

Abstract
Caffeic acid phenethyl ester (CAPE) inhibits the growth of tumor cells and is a known inhibitor of NF-κB that is constitutively active in cholangiocarcinoma (CCH) cells. We evaluated the effects of CAPE on CCH growth both in vitro and in vivo. Inhibition of NF-κB DNA-binding activity was confirmed in nuclear extracts treated with CAPE at 50, 40 and 20 μM. CAPE decreases the expression of NF-κB1 (p50) and RelA (p65). CAPE decreased the growth of a number of CCH cells but not normal cholangiocytes. Cell cycle decrease was seen by a decrease in PCNA protein expression and the number of BrdU-positive cells treated with CAPE at 20 μM compared to vehicle. Inhibition of growth and increased cell cycle arrest of Mz-ChA-1 cells by CAPE were coupled with increased apoptosis. Bax expression was increased, whereas Bcl-2 was decreased in cells treated with CAPE compared to vehicle. In vivo studies were performed in BALB/c nude (nu/nu) mice implanted subcutaneously with Mz-ChA-1 cells and treated with daily IP injections of DMSO or CAPE (10 mg/kg body weight in DMSO) for 77 days. Tumor growth was decreased and tumor latency was increased 2-fold in CAPE compared to vehicle-treated nude mice. In tumor samples, decreased CCH growth by CAPE was coupled with increased apoptosis. CAPE both in vivo and in vitro decreases the growth of cholangiocarcinoma cells by increasing apoptosis. These results demonstrate that CAPE might be an important therapeutic tool in the treatment of CCH.
Keywords: Biliary cancer, apoptosis, cell cycle, inhibition, proliferation
INTRODUCTION
Cholangiocarcinomas are cancers of both intrahepatic and extrahepatic origin that are growing in both prevalence and mortality rate 1–3. The greatest challenge put forward by these increasingly dangerous cancers is the limited ability to diagnose early, leaving the only hope for long-term survival being absolute surgical resection of the tumor 1–3. Alterations in apoptotic thresholds resulting from a chronic inflammatory state may be important in cholangiocarcinoma development 2. Cholangiocytes are inherently resistant to apoptosis, but are observed to undergo programmed cell death in primary biliary cirrhosis and primary sclerosing cholangitis (PSC), leading to bile duct loss 2. In PSC, the inflammatory environment may lead to a further dysregulation of apoptosis, thereby allowing genetically damaged cells to proliferate and perhaps escape immune recognition, ultimately leading to the frightening 10–20% incidence of bile duct malignancy observed in this condition 2.
Caffeic acid (3,4-dihydroxycinnamic acid) phenethyl ester (CAPE) is structurally related to flavonoids and is a biologically active component of propolis from honeybee hives. It has antiviral, anti-mitogenic, anti-inflammatory, and immunomodulatory properties 4–7 and has been shown to inhibit the growth of different types of transformed cells such as breast and colon cancer cells 8. In transformed cells, CAPE alters the redox state and induces apoptosis 9. It has been shown that CAPE suppresses lipid peroxidation 10 and displays antioxidant activity 11. CAPE can also inhibit phorbol ester-induced H2O2 production and tumor promotion 12. CAPE is a well-known and well-documented inhibitor of the transcription factor, nuclear factor kappa beta (NF-κB) 13–15.
The NF-κB signal transduction pathway is dysregulated in a variety of human cancers16. In most cancer cells, NF-κB is constitutively active and resides in the nucleus17–19. NF-κB activity not only protects cancer cells from apoptotic cell death, but may even enhance their growth activity 20. Inhibition of NF-κB activation produces a corresponding increase in apoptosis, indicating that the balance of cell viability versus cell death is preserved by the degree of NF-κB activation 20. Agents that can down-modulate the activation of NF-κB have potential for therapeutic intervention21, 22. One such agent may be the NF-κB inhibitor, CAPE, via induction of cell death.
Apoptotic cell death has been characterized by the progression of morphological and biochemical changes ranging from the manifestation of the phospholipid phosphatidylserine on cell surfaces, to proteolytic cleavage of numerous intracellular proteins, to nuclear condensation and fragmentation and the cleavage of DNA into nucleosomal fragments 23. Dysregulation of apoptosis can disrupt the equilibrium between cell growth and cell death and is critical in the development of cancer and tumor cell survival 24, 25. It is this understanding that has led researchers to explore the therapeutic activation of apoptosis in cancer cells as a potential cancer-fighting strategy.
All of these factors together have led us to propose the following hypothesis: CAPE inhibits the growth of cholangiocarcinoma via inhibition of NF-κB activation and increasing apoptosis.
METHODS AND MATERIALS
Materials
All high-quality reagents were obtained from Sigma Chemical Company (St. Louis, MO) unless otherwise indicated. Antibodies for immunoblotting and immunohistochemistry were all obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Cell culture media and other reagents were obtained from Gibco Invitrogen Corporation (Carlsbad, CA).
Cultured Cell Lines
For our study we used the following cell lines. Mz-ChA-1 cells, an extrahepatic biliary cancer cell line from human gallbladder 26–28, were a gift from Dr. G. Fitz (University of Texas Southwestern Medical Center, Dallas, TX) and were cultured as described 26. HuH-28 (from human intrahepatic bile duct) 26, 29, and TFK-1 (from human extrahepatic bile duct) 26, 30 cells were acquired from Cancer Cell Repository, Tohoku University, Japan and were cultured as described 26. HuCC-T1 and SG231 cells 31, 32, from intrahepatic bile ducts (obtained from Japanese Cancer Research Resources Bank) were a gift from Dr. A. J. Demetris (University of Pittsburgh, Pittsburgh, PA) and were cultured as described 31–34. The human immortalized, nonmalignant cholangiocyte cell line H69 (a gift from Dr. G. J Gores, Mayo Clinic) was cultured as described 35.
NF-κB Binding Activity
EMSA was performed in Mz-ChA-1 cells after CAPE treatment (20, 40 and 50 μM for 2 hours). Briefly, cells were scraped into 1 mL of 1× phosphate buffered saline (1× PBS) and pelleted by centrifugation (300 g) to remove any trace of media. The cellular pellet was resuspended in 400 μL of homogenization buffer (100 mM NaCl, 1.5 mM MgCl2, 0.5 mM EDTA, 0.7% igepal, 0.5 mM DTT, 10% (w/v) glycerol, 10 μg/ml leupeptin, 5 μg/ml aprotinin, 0.5 mM PMSF (phenylmethanesulphonylfluoride) in 20 mM HEPES, pH 7.9) and centrifuged for 10 min at 2000 g. The pellet was incubated for 30 min at 4°C on a rocker in 50 μL of high-salt buffer (500 mM KCl, 0.5 mM EDTA, 25% (w/v) glycerol, 0.5 mM DTT, 0.5 mM PMSF in 20 mM HEPES or (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), pH 7.9). Nuclear debris was removed by centrifugation at 14,000 g for 30 minutes at 4°C and the supernatant stored in aliquots at −70°C until further processing. Double-stranded oligonucleotides containing the consensus binding motif for NF-κB (5′–AGT TGA GGG GAC TTT CCC AGG C-3′, Promega, Madison, WI) were end labeled with 32P-dATP using T4 polynucleotide kinase for 10 minutes at room temperature. Nuclear protein (10 μg) was preincubated with 1 mg poly (dI:dC) for 10 min at 25°C in 10 mM Tris (pH 7.5) 100 mM KCl, 5 mM MgCl2, 5 mM EDTA, 1 mM DTT and 30 mg/mL bovine serum albumin (BSA) before incubation for a further 20 min at 25°C in the presence of labeled DNA. DNA–protein complexes were resolved by 4% non-denaturing polyacrylamide gel electrophoresis in 1 × Tris/Borate/EDTA buffer.
CAPE Effects on the Expression of NF-κB family members, NF-κB1 (p50) and RelA (p65)
To further evaluate the effects of CAPE on NF-κB inhibition, we performed immunoblotting and immunofluorescence for the proteins, NF-κB1 (p50) and RelA (p65). The antibody for NF-κB1 (p50) recognizes both p50 and p105. The p105 (p110) precursor contains p50 at its N-terminus and a C-terminal region that, when expressed as a separate molecule, binds to p50 and regulates its activity 36. For immunoblotting assays, cells were stimulated with 0.1% DMSO or CAPE (20 μM) with 0.1% DMSO for 48 hours. Immunoblotting was performed as previously described 37. The intensity of the bands was determined by scanning video densitometry using the phospho-imager, Storm 860, (GE Healthcare, Piscataway, NJ) and the ImageQuant TLV 2003.02 (Little Chalfont, Buckinghamshire, England).
To evaluate the expression of NF-κB1 (p50) and RelA (p65) by immunofluorescence, cells were seeded into 6-well plates containing a sterile coverslip on the bottom of each well. After adherence overnight, cells were stimulated with 0.1% DMSO or CAPE (20 μM) in 0.1% DMSO for 24 hours. Immunofluorescence was performed as previously described 38 using antibodies for NF-κB1 (p50) and RelA (p65) or non-immune serum for negative controls. Sections were visualized using the Olympus IX-71 inverted confocal microscope (Tokyo, Japan).
Effect of CAPE on Cholangiocarcinoma Growth
Evaluation of cholangiocarcinoma growth was performed as previously described using proliferation assays 39. For this, cells were trypsinized and seeded into 96-well plates (10,000 cells per well) in a final volume of 200 μL of medium. Cells were stimulated for 24, 48 and 72 hours with CAPE (0–50 μM dissolved in 0.1% DMSO) or 0.1% DMSO (vehicle) prior to evaluation of proliferation 39.
