrographs. Consequently many cryo-EM data sets contain large amounts of particles that do not contain a common underlying signal. This problem typically becomes more severe with the use of (semi-) automated particle selection procedures, which are still outperformed by expert human beings (Zhu et al., 2004). Currently, none of the available ML approaches can distinguish between genuine and artifactual particles, but one approach does aim to provide robustness against outliers in the data (Scheres & Carazo, 2009). This approach uses t-distributions, which have wider tails than Gaussians, to accomodate atypical observations. The resulting algorithm downweights images with relatively large residuals. However, application to cryo-EM images did not show clear advantages over the Gaussian algorithm, probably because typical artifacts in the data have relatively small residuals compared to the high levels of noise.
Reducing computational requirements
Perhaps the major obstacle for a more wide-spread use of the EM-ML approach in cryo-EM image processing is currently its computational load. For many applications the exhaustive integrations over the hidden variables in the expectation step require large amounts of computing time. For example, in single-particle analysis the integrations over the 3D rotations and in-plane translations span a five-dimensional space that is extended to even six dimensions in the multi-reference case. That is, for each 3D rotation (αi, βi,γi) and each class ki, all J possible in-plane translations (xi, yi) are to be considered. Conventional approaches typically divide this problem into several lower-dimensional ones, e.g. by searching in-plane translations only for a single 3D rotation. Although this strategy is not guaranteed to converge, it does provide major speed-ups, e.g. of two orders of magnitude, compared to the exhaustive integrals in the ML approach. Consequently, the latter has become known as a computationally expensive technique.
Several contributions have been made to reduce the computational requirements of ML approaches in cryo-EM. Two approaches for single-particle analysis are based on the observation that many of the alignment parameters give rise to near-zero probabilities, especially when the algorithm nears convergence (Tagare et al., 2008; Scheres et al., 2005b). Consequently, the ML approach can be speeded up considerably by domain reduction strategies, where the integrals over the hidden variables are restricted to those alignments that contribute significantly to the weighted averages.
The first proposal to domain reduction involved skipping part of the integrations over all in-plane translations (Scheres et al., 2005b). In this approach optimal translations from the previous iteration are used to calculate probabilities for pre-centered images as a function of the in-plane rotation for each 2D reference (or projection of the 3D references). Then, for those in-plane rotations where this probability is smaller than a cut-off value times its maximum, the integration over all translations is skipped. Although in a strict sense the resulting algorithm is not guaranteed to converge, in practice a 7-fold acceleration could be obtained for a small 2D problem without notably changing the convergence behavour. For larger 3D problems (Scheres et al., 2007a), the same approach was estimated to yield 10–20 fold accelerations (unpublished results).
Even higher speed-ups of 30–60 times are reported by combining a related domain reduction strategy with an additional grid interpolation strategy (Tagare et al., 2008). The latter is based on the observation that the squared difference terms in Eq (17) vary much more smoothly with φ than the probability terms themselves. Consequently, one may calculate the squared difference terms on a relatively coarse grid and use B-spline interpolation to evaluate the corresponding values on a much finer grid. A similar approach had previously been proposed for the maximum-seeking problem in conventional cross-correlation based strategies (Sander et al., 2003).
Whereas domain reduction and grid interpolation provide approximations of the original EM-ML algorithm, the ML approach for icosahedral viruses in (Doerschuk & Johnson, 2000) may be reformualted in an exact manner that is much faster (Lee et al., 2007). In the original approach, the spherical harmonics themselves already provide a highly efficient way to sample 3D rotations and to restrict the resolution of the model. Still, using an idea from Navaza (2003), the same problem may be expressed more efficiently by application of a linear transformation to the observed data, which allows expressing the expectation step in terms of lower-dimensional integrations over the hidden variables. Although the resulting approach thus involves an additional precomputation step, each expectation step itself is accelerated up to 25 times and has reduced storage complexity.