CAPE Effects on Cell Cycle Progression
BrdU labeling was used to evaluate the effects of CAPE on cell cycle progression. Cells were stimulated with 0.1% DMSO or CAPE (20 μM) with 0.1% DMSO for 48 hours and staining was performed as previously described 38. The number of BrdU-positive nuclei were counted and expressed as a percentage of total cells in five random fields for each treatment group.
PCNA protein expression was evaluated by immunoblotting using Mz-ChA-1 cell lysates stimulated with either 0.1% DMSO or CAPE (20 μM with 0.1% DMSO) for 48 hours. After stimulation, cells were lysed and scraped and immunoblotting was performed as previously described 37. Alpha-tubulin was used to normalize the amount of protein used. Densitometric analysis was performed as described above.
Effect of CAPE on Apoptosis
Apoptosis was evaluated using Annexin-V labeling as described 38. Cells were seeded into 6 well plates (500,000 cells/well) containing sterile coverslips on the bottom of each well and allowed to adhere overnight. Cells were stimulated with 0.1% DMSO or CAPE (20 μM with 0.1% DMSO) for 48 hours and processed as described 38. The number of Annexin-V-positive cells were counted and expressed as a percentage of total cells in five random fields for each treatment group.
Immunoblotting analysis was performed on selected proteins to evaluate the mechanisms by which CAPE activates apoptosis thus decreasing cholangiocarcinoma growth. We measured the effects of CAPE on the pro-apoptotic protein BAX 40 and the anti-apoptotic protein Bcl-2 41, 42. Briefly, Mz-ChA-1 cells were seeded into 6-well plates and allowed to adhere and become confluent overnight. Cells were stimulated with 0.1% DMSO or CAPE (20 μM) with 0.1% DMSO for 48 hours. Immunoblotting was performed as previously described 37.
CAPE Effects on In Vivo Xenograft Studies
We next evaluated the effects of CAPE on cholangiocarcinoma growth using an in vivo animal model 39. Male balb/c 8-week-old nude (nu/nu) mice were kept in a temperature-controlled environment (20–22°C) with a 12-hour light-dark cycle and with free access to drinking water and mouse chow. Mz-ChA-1 cells (5 × 106) were suspended in 0.25 mL of extracellular matrix gel and injected subcutaneously into the hind flanks of the animals. After the establishment of the tumors, mice received the following treatments: 0.1% DMSO or CAPE (10 mg/Kg BW) 43 dissolved in 1:1 DMSO:NaCl and injected IP three times per week. Tumor parameters were measured three times a week by an electronic calipers and volume was determined as: tumor volume (mm3) = 0.5 × [length (mm) × width (mm) × height (mm)]. Tumor latency was also evaluated 39, 43. Latency represents the time for the tumor to increase to 150% of the initial volume 39, 43. After 77 days, mice were anaesthetized with sodium pentobarbital (50 mg/kg IP) and sacrificed according to the institutional guidelines. Small tumor samples were excised from the flanks of these mice, fixed in 10% buffered formalin for 2 to 4 hours and embedded in low temperature fusion paraffin (55 to 57°C), and 4-μm sections were stained with hematoxylin and eosin (for evaluation of necrosis and inflammation) 39 and Masson’s trichrome (for evaluation of fibrosis) 39. For immunohistochemistry, sections (4 μm) were mounted on glass slides coated with 0.1% poly-L-lysine. After deparaffination, endogenous peroxidase activity was blocked by a 20-minute incubation in methanolic hydrogen peroxide (2.5%). The endogen biotin was blocked by the Biotin Blocking System (DAKO, Copenhagen, Denmark) according to the instructions supplied by the vendor. Sections were hydrated in graded alcohol and rinsed in 1× PBS, pH 7.4 before applying the primary antibody. Sections were incubated overnight at 4°C with antibodies for cytokeratin-7 (CK-7), proliferating cellular nuclear antigen (PCNA), vascular endothelial growth factor-A (VEGF-A), VEGF-C, VEGFR-2 and VEGFR-3. Antibodies for the NF-κB family members, NF-κB1 (p50) and RelA (p65) were also utilized for immunoblotting in tumor lysates and immunostaining in tumor sections. Samples were rinsed with 1× PBS for 5 minutes, incubated for 20 minutes at room temperature with a secondary biotinylated antibody (DAKO LSAB Plus System, HRP, Milan, Italy), incubated with DAKO ABC (DAKO LSAB Plus System, HRP), and developed with 3-3′diaminobenzidine. For the detection of apoptosis on single cells, the terminal deoxynucleotide transferase end labeling (TUNEL) kit (ApopTag; Oncor, Gaithersburg, MD) was used. For all immunoreactions, negative controls (the primary antibody was replaced -same dilution- with normal serum from the same species) were also included. Light microscopy and immunohistochemistry observations were taken by BX-51 light microscopy (Olympus) with a videocam (Spot Insight; Diagnostic Instrument, Inc., Sterling Heights, MI) and evaluated with an Image Analysis System (IAS; Delta Sistemi, Rome, Italy). Light microscopy and immunohistochemical observations were independently performed by three morphologists in a blinded manner. The numbers of PCNA, CK-7, TUNEL, VEGF-A, VEGF-C, VEGFR-2, and VEFGR-3 cells were assessed in six slides for each group. Positive cells were counted in six non-overlapping fields for each slide and the data expressed as percentage of positive cells.
The degree of inflammation and fibrosis was evaluated in five randomly non-overlapping fields for each slide using light microscopy of Masson’s-stained sections as previously described 44. The necrotic mass was evaluated by quantitative morphometry on light microscopic images as described 45 and expressed as area of necrosis/total area of tumour × 100. For each sample, more than 5 non-overlapping fields were studied. Organ damage was also assessed in other organs from vehicle- and CAPE- treated mice.
RESULTS
CAPE inhibits NF-κB Binding Activity and Decreases Expression of NF-κB1 (p50) and RelA (p65)
As seen in other cell systems 46, 47, CAPE decreased NF-κB DNA-binding activity in Mz-ChA-1 cells treated with CAPE compared to their corresponding basal value (Figure 1a). Decreased binding activity was seen after 20, 40 and 50 μM of CAPE treatment (Figure 1a). By confocal microscopy, we found a change in the localization of the expression of NF-κB1 (p50) and RelA (p65) in Mz-ChA-1 cells stimulated with CAPE compared to basal, DMSO- treated cells (Figure 1b). Staining for NF-κB1 (p50) under basal conditions was found in both nuclear and cytoplasmic regions, whereas following CAPE treatment, staining was localized outside of the nucleus. RelA (p65) staining under basal conditions was almost purely nuclear and after CAPE treatment, staining was confined to the cytosol/membrane area (Figure 1b). Negative controls performed with non-immune serum are also shown (Figure 1b). By immunoblots, we found that Mz-ChA-1 cells stimulated with CAPE (20 μM) exhibited a marked decrease in the protein expression of both NF-κB1 (p50) and RelA (p65) compared to cells treated with 0.1% DMSO (Figure 1c). Expression of p105 (that, when expressed as a separate molecule, binds to p50 to regulate the activity) 36 was also decreased in CAPE stimulated cells compared to basal (not shown). Alpha-tubulin levels were unchanged in DMSO- and CAPE- treated samples (Figure 1c).
Figure 1.
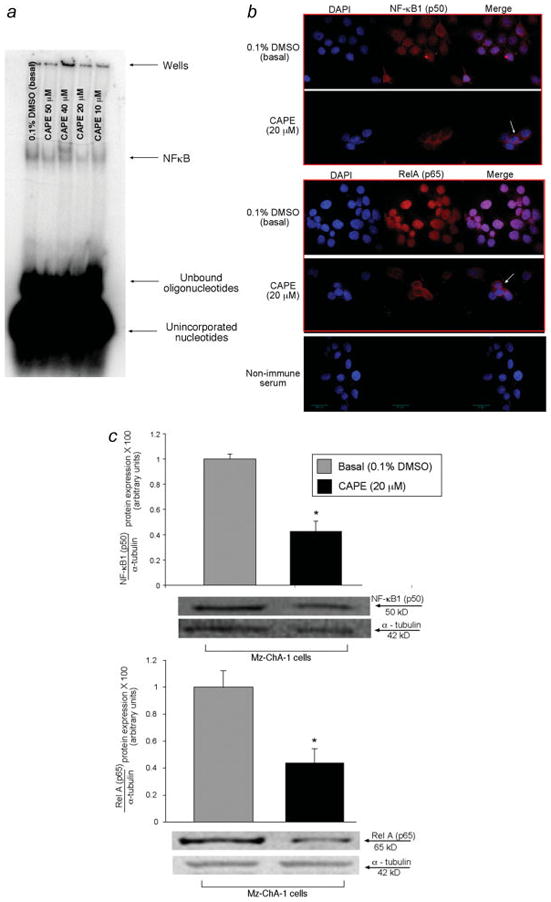
[a] CAPE inhibited NF-κB DNA-binding activity in Mz-ChA-1 cells at 20, 40 and 50 μM compared to the corresponding basal treatment. A representative radiograph is shown. [b] Immunofluorescence staining for NF-κB1 (p50) and RelA (p65) reveals that positive staining appears to be nuclear and cytoplasmic and is decreased in Mz-ChA-1 cells stimulated with CAPE compared to DMSO- stimulated cells. NF-κB1 (p50) and RelA (p65) expression is seen in red with nuclear counterstain seen in blue (DAPI) (Figure 1b). Original magnification for immunofluorescence is × 60. [c] Immunoblotting analysis demonstrates that CAPE inhibits the expression of the NF-κB family members, NF-κB1 (p50) and RelA (p65) compared to basal levels. Representative blots are shown for the expression of p50 and p65 (Figure 1c). Alpha-tubulin levels were unchanged in both DMSO and CAPE stimulated cells. Data are ± SEM of 6 experiments. *p < 0.05 compared to basal (0.1% DMSO).
Effects of CAPE on Cholangiocarcinoma Cell Growth
Using MTS assays, we showed that CAPE inhibits the growth of numerous intra- and extra-hepatic cholangiocarcinoma cells lines, but has no effect on normal cells. Specifically, we demonstrated that CAPE significantly decreased the growth of Mz-ChA-1 cells in a time (24 and 48 hours) and dose- dependent (0–50 μM) manner (Figure 2, top left panel). To expand our studies, we evaluated the effects of CAPE on other cholangiocarcinoma cell lines. These studies demonstrated that in cholangiocarcinoma (both intra- and extra-hepatic) cell lines CAPE decreases the growth of these cells (Figure 2). Proliferation in all cell lines was decreased with stimulations of 40 and 50 μM of CAPE for 48 hours (Figure 2) compared to cholangiocarcinoma cell lines stimulated with 0.1% DMSO (basal, vehicle for CAPE). Treatment of normal, H69 cells with CAPE had no effect on growth (data not shown).
Figure 2.

MTS assay for Mz-ChA-1 cells shows the time (24 and 48 hours) and dose (0–50 μM) dependent effects of CAPE on the growth of cholangiocarcinoma. Figure 2 shows a significant decrease in Mz-ChA-1 growth at both 24 and 48 hours at 20, 40 and 50 μM. By MTS assay, CAPE significantly decreased the growth of a number of cholangiocarcinoma cell lines similar to that seen in Mz-ChA-1 cells. Graphs shown for other cell lines in Figure 2 represent 48 hours. CAPE had no effect on H69 cells compared to basal levels (data not shown). Data are ± SEM of 12 experiments. *p < 0.05 compared to basal (0.1% DMSO).
CAPE Effects on Cell Cycle
We used BrdU incorporation and PCNA protein expression to determine if CAPE alters cell cycle progression. BrdU is incorporated into newly synthesized DNA in replicating cells during the S phase of the cell cycle, and thus is used as a marker for cell replication48. Results from BrdU staining revealed that CAPE slows cell cycle progression. Specifically, ~50% of Mz-ChA-1 cells treated with 0.1% DMSO incorporated BrdU (after 2 hours incubation) into their DNA compared to only ~10% of cells stimulated with CAPE (20 μM) (Figure 3a), thus demonstrating decreased cell cycle progression induced by CAPE. PCNA is also expressed during the S phase of the cell cycle and allows detection of cell proliferation and replication 49. Using immunoblotting analysis for PCNA protein expression, we found that CAPE induced a significant decrease in PCNA protein expression compared to basal- stimulated cells (Figure 3b). Levels of alpha-tubulin (used as a loading control) were unchanged in both basal- and CAPE- stimulated cells (Figure 3b). A representative immunoblot is shown. These results combined allow us to conclude that CAPE interrupts the normal progression of cell replication.
Figure 3.

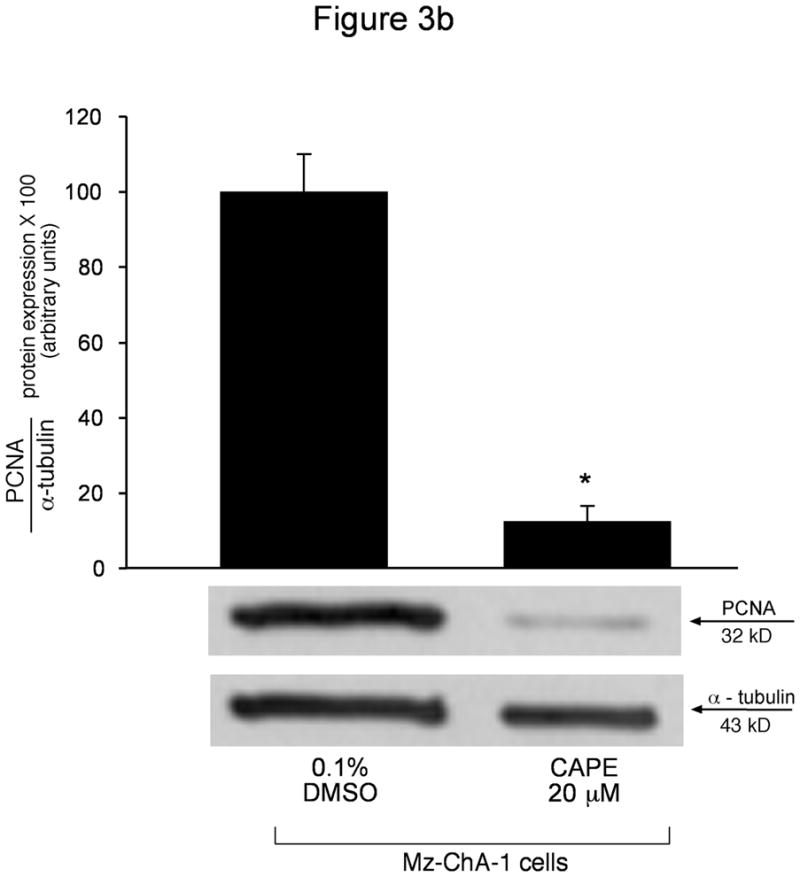
[a] BrdU incorporation for measuring cell cycle progression in Mz-ChA-1 cells stimulated with 0.1% DMSO (basal) or CAPE (20 μM with 0.1% DMSO) for 48 hours demonstrated that in basal conditions the number of BrdU positive cells was approximately 50%. However, when Mz-ChA-1 cells were stimulated with CAPE, the number of BrdU positive cells decreased to approximately 10% thus decreasing cell cycle progression. The number of BrdU-positive nuclei were counted and expressed as a percentage of total cells in 5 random fields for each treatment group. *p<0.01 compared to basal (0.1% DMSO). [b] PCNA protein expression is markedly decreased by CAPE treatment (20 μM for 48 hours) compared to basal (0.1% DMSO). Alpha-tubulin levels were unchanged between basal- and CAPE- stimulated cell lysates. Data are ± SEM of 6 experiments. *p < 0.05 compared to basal (0.1% DMSO). A representative blot is shown.
CAPE effects on Apoptosis
We used Annexin-V staining and immunoblotting for the apoptotic markers, Bax and Bcl-2 40–42, to determine the effects induced by CAPE on apoptosis. By Annexin-V staining, we found that CAPE induces a significant amount of cell death (almost 100%) in Mz-ChA-1 cells stimulated with CAPE (Figure 4a). Approximately 10% of Mz-ChA-1 cells treated with 0.1% DMSO stained positive for Annexin-V, whereas ~99% of Mz-ChA-1 cells stimulated with CAPE (20 μM) were positive for Annexin-V (Figure 4a).
Figure 4.
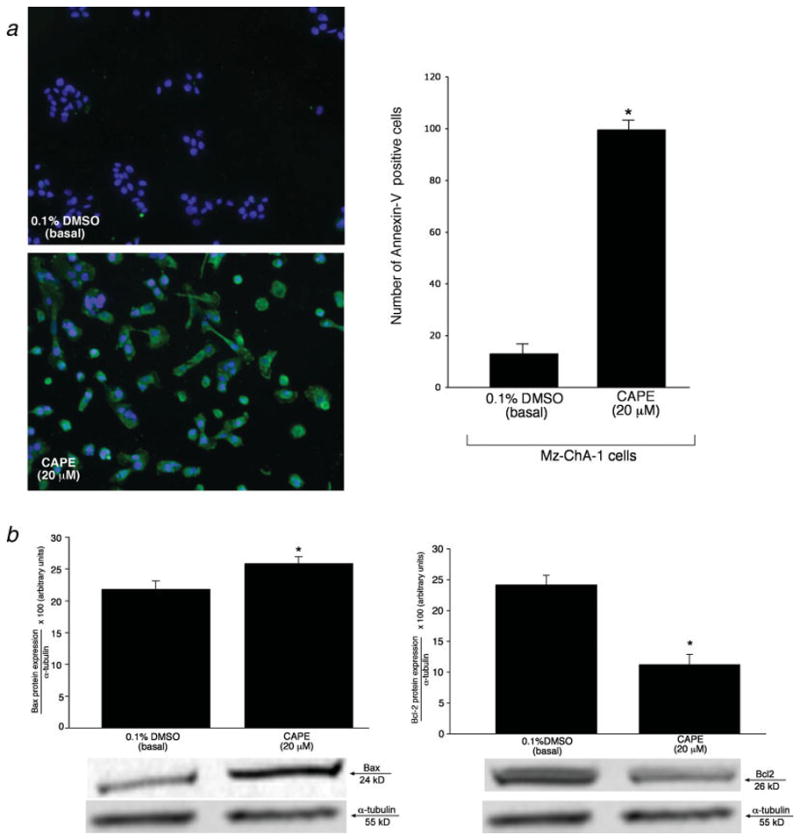
[a] Evaluation of apoptosis (by Annexin-V staining) in Mz-ChA-1 cells treated with 0.1% DMSO (basal) or CAPE (20 μM) with 0.1% DMSO. There was an approximate 10% positivity in Annexin-V staining in Mz-ChA-1 cells treated with 0.1% DMSO, whereas when cells were stimulated with CAPE there was almost a 100% positive staining for Annexin-V. The number of Annexin-V-positive cells were counted and expressed as a percentage of total cells in 5 random fields for each treatment group. *p<0.01 compared to the corresponding basal value. [b] Immunoblotting analysis for Bax and Bcl-2 in Mz-ChA-1 cells treated with 0.1% DMSO (basal) or CAPE (20 μM, 48 hours) with 0.1% DMSO. CAPE induced an increase in Bax and a decrease in Bcl-2 protein expression compared to Mz-ChA-1 cells stimulated with 0.1% DMSO (basal). Alpha-tubulin was used as an internal control and no changes were observed with this gene. Data are ± SEM of 6 experiments. *p < 0.05 compared to basal (0.1% DMSO).