Outlook
With the rapid increase of available computing power and continuing efforts to accelerate EM-ML approaches, the applicability of ML methods is expected to increase significantly in the near future. Already with relatively modest computer clusters, which soon will be replaced by multi-core desktop computers, medium-resolution single-particle multi-reference refinements are feasible within the time span of only a few days (also see part II of this contribution).
Preliminary investigations further indicate that the EM-ML approach may be particularly suitable for acceleration by general purpose computing on the graphical processing unit (GPU), which provides high computing power at relatively low cost (Tagare, Sigworth et al, in preparation).
In addition, apart from continuing developments in domain reduction and grid interpolation strategies, one may employ algorithms to optimize log-likelihood targets with faster convergence rates than the classical EM-ML algorithm. For example, similar to block-Kaczmarz methods for iterative reconstruction (Eggermont et al., 1981), one may perform partial expectation steps by processing the experimental images in subsets, or blocks. Thereby, convergence is speeded by incorporating new information faster into the model (Neal & Hinton, 1998). In single-photon (SPECT) and positron emission tomography (PET) this approach has resulted in an order of magnitude speed-up (Hudson et al., 1994), and investigations into the efficiency of a similar approach for cryo-EM single-particle analysis are currently underway (Scheres et al, in preparation).
However, the most important future contributions are not expected from making ML methods faster but from efforts to make them better. As explained above, the statistical data model and in particular the assumptions about the noise in the data remain apt for improvement and also the robustness to artifactual particles in the data still needs to be improved. Moreover, there are various aspects of ML theory for which applicability to cryo-EM image processing remains open for more in-depth investigations. While structure validation and resolution assessments remain critical areas of research in the field, ML theory promises reliable error estimates on the model parameters through the use of the Hessian to the log-likelihood. The first indication that these estimates may indeed be useful is the successful definition of a ML equivalent of the FSC as recently explored by Prust et al. (2009). In addition, Cramer-Rao lower bounds to the log-likelihood function may be employed to design optimal experimental setups, as was tentatively explored in (Doerschuk & Johnson, 2000).
Finally, there may be improvements to be gained from using maximum a posteriori (MAP) estimation instead of ML estimation. As was mentioned in conjunction with Eq. (1), the inclusion of prior information in the form of a prior probability P(Θ) should improve the quality of final models. The advantage of MAP estimation appears when the amount of experimental data is limited, so that the prior probability term makes a substantial contribution. An important example of a prior probability function, well known in X-ray crystal structure determination, is one that penalizes all 3D models in which the density outside the particle boundary (the “solvent” or “ice” density of the map) is nonzero. This choice of a prior probability function is the formal basis of the well known “solvent flattening” process.
In summary, we have seen that the theory of maximum-likelihood estimation provides improved methods for the processing of cryo-EM data. Of particular interest is its flexible theoretical framework, which allows the limitations of experimental data—heterogeneity, noise, missing wedges, and so on—to be readily and rigorously modeled. With these data models, the process of determining macromolecuar structures is both improved and placed on a firmer statistical foundation.
Acknowledgments
FS was supported by NIH grant P01-GM062580; PCD by the US National Science Foundation (grant 0735297); and JMC and SHWS by the Spanish Ministry of Science and Technology (grants CDS2006-0023, BIO2007-67150-C01 and ACI2009-1022) and the National Heart, Lung and Blood Institute (grant R01HL070472).
Contributor Information
Fred J. Sigworth, Email: fred.sigworth@yale.edu.
Peter C. Doerschuk, Email: pd83@cornell.edu.
Jose-Maria Carazo, Email: carazo@cnb.csic.es.
Sjors H.W. Scheres, Email: scheres@mrc-lmb.cam.ac.uk.