To further confirm our hypothesis that CAPE acts through a pro-apoptotic pathway (evidenced by increased Annexin-V staining, Figure 4a), we studied the expression of Bax and Bcl-2 that are important players in the apoptotic pathway 40–42. Immunoblotting seen in Figure 4b shows a significant increase in protein expression for the pro-apoptotic protein, Bax (Figure 4b, upper panel) in Mz-ChA-1 cells treated with CAPE (20 μM) compared to basal (0.1% DMSO) treated cells. Similar to experiments in other cell types, the anti-apoptotic protein Bcl-2 was decreased in CAPE-treated cells (20 μM) compared to the corresponding basal value (lower panel, Figure 4b). Alpha-tubulin levels were unchanged in basal-and DMSO- stimulated cells (Figure 4b, upper and lower panels).
Xenograft Studies
In xenograft studies in nude mice we found that chronic treatment with CAPE (10 mg/Kg BW) 43 decreased tumor growth compared to mice treated with vehicle (0.1% DMSO) alone (Figure 5a). The time period for CAPE-treated tumors to reach 150% of their growth was almost 3 times longer than in vehicle-treated mice (Table 1). Body weight and liver weight were similar in both vehicle- and CAPE-treated mice (Table 1). Histologically, no changes were noted in fibrosis or inflammation in tumors from mice treated with either CAPE or vehicle (data not shown). However, there was a slight change in necrosis in DMSO versus CAPE- treated mice (35.79± 2.56 [DMSO] vs. 48.64 ± 4.05 [CAPE]). Almost all tumor cells were positive for CK-7 in both vehicle and CAPE-treated mice (data not shown). PCNA staining was decreased in CAPE-treated animals compared to vehicle-treated mice (Figure 5b). By TUNEL analysis, apoptotic cells were increased in tumors from CAPE-treated mice compared to vehicle (Figure 5b). Quantitative data for PCNA and TUNEL analysis is found in Table 2. Expression for VEGF-A, VEGF-C, VEGFR-2 and VEGFR-3 was unchanged in samples from CAPE-treated animals compared to vehicle treated mice (data not shown). Quantitative data for VEGF protein and receptor expression is seen in Table 2. Further evidence of NF-κB inhibition by CAPE was seen in immunohistochemical analysis (Figure 6a) and immunoblotting (Figure 6b) for NF-κB family members, NF-κB1 (p50) and RelA (p65) in tumor sections from CAPE and vehicle treated mice. Both protein expression (Figure 6b) and staining (Figure 6a) for these markers were decreased in CAPE-treated tumors compared to tumors from vehicle-treated mice thus supporting the concept that CAPE treatment decreases NF-κB in vivo. Evaluation of other organs demonstrated no histological changes in CAPE-treated animals compared to vehicle (not shown) indicating that the dosage of CAPE was not toxic to other organ systems and was well tolerated.
Figure 5.

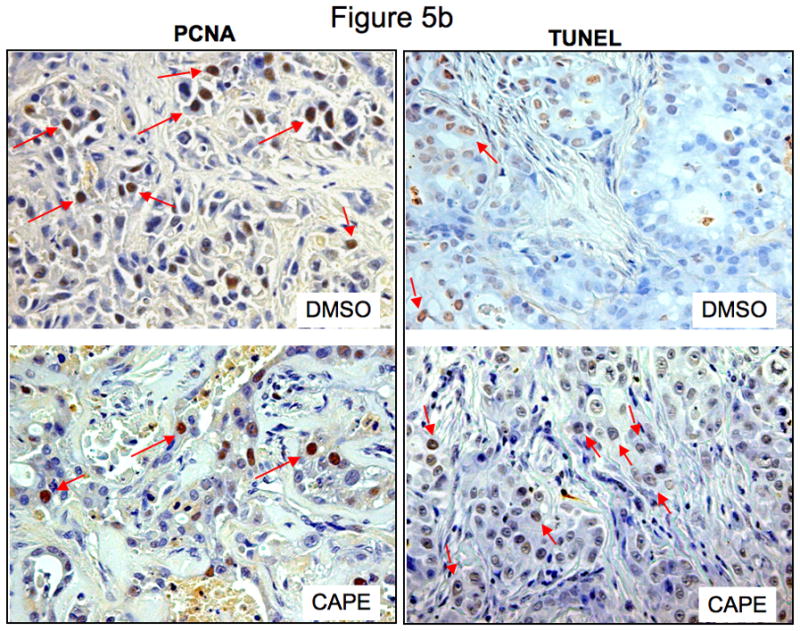
Effect of chronic administration of CAPE (10 mg/Kg BW with 0.1% DMSO) or vehicle (1:1/DMSO: NaCl) on [a] the growth of Mz-ChA-1 cells subcutaneously implanted into BALB/c nude (nu/nu) mice; the number of malignant cholangiocytes positive for [b] PCNA and TUNEL.
Figure 5a = CAPE decreases tumor growth after chronic treatment compared to vehicle-treated mice. Data shown is average tumor values from 3 mice (6 tumors in total) for each treatment group. At day 77 the cholangiocarcinoma xenograft volume of mice injected with CAPE (10 mg/kg) was 139.49 ± 57.05 versus 274.10 ± 66.49 mm3 for the control mice. Representative images for vehicle- and CAPE- treated tumors are shown in Figure 5a. Points = mean tumor size (mm3) and bars = SE. *p < 0.05 compared to vehicle. **p < 0.01 compared to vehicle.
Figure 5b = Immunohistochemical staining for the number of PCNA- and TUNEL-positive cells in tumors from both DMSO- and CAPE- treated mice. CAPE induced a decrease in the number of PCNA- positive cells coupled with an increase in the amount of TUNEL- positive cells compared to DMSO treatment (see Table 2 for quantitative data). Orig. magnification × 20.
Table 1.
Liver weight, body weight and liver to body weight ratio in nude mice treated with vehicle (0.1% DMSO) or CAPE (10 mg/Kg/BW) and tumor latency
| Treatment | Liver Weight (gm) | Body Weight (gm) | Liver to Body Weight Ratio (%) | Tumor Latency (days) |
|---|---|---|---|---|
| Vehicle (0.1% DMSO) | 2.25 ± 0.086 | 32.27 ± .691 | 6.97 ± 0.28 | 27 ± 2.09 |
| CAPE (10 mg/Kg/BW) | 2.1 ± 0.177 | 31.5 ± 1.85 | 6.66 ± 0.72 | 66 ± 1.13* |
Values are mean ± SE of 4 mice per group.
p < 0.05 compared to vehicle.
Table 2.
Quantitative data for the number of PCNA, TUNEL, VEGF-A, VEGF-C, VEGFR-1 and VEGFR-2- positive cells in nude mice treated with vehicle (0.1% DMSO) or CAPE (10 mg/Kg BW)
| Parameter | Vehicle (0.1% DMSO) | CAPE (10 mg/Kg BW) |
|---|---|---|
| PCNA | 30.60 ± 2.46 | 22.40 ± 2.04* |
| TUNEL | 30.80 ± 3.48 | 41.00 ± 2.85* |
| VEGF-A | 84.00 ± 2.45 | 88.00 ± 2.00 |
| VEGF-C | 20.00 ± 2.73 | 23.00 ± 2.61 |
| VEGFR-2 | 15.00 ± 3.53 | 19.00 ± 3.31 |
| VEGFR-3 | 53.00 ± 3.39 | 51.00 ± 1.87 |
Values are mean ± SE of 4 mice per group.
p < 0.05 vs. vehicle.
Figure 6.