References
- Al-Amoudi A, Diez DC, Betts MJ, Frangakis AS. The molecular architecture of cadherins in native epidermal desmosomes. Nature. 2007;450:832–837. doi: 10.1038/nature05994. [DOI] [PubMed] [Google Scholar]
- Baxter WT, Grassucci RA, Gao H, Frank J. Determination of signal-to-noise ratios and spectral SNRs in cryo-EM low-dose imaging of molecules. J Struct Biol. 2009;166:126–132. doi: 10.1016/j.jsb.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q. Master’s thesis. School of Electrical and Computer Engineering, Cornell University; 2008. Nonlinear stochastic tomography reconstruction algorithms for objects with helical symmetry and applications to virus structures. [Google Scholar]
- Cheng Y, Walz T. The advent of Near-Atomic resolution in Single-Particle electron microscopy. Annu Rev Biochem. 2009;78:723–742. doi: 10.1146/annurev.biochem.78.070507.140543. [DOI] [PubMed] [Google Scholar]
- Dempster AP, Laird NM, Rubin DB. Maximum-likelihood from incomplete data via the EM algorithm. J Royal Statist Soc Ser B. 1977;39:1–38. [Google Scholar]
- Doerschuk PC, Johnson JE. Ab initio reconstruction and experimental design for cryo electron microscopy. IEEE Trans Inf Theory. 2000;46:1714–1729. [Google Scholar]
- Efron B, Hinkley DV. Assessing the accuracy of the maximum likelihood estimator: Observed versus expected fisher information. Biometrika. 1978;65:457–487. [Google Scholar]
- Eggermont PP, Herman GT, Lent A. Iterative algorithms for large partitioned linear systems, with applications to image reconstruction. Linear Algebra and Its Applications. 1981;40:37–67. [Google Scholar]
- Frank J. Three-dimensional Electron Microscopy of Macromolecular Assemblies. New York: Oxford University Press; 2006. [Google Scholar]
- Harauz G, van Heel M. Exact filters for general geometry three dimensional reconstruction. Optik. 1986;73:146–156. [Google Scholar]
- Hudson M, Larkin R. Accelerated image reconstruction using ordered subsets of projection data. IEEE Trans Med Imag. 1994;13:601–609. doi: 10.1109/42.363108. such As Expectation Maximization, A. [DOI] [PubMed] [Google Scholar]
- Julian P, Konevega AL, Scheres SHW, Lazaro M, Gil D, Wintermeyer W, Rodnina MV, Valle M. Structure of ratcheted ribosomes with tRNAs in hybrid states. Proc Natl Acad Sci U S A. 2008;105:16924–16927. doi: 10.1073/pnas.0809587105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam Z, Gafni I. Three-dimensional reconstruction of the shape of human wart virus using spatial correlations. Ultramicroscopy. 1985;17:251–262. doi: 10.1016/0304-3991(85)90092-0. [DOI] [PubMed] [Google Scholar]
- Laporte O. Polyhedral harmonics. Naturforschung. 1948;3a:447–456. [Google Scholar]
- Lee J, Doerschuk PC, Johnson JE. Exact reduced-complexity maximum likelihood reconstruction of multiple 3-D objects from unlabeled unoriented 2-D projections and electron microscopy of viruses. IEEE Trans Image Process. 2007;16:2865–2878. doi: 10.1109/tip.2007.908298. [DOI] [PubMed] [Google Scholar]
- Lee S. PhD thesis. School of Electrical and Computer Engineering, Purdue University West Lafayette; Indiana, USA: 2009. Maximum likelihood reconstruction of 3D objects with helical symmetry from 2D projections of unknown orientation and application to electron microscope images of viruses. [Google Scholar]
- Lee S, Doerschuk PC, Johnson JE. Reciprocal space representations of helical objects and their projection images for helices constructed from motifs without spherical symmetry. Ultramicroscopy. 2009;109:253–263. doi: 10.1016/j.ultramic.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marabini R, Herman GT, Carazo JM. 3D reconstruction in electron microscopy using ART with smooth spherically symmetric volume elements (blobs) Ultramicroscopy. 1998;72:53–65. doi: 10.1016/s0304-3991(97)00127-7. [DOI] [PubMed] [Google Scholar]