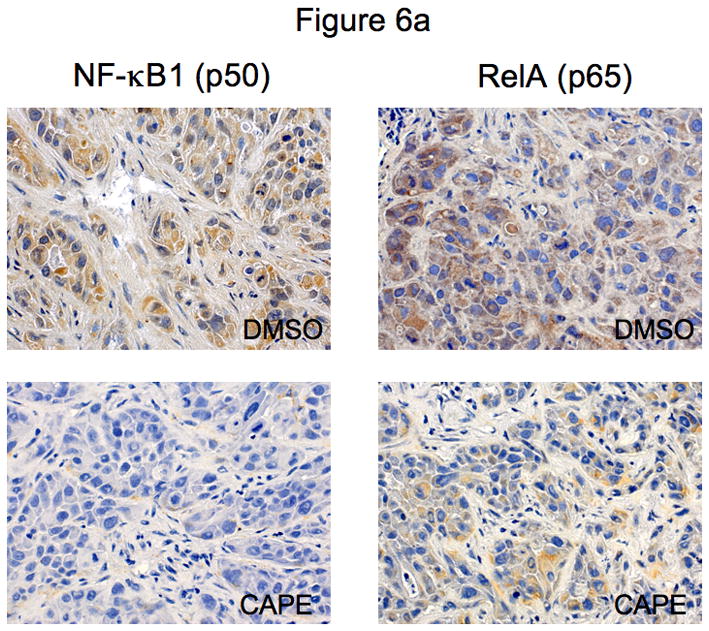

[a] Evaluation of NF-κB family members, NF-κB1 (p50) and RelA (p65) in tumor sections from mice treated with CAPE (10 mg/Kg BW with 0.1% DMSO) or vehicle (1:1/DMSO: NaCl). Immunohistochemical analysis shows that both NF-κB1 (p50) and RelA (p65) are downregulated after CAPE treatment compared to vehicle. Orig. magnification × 20. [b] Immunoblotting analysis for NF-κB1 (p50) and RelA (p65) in tumor sections from from mice treated with CAPE (10 mg/Kg BW with 0.1% DMSO) or vehicle (1:1/DMSO: NaCl). Protein expression was downregulated for both p50 and p65 in CAPE- treated mice samples compared to vehicle. Alpha-tubulin levels were unchanged in vehicle- and CAPE- treated tumor samples.
Data are ± SEM of 4 experiments. *p< 0.05 compared to basal (0.1% DMSO).
DISCUSSION
In this study, we explored the role of the NF-κB inhibitor 15, 50, CAPE, on the regulation of CCH growth. While it has been shown that CAPE inhibits the growth of numerous other cancers 43, 51, 52 there is no information on the role of CAPE in the inhibition of CCH growth. Also, CAPE has been demonstrated to be a potent inhibitor of NF-κB in several cell systems 15, 50, however, there is no information on the ability of CAPE to inhibit NF-κB signalling in CCH cells. Here, we have demonstrated that CAPE inhibits NF-κB DNA-binding activity and the growth of numerous CCH cell lines by changes in apoptotic signalling pathways. In vitro, CAPE induced inhibition of cell cycle progression evaluated by BrdU staining and PCNA protein expression in CCH cells. Cell death in CCH cell lines was demonstrated by Annexin-V staining, and by immunoblotting for certain apoptotic markers: the pro-apoptotic protein, Bax 53, and the anti-apoptotic protein, Bcl-2 54. As expected, CAPE increased Annexin-V labelling and the expression of Bax while decreasing the expression of Bcl-2 in Mz-ChA-1 cells. Lastly, our studies were taken into an in vivo model whereby Mz-ChA-1 cells were injected into the hind flanks of nude mice and treated with CAPE or 0.1% DMSO for 77 days. In vivo studies revealed that: (i) CAPE decreased the growth of tumors in nude mice with increased tumor latency; (ii) CAPE decreased PCNA expression with increased TUNEL expression; (iii) expression for CK-7, VEGF-A, VEGF-C, VEGFR-2 and VEGFR-3 was unchanged in CAPE- compared to vehicle-treated animals and (iv) no differences were observed in fibrosis or inflammation in either the vehicle or CAPE-treated animals with no other tissue/organ damage. Further, CAPE decreased the expression of both NF-κB1 (p50) and RelA (p65) compared to vehicle-treated mice in tumor sections from these animals demonstrated by both immunoblotting and immunohistochemistry.
The transcription factor, NF-κB, plays a critical regulatory role in the expression of numerous target genes that control cell death, proliferation, differentiation and immune and inflammatory responses 55. There are numerous members of the NF-κB family including: NF-κB1 (p50), NF-κB2 (p52), RelA (p65), RelB and c-Rel 56, 57. Blocking NF-κB has been shown to cause tumor cells to stop proliferating, to die, or to become more sensitive to the action of anti-tumor agents 58. To determine the effects of CAPE on NF-κB activity we demonstrated that CAPE suppresses the function of NF-κB by blocking the nuclear translocation of NF-κB in human cholangiocarcinoma cells as shown by EMSA. In support of our studies, it has recently been shown that rottlerin blocks the NF-κB/cyclin D cascade in human breast cancer cell lines 59 and that the dietary anti-oxidant, curcumin, inhibits NF-κB activation thus decreasing metastasis in breast cancer cells 60. In lung cancer cells, it has been demonstrated that nutlin, a potent Mdm2 antagonist that blocks the p53-Mdm2 interaction, suppresses NF-κB activity that may lead to down-regulation or tumor formation and metastasis 61. In addition to EMSA data demonstrating that CAPE induces an inhibition of NF-κB DNA-binding activity, we have also shown that two members of the NF-κB family, NF-κB1 (p50) and RelA (p65), were decreased after CAPE treatment both in vitro and in vivo. These studies support the role that NF-κB plays in tumorigenesis and further validates our studies herein. While the effects of CAPE on NF-κB inhibition and p50 and p65 suppression are seen mostly at the activity level, there are have been some preliminary studies 62 suggesting that the effects of CAPE can also occur at a translational level. Indeed, this preliminary study has shown that CAPE suppressed p50 expression both at the message and protein level in osteosarcoma cells 62. Further, a study involving hepatocellular carcinoma cells (HCC) has shown that the protein expression of both p50 and p65 was increased in HCC cells compared to normal liver cells 17–19, 63. This study supports the notion that NF-κB activity may be increased in cancer cells compared to normal healthy cells 17–19. Therefore, inhibiting or blocking NF-κB activity or repressing the expression of the family members of NF-κB may represent important targets in cancer research and possible therapeutic developments. Our data showing the decreased DNA binding activity of NF-κB and decreased protein expression of p50 and p65 (both in vitro and in vivo) supports the concept that CAPE may act at both transcriptional and post-transcriptional levels to suppress cholangiocarcinoma growth.
CAPE has been shown to possess antitumor activity 64 and anti-inflammatory properties 6, as well as being a potent and specific NF-κB inhibitor 6. Consistent within the modality that phenolic compounds decrease carcinogenesis and tumor growth, in our study we have demonstrated that the proliferation of various cholangiocarcinoma cells lines is strongly reduced by in vitro treatment with CAPE. In support of our study, it has been shown that proliferation in human HeLa cervical carcinoma cells was greatly inhibited by administration of both CA (caffeic acid) and CAPE 65. In our study, the anti-proliferative effects of CAPE on CCH growth were also coupled with increased cell cycle arrest and apoptosis. These results are consistent with other studies demonstrating the ability of CAPE to induce growth inhibition coupled with cell cycle arrest and increased apoptosis 66, 67. The growth of human colorectal cancer cells was halted by CAPE treatment in vitro in a dose-dependent manner 68 similar to what was seen in our study in cholangiocarcinoma cells from different origins. Likewise, colorectal cancer cells treated with CAPE exhibited increased cell cycle arrest and induction of apoptosis 68.
Markers to evaluate the effects of apoptosis are numerous. In our study, we evaluated the effects of CAPE on two particular apoptotic markers, Bax and Bcl-2. Bcl-2 is the prototype member for a family of genes and proteins that regulate mitochondrial outer membrane permeabilization and are either anti-apoptotic (i.e. Bcl-2) or pro-apoptotic (Bax) 53, 54. Bcl-2 has been implicated in a number of diseases such as autoimmunity 69 as well as in many cancers including breast, prostate 70 and lung 71. Deviations in gene expression of anti-apoptotic and pro-apoptotic proteins may play a part in the cause of the many forms of cancer 72. Bcl-2 is imperative to the process and progression of apoptosis as it inhibits the initiation of the cell-death process. In our study, we found that cholangiocarcinoma cells treated with CAPE in vitro demonstrated a decrease in protein expression of the anti-apoptotic maker, Bcl-2 coupled with a significant increase in the pro-apoptotic marker, Bax. In agreement with our findings, studies have offered evidence of a dysregulation of the Bcl-2/Bax ratio including a study showing that leutolin increased Bax protein expression with a concurrent decrease in Bcl-2 protein expression in human hepatoma cell lines 73. Similarly, IGF-1 has been shown to regulate the expression of Bax and Bcl-2 in human breast cancer cells 74.
We further expanded our study by examining the effects of CAPE on an in vivo model. As we (and others) have shown, the model system used herein allows for the evaluation of a compound on in vivo tumor growth. Recently we have shown that GABA inhibits the tumor growth of CCH in a nude mouse model 39. Using this model, we demonstrate that chronic administration of CAPE to tumor-induced mice significantly decreased tumor growth and increased tumor latency compared to vehicle-treated mice. Immunohistochemical analysis of inflammation and fibrosis showed that there was relatively no change in these parameters between vehicle- and CAPE-treated mice. In vivo CAPE decreased tumor growth and proliferation as seen by the decrease in the number of PCNA-positive cells by immunohistochemistry in tumor samples. The decrease in tumor growth and proliferation is likely regulated by apoptosis which is demonstrated by the significant increase in TUNEL-positive cells from CAPE-treated tumors compared to vehicle. Changes in cell death that were consistent with in vitro data were coupled with no observable changes in VEGF expression between the two groups indicating an angiogenic-independent regulation of growth inhibition. In addition, NF-κB1 (p50) and RelA (p65) expression was decreased in tumors from CAPE-treated mice demonstrating that in vivo treatment of CAPE inhibits NF-κB signaling. Evaluation of other organs (lung, heart, kidney, stomach and intestine) showed that chronic treatment with CAPE had no toxic or secondary effects and was well tolerated throughout the study. In support of our study using an in vivo xenograft model, CAPE has previously been shown to suppress the growth of HepG2 tumor xenografts in nude mice treated with CAPE both subcutaneously and orally 51.