- Marzetta T. A simple derivation of the constrained multiple parameter Cramer-Rao bound. IEEE Trans Signal Process. 1993;41:2247–2249. [Google Scholar]
- Moody M. Image analysis of electron micrographs. In: Hawkes PW, Valdre U, editors. Biophysical Electron Microscopy: Basic Concepts and Modern Techniques. Academic Press; 1990. pp. 145–287. [Google Scholar]
- Navaza J. On the three-dimensional reconstruction of icosahedral particles. J Struct Biol. 2003;144:13–23. doi: 10.1016/j.jsb.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Neal R, Hinton GE. A view of the em algorithm that justifies incremental, sparse, and other variants. Learning in graphical models. 1998:355–368. [Google Scholar]
- Pascual-Montano A, Donate LE, Valle M, Barcena M, Pascual-Marqui RD, Carazo JM. A novel neural network technique for analysis and classification of EM single-particle images. J Struct Biol. 2001;133:233–245. doi: 10.1006/jsbi.2001.4369. [DOI] [PubMed] [Google Scholar]
- Pascual-Montano A, Taylor KA, Winkler H, Pascual-Marqui RD, Carazo J. Quantitative self-organizing maps for clustering electron tomograms. J Struct Biol. 2002;138:114–122. doi: 10.1016/s1047-8477(02)00008-4. [DOI] [PubMed] [Google Scholar]
- Provencher SW, Vogel RH. Three-dimensional reconstruction from electron micrographs of disordered specimens i. method. Ultramicroscopy. 1988;25:209–221. doi: 10.1016/0304-3991(88)90016-2. [DOI] [PubMed] [Google Scholar]
- Prust CJ, Doerschuk PC, Lander GC, Johnson JE. Ab initio maximum likelihood reconstruction from cryo electron microscopy images of an infectious virion of the tailed bacteriophage p22 and maximum likelihood versions of fourier shell correlation appropriate for measuring resolution of spherical or cylindrical objects. J Struct Biol. 2009;167:185–199. doi: 10.1016/j.jsb.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redner R, Walker H. mixture densities, maximum likelihood and the EM algorithm. SIAM Review. 1984;26:239, 195. [Google Scholar]
- Sander B, Golas MM, Makarov EM, Brahms H, Kastner B, Lhrmann R, Stark H. Organization of core spliceosomal components u5 snRNA loop i and U4/U6 Di-snRNP within U4/U6. U5 Tri-snRNP as revealed by electron cryomicroscopy. Mol Cell. 2006;24:267–278. doi: 10.1016/j.molcel.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Sander B, Golas MM, Stark H. Corrim-based alignment for improved speed in single-particle image processing. J Struct Biol. 2003;143:219–228. doi: 10.1016/j.jsb.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Scheres SHW, Carazo JM. Introducing robustness to maximum-likelihood refinement of electron-microscopy data. Acta Cryst D. 2009;65:672–678. doi: 10.1107/S0907444909012049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SHW, Gao H, Valle M, Herman GT, Eggermont PPB, Frank J, Carazo JM. Disentangling conformational states of macromolecules in 3D-EM through likelihood optimization. Nat Methods. 2007a;4:27–29. doi: 10.1038/nmeth992. [DOI] [PubMed] [Google Scholar]
- Scheres SHW, Melero R, Valle M, Carazo J. Averaging of electron subtomograms and random conical tilt reconstructions through likelihood optimization. Structure. 2009a;17:1563–1572. doi: 10.1016/j.str.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SHW, Nunez-Ramirez R, Gomez-Llorente Y, San Martin S, Eggermont PPB, Carazo JM. Modeling experimental image formation for likelihood-based classification of electron microscopy data. Structure. 2007b;15:1167–1177. doi: 10.1016/j.str.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SHW, Valle M, Carazo JM. Fast maximum-likelihood refinement of electron microscopy images. Bioinformatics. 2005a;21(Suppl 2):ii243–ii244. doi: 10.1093/bioinformatics/bti1140. [DOI] [PubMed] [Google Scholar]