In conclusion, we have provided evidence that in vitro stimulation of cholangiocarcinoma by CAPE produces significant suppression of CCH growth via inhibition of NF-κB DNA-binding, downregulation of the family members, NF-κB1 and RelA along with activated apoptosis coupled with increased cell cycle arrest. Further, the in vivo studies strengthen and validate our in vitro findings demonstrating that CAPE should be considered a valuable and resourceful tool in the fight against cholangiocarcinoma. Our studies are both novel and important in that finding alternative therapies to battle cholangiocarcinoma is an avenue that is becoming more explored as traditional therapies continue to fail to provide therapeutic relief to suffering patients. While the effects of CAPE in other cancerous tumors have been shown 65–68, our study provides the first evidence of the inhibitory effects of CAPE in cholangiocarcinoma. In addition to modulation of the well-known NF-κB signaling pathway, we have also shown that CAPE has translational effects by down-regulating the protein expression of NF-κB family members. This concept has not been shown in cholangiocarcinoma. Targeting the NF-κB signaling pathway at either the level of protein expression or its ability to regulate gene transcription could provide important therapeutic answers to a complicated problem.
Acknowledgments
We acknowledge the Texas A&M Health Science Center Microscopy Imaging Center for assistance with the confocal microscopy imaging.
The study was supported by the Dr. Nicholas C. Hightower Centennial Chair of Gastroenterology from Scott & White, the VA Research Scholar Award and a VA Merit Award and the NIH grants DK054811, DK062975 and DK76898 to Dr. Alpini, an NIH K01 grant award (DK078532) to Dr. DeMorrow and a grant award from Health and Labor Sciences Research Grants for the Research on Measures for Intractable Diseases (from the Ministry of Health, Labor and Welfare of Japan), from a Grant-in-Aid for Scientific Research C (19590744) from JSPS to Dr. Ueno and by University funds to Dr. Onori and PRIN 2007 and Federate Athenaeum funds from University of Rome “La Sapienza” to Prof. Gaudio.
Abbreviations
- Bax
Bcl2- associated X protein
- Bcl-2
B-cell lymphoma 2
- BrdU
bromodeoxyuridine
- BSA
bovine serum albumin
- CA
caffeic acid
- CAPE
caffeic acid phenethyl ester
- CCH
cholangiocarcinoma
- CK-7
cytokeratin-7
- DTT
1,4-Dithio-DL-threitol dithiothreitol
- DMSO
dimethyl sulfoxide
- EDTA
ethylene diamine tetraacetic acid
- EMSA
electrophoretic mobility shift assay
- HEPES
(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid)
- MTS
3- (4, 5- dimethyl thiazol - 2- yl) -5- (3- carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H -tetrazolium
- NF-κB
nuclear factor kappa beta
- PBS
phosphate buffered saline
- PCNA
proliferating cell nuclear antigen
- PMSF
phenylmethanesulphonylfluoride
- PSC
primary sclerosing cholangitis
- TUNEL
terminal deoxynucleotide transferase end labeling
References
- 1.Alpini G, Prall RT, LaRusso NF. In: The pathobiology of biliary epithelia. The Liver; Biology & Pathobiology, 4E. Arias IM, Boyer JL, Chisari FV, Fausto N, Jakoby W, Schachter D, Shafritz DA, editors. Philadelphia, PA: Lippincott Williams & Wilkins; 2001. pp. 421–35. [Google Scholar]
- 2.Sirica AE. Cholangiocarcinoma: molecular targeting strategies for chemoprevention and therapy. Hepatology. 2005;41:5–15. doi: 10.1002/hep.20537. [DOI] [PubMed] [Google Scholar]
- 3.Blechacz BR, Gores GJ. Cholangiocarcinoma. Clin Liver Dis. 2008;12:131–50. doi: 10.1016/j.cld.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Son S, Lewis BA. Free radical scavenging and antioxidative activity of caffeic acid amide and ester analogues: structure-activity relationship. J Agric Food Chem. 2002;50:468–72. doi: 10.1021/jf010830b. [DOI] [PubMed] [Google Scholar]
- 5.Koltuksuz U, Mutus HM, Kutlu R, Ozyurt H, Cetin S, Karaman A, Gurbuz N, Akyol O, Aydin NE. Effects of caffeic acid phenethyl ester and epidermal growth factor on the development of caustic esophageal stricture in rats. J Pediatr Surg. 2001;36:1504–9. doi: 10.1053/jpsu.2001.27032. [DOI] [PubMed] [Google Scholar]
- 6.Michaluart P, Masferrer JL, Carothers AM, Subbaramaiah K, Zweifel BS, Koboldt C, Mestre JR, Grunberger D, Sacks PG, Tanabe T, Dannenberg AJ. Inhibitory effects of caffeic acid phenethyl ester on the activity and expression of cyclooxygenase-2 in human oral epithelial cells and in a rat model of inflammation. Cancer Res. 1999;59:2347–52. [PubMed] [Google Scholar]
- 7.Borrelli F, Izzo AA, Di Carlo G, Maffia P, Russo A, Maiello FM, Capasso F, Mascolo N. Effect of a propolis extract and caffeic acid phenethyl ester on formation of aberrant crypt foci and tumors in the rat colon. Fitoterapia. 2002;73 (Suppl 1):S38–43. doi: 10.1016/s0367-326x(02)00189-2. [DOI] [PubMed] [Google Scholar]
- 8.Burke TR, Jr, Fesen MR, Mazumder A, Wang J, Carothers AM, Grunberger D, Driscoll J, Kohn K, Pommier Y. Hydroxylated aromatic inhibitors of HIV-1 integrase. J Med Chem. 1995;38:4171–8. doi: 10.1021/jm00021a006. [DOI] [PubMed] [Google Scholar]
- 9.Chiao C, Carothers AM, Grunberger D, Solomon G, Preston GA, Barrett JC. Apoptosis and altered redox state induced by caffeic acid phenethyl ester (CAPE) in transformed rat fibroblast cells. Cancer Res. 1995;55:3576–83. [PubMed] [Google Scholar]
- 10.Laranjinha J, Vieira O, Madeira V, Almeida L. Two related phenolic antioxidants with opposite effects on vitamin E content in low density lipoproteins oxidized by ferrylmyoglobin: consumption vs regeneration. Arch Biochem Biophys. 1995;323:373–81. doi: 10.1006/abbi.1995.0057. [DOI] [PubMed] [Google Scholar]
- 11.Kimura Y, Okuda H, Okuda T, Hatano T, Agata I, Arichi S. Studies on the activities of tannins and related compounds from medicinal plants and drugs. VII. Effects of extracts of leaves of Artemisia species, and caffeic acid and chlorogenic acid on lipid metabolic injury in rats fed peroxidized oil. Chem Pharm Bull (Tokyo) 1985;33:2028–34. doi: 10.1248/cpb.33.2028. [DOI] [PubMed] [Google Scholar]
- 12.Frenkel K, Wei H, Bhimani R, Ye J, Zadunaisky JA, Huang MT, Ferraro T, Conney AH, Grunberger D. Inhibition of tumor promoter-mediated processes in mouse skin and bovine lens by caffeic acid phenethyl ester. Cancer Res. 1993;53:1255–61. [PubMed] [Google Scholar]
- 13.Fitzpatrick LR, Wang J, Le T. Caffeic acid phenethyl ester, an inhibitor of nuclear factor-kappaB, attenuates bacterial peptidoglycan polysaccharide-induced colitis in rats. J Pharmacol Exp Ther. 2001;299:915–20. [PubMed] [Google Scholar]
- 14.Orban Z, Mitsiades N, Burke TR, Jr, Tsokos M, Chrousos GP. Caffeic acid phenethyl ester induces leukocyte apoptosis, modulates nuclear factor-kappa B and suppresses acute inflammation. Neuroimmunomodulation. 2000;7:99–105. doi: 10.1159/000026427. [DOI] [PubMed] [Google Scholar]
- 15.Natarajan K, Singh S, Burke TR, Jr, Grunberger D, Aggarwal BB. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc Natl Acad Sci U S A. 1996;93:9090–5. doi: 10.1073/pnas.93.17.9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Jin B, Huang C. The PI3K/Akt pathway and its downstream transcriptional factors as targets for chemoprevention. Curr Cancer Drug Targets. 2007;7:305–16. doi: 10.2174/156800907780809741. [DOI] [PubMed] [Google Scholar]
- 17.Lu T, Stark GR. Cytokine overexpression and constitutive NFkappaB in cancer. Cell Cycle. 2004;3:1114–7. [PubMed] [Google Scholar]