- Scheres SHW, Valle M, Grob P, Nogales E, Carazo J. Maximum likelihood refinement of electron microscopy data with normalization errors. J Struct Biol. 2009b;166:234–240. doi: 10.1016/j.jsb.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SHW, Valle M, Nunez R, Sorzano COS, Marabini R, Herman GT, Carazo JM. Maximum-likelihood multi-reference refinement for electron microscopy images. J Mol Biol. 2005b;348:139–149. doi: 10.1016/j.jmb.2005.02.031. [DOI] [PubMed] [Google Scholar]
- Sigworth FJ. A maximum-likelihood approach to single-particle image refinement. J Struct Biol. 1998;122:328–339. doi: 10.1006/jsbi.1998.4014. [DOI] [PubMed] [Google Scholar]
- Sigworth FJ. Classical detection theory and the cryo-EM particle selection problem. J Struct Biol. 2004;145:111–122. doi: 10.1016/j.jsb.2003.10.025. [DOI] [PubMed] [Google Scholar]
- Tagare HD, Sigworth F, Barthel A. Fast, adaptive expectation-maximization alignment for Cryo-EM. Med Imag Comput Comput Assist Interv. 2008;11:855–862. doi: 10.1007/978-3-540-85990-1_103. [DOI] [PubMed] [Google Scholar]
- Vogel RH, Provencher SW. Three-dimensional reconstruction from electron micrographs of disordered specimens. II. implementation and results. Ultramicroscopy. 1988;25:223–239. doi: 10.1016/0304-3991(88)90017-4. [DOI] [PubMed] [Google Scholar]
- Vogel RH, Provencher SW, von Bonsdorff C, Adrian M, Dubochet J. Envelope structure of semliki forest virus reconstructed from cryo-electron micrographs. Nature. 1986;320:533–535. doi: 10.1038/320533a0. [DOI] [PubMed] [Google Scholar]
- Walz J, Typke D, Nitsch M, Koster AJ, Hegerl R, Baumeister W. Electron tomography of single Ice-Embedded macromolecules: Three-Dimensional alignment and classification. J Struct Biol. 1997;120:387–395. doi: 10.1006/jsbi.1997.3934. [DOI] [PubMed] [Google Scholar]
- Yin Z, Doerschuk PC, Gelfand SB. Model calculations for joint pattern recognition and signal reconstruction in cyro electron microscopy. Communications in Information Systems. 2004;4:73–88. [Google Scholar]
- Yin Z, Zheng Y, Doerschuk PC. An ab initio algorithm for low-resolution 3-D reconstructions from cryoelectron microscopy images. J Struct Biol. 2001;133:132–142. doi: 10.1006/jsbi.2001.4356. [DOI] [PubMed] [Google Scholar]
- Yin Z, Zheng Y, Doerschuk PC, Natarajan P, Johnson JE. A statistical approach to computer processing of cryo-electron microscope images: virion classification and 3-D reconstruction. J Struct Biol. 2003;144:24–50. doi: 10.1016/j.jsb.2003.09.023. [DOI] [PubMed] [Google Scholar]
- Zeng X, Stahlberg H, Grigorieff N. A maximum likelihood approach to two-dimensional crystals. J Struct Biol. 2007;160:362–374. doi: 10.1016/j.jsb.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Doerschuk PC. Explicit orthonormal fixed bases for spaces of functions that are totally symmetric under the rotational symmetries of a platonic solid. Acta Cryst A. 1996;52:221–235. [Google Scholar]
- Zhu Y, Carragher B, Glaeser RM, Fellmann D, Bajaj C, Bern M, Mouche F, de Haas F, Hall RJ, Kriegman DJ, Ludtke SJ, Mallick SP, Penczek PA, Roseman AM, Sigworth FJ, Volkmann N, Potter CS. Automatic particle selection: results of a comparative study. J Struct Biol. 2004;145:3–14. doi: 10.1016/j.jsb.2003.09.033. [DOI] [PubMed] [Google Scholar]