- 18.Weisz L, Damalas A, Liontos M, Karakaidos P, Fontemaggi G, Maor-Aloni R, Kalis M, Levrero M, Strano S, Gorgoulis VG, Rotter V, Blandino G, et al. Mutant p53 enhances nuclear factor kappaB activation by tumor necrosis factor alpha in cancer cells. Cancer Res. 2007;67:2396–401. doi: 10.1158/0008-5472.CAN-06-2425. [DOI] [PubMed] [Google Scholar]
- 19.Fabre C, Grosjean J, Tailler M, Boehrer S, Ades L, Perfettini JL, de Botton S, Fenaux P, Kroemer G. A novel effect of DNA methyltransferase and histone deacetylase inhibitors: NFkappaB inhibition in malignant myeloblasts. Cell Cycle. 2008;7:2139–45. doi: 10.4161/cc.7.14.6268. [DOI] [PubMed] [Google Scholar]
- 20.Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–4. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 21.Bachmeier B, Nerlich AG, Iancu CM, Cilli M, Schleicher E, Vene R, Dell’Eva R, Jochum M, Albini A, Pfeffer U. The chemopreventive polyphenol Curcumin prevents hematogenous breast cancer metastases in immunodeficient mice. Cell Physiol Biochem. 2007;19:137–52. doi: 10.1159/000099202. [DOI] [PubMed] [Google Scholar]
- 22.Fister S, Schlotawa L, Gunthert AR, Emons G, Grundker C. Increase of doxorubicin-induced apoptosis after knock-down of gonadotropin-releasing hormone receptor expression in human endometrial, ovarian and breast cancer cells. Gynecol Endocrinol. 2008;24:24–9. doi: 10.1080/09513590701668882. [DOI] [PubMed] [Google Scholar]
- 23.Wyllie AH. Apoptosis and carcinogenesis. Eur J Cell Biol. 1997;73:189–97. [PubMed] [Google Scholar]
- 24.Fadeel B, Orrenius S. Apoptosis: a basic biological phenomenon with wide-ranging implications in human disease. J Intern Med. 2005;258:479–517. doi: 10.1111/j.1365-2796.2005.01570.x. [DOI] [PubMed] [Google Scholar]
- 25.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 26.Kanno N, Glaser S, Chowdhury U, Phinizy JL, Baiocchi L, Francis H, LeSage G, Alpini G. Gastrin inhibits cholangiocarcinoma growth through increased apoptosis by activation of Ca2+-dependent protein kinase C-alpha. J Hepatol. 2001;34:284–91. doi: 10.1016/s0168-8278(00)00025-8. [DOI] [PubMed] [Google Scholar]
- 27.Kanno N, LeSage G, Phinizy JL, Glaser S, Francis H, Alpini G. Stimulation of alpha2-adrenergic receptor inhibits cholangiocarcinoma growth through modulation of Raf-1 and B-Raf activities. Hepatology. 2002;35:1329–40. doi: 10.1053/jhep.2002.33330. [DOI] [PubMed] [Google Scholar]
- 28.Knuth A, Gabbert H, Dippold W, Klein O, Sachsse W, Bitter-Suermann D, Prellwitz W, Meyer zum Buschenfelde KH. Biliary adenocarcinoma. Characterisation of three new human tumor cell lines. J Hepatol. 1985;1:579–96. doi: 10.1016/s0168-8278(85)80002-7. [DOI] [PubMed] [Google Scholar]
- 29.Kusaka Y, Tokiwa T, Sato J. Establishment and characterization of a cell line from a human cholangiocellular carcinoma. Res Exp Med (Berl) 1988;188:367–75. doi: 10.1007/BF01851205. [DOI] [PubMed] [Google Scholar]
- 30.Saijyo S, Kudo T, Suzuki M, Katayose Y, Shinoda M, Muto T, Fukuhara K, Suzuki T, Matsuno S. Establishment of a new extrahepatic bile duct carcinoma cell line, TFK-1. Tohoku J Exp Med. 1995;177:61–71. doi: 10.1620/tjem.177.61. [DOI] [PubMed] [Google Scholar]
- 31.Miyagiwa M, Ichida T, Tokiwa T, Sato J, Sasaki H. A new human cholangiocellular carcinoma cell line (HuCC-T1) producing carbohydrate antigen 19/9 in serum-free medium. In Vitro Cell Dev Biol. 1989;25:503–10. doi: 10.1007/BF02623562. [DOI] [PubMed] [Google Scholar]
- 32.Storto PD, Saidman SL, Demetris AJ, Letessier E, Whiteside TL, Gollin SM. Chromosomal breakpoints in cholangiocarcinoma cell lines. Genes Chromosomes Cancer. 1990;2:300–10. doi: 10.1002/gcc.2870020408. [DOI] [PubMed] [Google Scholar]
- 33.Shimizu Y, Demetris AJ, Gollin SM, Storto PD, Bedford HM, Altarac S, Iwatsuki S, Herberman RB, Whiteside TL. Two new human cholangiocarcinoma cell lines and their cytogenetics and responses to growth factors, hormones, cytokines or immunologic effector cells. Int J Cancer. 1992;52:252–60. doi: 10.1002/ijc.2910520217. [DOI] [PubMed] [Google Scholar]
- 34.Wu T, Leng J, Han C, Demetris AJ. The cyclooxygenase-2 inhibitor celecoxib blocks phosphorylation of Akt and induces apoptosis in human cholangiocarcinoma cells. Mol Cancer Ther. 2004;3:299–307. [PubMed] [Google Scholar]
- 35.Grubman SA, Perrone RD, Lee DW, Murray SL, Rogers LC, Wolkoff LI, Mulberg AE, Cherington V, Jefferson DM. Regulation of intracellular pH by immortalized human intrahepatic biliary epithelial cell lines. Am J Physiol. 1994;266:G1060–70. doi: 10.1152/ajpgi.1994.266.6.G1060. [DOI] [PubMed] [Google Scholar]
- 36.Basak S, Shih VF, Hoffmann A. Generation and activation of multiple dimeric transcription factors within the NF-kappaB signaling system. Mol Cell Biol. 2008;28:3139–50. doi: 10.1128/MCB.01469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glaser S, Benedetti A, Marucci L, Alvaro D, Baiocchi L, Kanno N, Caligiuri A, Phinizy JL, Chowdhury U, Papa E, LeSage G, Alpini G. Gastrin inhibits cholangiocyte growth in bile duct-ligated rats by interaction with cholecystokinin-B/Gastrin receptors via D-myo-inositol 1,4,5-triphosphate-, Ca(2+)-, and protein kinase C alpha-dependent mechanisms. Hepatology. 2000;32:17–25. doi: 10.1053/jhep.2000.8265. [DOI] [PubMed] [Google Scholar]
- 38.DeMorrow S, Glaser S, Francis H, Venter J, Vaculin B, Vaculin S, Alpini G. Opposing actions of endocannabinoids on cholangiocarcinoma growth: recruitment of Fas and Fas ligand to lipid rafts. J Biol Chem. 2007;282:13098–113. doi: 10.1074/jbc.M608238200. [DOI] [PubMed] [Google Scholar]
- 39.Fava G, Marucci L, Glaser S, Francis H, De Morrow S, Benedetti A, Alvaro D, Venter J, Meininger C, Patel T, Taffetani S, Marzioni M, et al. gamma-Aminobutyric acid inhibits cholangiocarcinoma growth by cyclic AMP-dependent regulation of the protein kinase A/extracellular signal-regulated kinase 1/2 pathway. Cancer Res. 2005;65:11437–46. doi: 10.1158/0008-5472.CAN-05-1470. [DOI] [PubMed] [Google Scholar]
- 40.Sheng G, Guo J, Warner BW. Epidermal Growth Factor Receptor Signaling Modulates Apoptosis via p38{alpha} MAPK-Dependent Activation of Bax in Intestinal Epithelial Cells. Am J Physiol Gastrointest Liver Physiol. 2007 doi: 10.1152/ajpgi.00182.2007. [DOI] [PubMed] [Google Scholar]
- 41.Kobayashi S, Lee SH, Meng XW, Mott JL, Bronk SF, Werneburg NW, Craig RW, Kaufmann SH, Gores GJ. Serine 64 phosphorylation enhances the antiapoptotic function of Mcl-1. J Biol Chem. 2007;282:18407–17. doi: 10.1074/jbc.M610010200. [DOI] [PubMed] [Google Scholar]
- 42.Werneburg NW, Guicciardi ME, Bronk SF, Kaufmann SH, Gores GJ. Tumor necrosis factor-related apoptosis-inducing ligand activates a lysosomal pathway of apoptosis that is regulated by Bcl-2 proteins. J Biol Chem. 2007;282:28960–70. doi: 10.1074/jbc.M705671200. [DOI] [PubMed] [Google Scholar]
- 43.Liao HF, Chen YY, Liu JJ, Hsu ML, Shieh HJ, Liao HJ, Shieh CJ, Shiao MS, Chen YJ. Inhibitory effect of caffeic acid phenethyl ester on angiogenesis, tumor invasion, and metastasis. J Agric Food Chem. 2003;51:7907–12. doi: 10.1021/jf034729d. [DOI] [PubMed] [Google Scholar]
- 44.Taffetani S, Glaser S, Francis H, DeMorrow S, Ueno Y, Alvaro D, Marucci L, Marzioni M, Fava G, Venter J, Vaculin S, Vaculin B, et al. Prolactin stimulates the proliferation of normal female cholangiocytes by differential regulation of Ca2+-dependent PKC isoforms. BMC Physiol. 2007;7:6. doi: 10.1186/1472-6793-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gaudio E, Onori P, Pannarale L, Alvaro D. Hepatic microcirculation and peribiliary plexus in experimental biliary cirrhosis: a morphological study. Gastroenterology. 1996;111:1118–24. doi: 10.1016/s0016-5085(96)70081-1. [DOI] [PubMed] [Google Scholar]
- 46.Watabe M, Hishikawa K, Takayanagi A, Shimizu N, Nakaki T. Caffeic acid phenethyl ester induces apoptosis by inhibition of NFkappaB and activation of Fas in human breast cancer MCF-7 cells. J Biol Chem. 2004;279:6017–26. doi: 10.1074/jbc.M306040200. [DOI] [PubMed] [Google Scholar]
- 47.Montpied P, de Bock F, Rondouin G, Niel G, Briant L, Courseau AS, Lerner-Natoli M, Bockaert J. Caffeic acid phenethyl ester (CAPE) prevents inflammatory stress in organotypic hippocampal slice cultures. Brain Res Mol Brain Res. 2003;115:111–20. doi: 10.1016/s0169-328x(03)00178-5. [DOI] [PubMed] [Google Scholar]
- 48.Schubart UK, Xu J, Fan W, Cheng G, Goldstein H, Alpini G, Shafritz DA, Amat JA, Farooq M, Norton WT, et al. Widespread differentiation stage-specific expression of the gene encoding phosphoprotein p19 (metablastin) in mammalian cells. Differentiation. 1992;51:21–32. doi: 10.1111/j.1432-0436.1992.tb00676.x. [DOI] [PubMed] [Google Scholar]
- 49.Lesage G, Glaser S, Ueno Y, Alvaro D, Baiocchi L, Kanno N, Phinizy JL, Francis H, Alpini G. Regression of cholangiocyte proliferation after cessation of ANIT feeding is coupled with increased apoptosis. Am J Physiol Gastrointest Liver Physiol. 2001;281:G182–90. doi: 10.1152/ajpgi.2001.281.1.G182. [DOI] [PubMed] [Google Scholar]
- 50.Haddad JJ, Fahlman CS. Nuclear factor-kappa B-independent regulation of lipopolysaccharide-mediated interleukin-6 biosynthesis. Biochem Biophys Res Commun. 2002;291:1045–51. doi: 10.1006/bbrc.2002.6556. [DOI] [PubMed] [Google Scholar]
- 51.Chung TW, Moon SK, Chang YC, Ko JH, Lee YC, Cho G, Kim SH, Kim JG, Kim CH. Novel and therapeutic effect of caffeic acid and caffeic acid phenyl ester on hepatocarcinoma cells: complete regression of hepatoma growth and metastasis by dual mechanism. Faseb J. 2004;18:1670–81. doi: 10.1096/fj.04-2126com. [DOI] [PubMed] [Google Scholar]
- 52.Hwang HJ, Park HJ, Chung HJ, Min HY, Park EJ, Hong JY, Lee SK. Inhibitory effects of caffeic acid phenethyl ester on cancer cell metastasis mediated by the down-regulation of matrix metalloproteinase expression in human HT1080 fibrosarcoma cells. J Nutr Biochem. 2006;17:356–62. doi: 10.1016/j.jnutbio.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 53.Pawlowski J, Kraft AS. Bax-induced apoptotic cell death. Proc Natl Acad Sci U S A. 2000;97:529–31. doi: 10.1073/pnas.97.2.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Werner AB, de Vries E, Tait SW, Bontjer I, Borst J. Bcl-2 family member Bfl-1/A1 sequesters truncated bid to inhibit is collaboration with pro-apoptotic Bak or Bax. J Biol Chem. 2002;277:22781–8. doi: 10.1074/jbc.M201469200. [DOI] [PubMed] [Google Scholar]
- 55.Kucharczak J, Simmons MJ, Fan Y, Gelinas C. To be, or not to be: NF-kappaB is the answer--role of Rel/NF-kappaB in the regulation of apoptosis. Oncogene. 2003;22:8961–82. doi: 10.1038/sj.onc.1207230. [DOI] [PubMed] [Google Scholar]
- 56.Bours V, Burd PR, Brown K, Villalobos J, Park S, Ryseck RP, Bravo R, Kelly K, Siebenlist U. A novel mitogen-inducible gene product related to p50/p105-NF-kappa B participates in transactivation through a kappa B site. Mol Cell Biol. 1992;12:685–95. doi: 10.1128/mcb.12.2.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rayet B, Gelinas C. Aberrant rel/nfkb genes and activity in human cancer. Oncogene. 1999;18:6938–47. doi: 10.1038/sj.onc.1203221. [DOI] [PubMed] [Google Scholar]
- 58.Escarcega RO, Fuentes-Alexandro S, Garcia-Carrasco M, Gatica A, Zamora A. The transcription factor nuclear factor-kappa B and cancer. Clin Oncol (R Coll Radiol) 2007;19:154–61. doi: 10.1016/j.clon.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 59.Torricelli C, Fortino V, Capurro E, Valacchi G, Pacini A, Muscettola M, Soucek K, Maioli E. Rottlerin inhibits the nuclear factor kappaB/Cyclin-D1 cascade in MCF-7 breast cancer cells. Life Sci. 2008;82:638–43. doi: 10.1016/j.lfs.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 60.Bachmeier BE, Mohrenz IV, Mirisola V, Schleicher E, Romeo F, Hohneke C, Jochum M, Nerlich AG, Pfeffer U. Curcumin downregulates the inflammatory cytokines CXCL1 and -2 in breast cancer cells via NFkappaB. Carcinogenesis. 2008;29:779–89. doi: 10.1093/carcin/bgm248. [DOI] [PubMed] [Google Scholar]
- 61.Dey A, Wong ET, Bist P, Tergaonkar V, Lane DP. Nutlin-3 inhibits the NFkappaB pathway in a p53-dependent manner: implications in lung cancer therapy. Cell Cycle. 2007;6:2178–85. doi: 10.4161/cc.6.17.4643. [DOI] [PubMed] [Google Scholar]
- 62.Frenkel K, Wu J, Yang C. Prevention of human cell transformation by CAPE (caffeic acid phenethyl ester): Role of NF-B factors. AACR Meeting. 2006:B105. Abstract. [Google Scholar]
- 63.Wang J, Huang Q, Chen M. The role of NF-kappaB in hepatocellular carcinoma cell. Chin Med J (Engl) 2003;116:747–52. [PubMed] [Google Scholar]
- 64.Tanaka T, Kojima T, Kawamori T, Wang A, Suzui M, Okamoto K, Mori H. Inhibition of 4-nitroquinoline-1-oxide-induced rat tongue carcinogenesis by the naturally occurring plant phenolics caffeic, ellagic, chlorogenic and ferulic acids. Carcinogenesis. 1993;14:1321–5. doi: 10.1093/carcin/14.7.1321. [DOI] [PubMed] [Google Scholar]
- 65.Orsolic N, Terzic S, Mihaljevic Z, Sver L, Basic I. Effects of local administration of propolis and its polyphenolic compounds on tumor formation and growth. Biol Pharm Bull. 2005;28:1928–33. doi: 10.1248/bpb.28.1928. [DOI] [PubMed] [Google Scholar]
- 66.He YJ, Liu BH, Xiang DB, Qiao ZY, Fu T, He YH. Inhibitory effect of caffeic acid phenethyl ester on the growth of SW480 colorectal tumor cells involves beta-catenin associated signaling pathway down-regulation. World J Gastroenterol. 2006;12:4981–5. doi: 10.3748/wjg.v12.i31.4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kuo HC, Kuo WH, Lee YJ, Wang CJ, Tseng TH. Enhancement of caffeic acid phenethyl ester on all-trans retinoic acid-induced differentiation in human leukemia HL-60 cells. Toxicol Appl Pharmacol. 2006;216:80–8. doi: 10.1016/j.taap.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 68.Wang D, Xiang DB, He YJ, Li ZP, Wu XH, Mou JH, Xiao HL, Zhang QH. Effect of caffeic acid phenethyl ester on proliferation and apoptosis of colorectal cancer cells in vitro. World J Gastroenterol. 2005;11:4008–12. doi: 10.3748/wjg.v11.i26.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Opferman JT. Apoptosis in the development of the immune system. Cell Death Differ. 2008;15:234–42. doi: 10.1038/sj.cdd.4402182. [DOI] [PubMed] [Google Scholar]
- 70.Friedman AE. Can a single model explain both breast cancer and prostate cancer? Theor Biol Med Model. 2007;4:28. doi: 10.1186/1742-4682-4-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tahir SK, Yang X, Anderson MG, Morgan-Lappe SE, Sarthy AV, Chen J, Warner RB, Ng SC, Fesik SW, Elmore SW, Rosenberg SH, Tse C. Influence of Bcl-2 family members on the cellular response of small-cell lung cancer cell lines to ABT-737. Cancer Res. 2007;67:1176–83. doi: 10.1158/0008-5472.CAN-06-2203. [DOI] [PubMed] [Google Scholar]
- 72.Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–2. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 73.Chang J, Hsu Y, Kuo P, Kuo Y, Chiang L, Lin C. Increase of Bax/ Bcl-XL ratio and arrest of cell cycle by luteolin in immortalized human hepatoma cell line. Life Sci. 2005;76:1883–93. doi: 10.1016/j.lfs.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 74.Butt AJ, Firth SM, King MA, Baxter RC. Insulin-like growth factor-binding protein-3 modulates expression of Bax and Bcl-2 and potentiates p53-independent radiation-induced apoptosis in human breast cancer cells. J Biol Chem. 2000;275:39174–81. doi: 10.1074/jbc.M908888199. [DOI] [PubMed] [Google Scholar]