

Abstract
Here, we define dynamic reciprocity (DR) as an ongoing, bidirectional interaction amongst cells and their surrounding microenvironment. In the review, we posit that DR is especially meaningful during wound healing as the DR-driven biochemical, biophysical and cellular responses to injury play pivotal roles in regulating tissue regenerative responses. Such cell-extracellular matrix interactions not only guide and regulate cellular morphology, but cellular differentiation, migration, proliferation, and survival during tissue development, including e.g. embryogenesis, angiogenesis, as well as during pathologic processes including cancer diabetes, hypertension and chronic wound healing. Herein, we examine DR within the wound microenvironment while considering specific examples across acute and chronic wound healing. This review also considers how a number of hypotheses that attempt to explain chronic wound pathophysiology, which may be understood within the DR framework. The implications of applying the principles of dynamic reciprocity to optimize wound care practice and future development of innovative wound healing therapeutics are also briefly considered.
Keywords: dynamic reciprocity, chronic wounds, extracellular matrix
INTRODUCTION
Normal wound healing is characterized by a well-coordinated, progressive series of events designed to restore the barrier function and mechanical integrity of the skin. Like other developmental processes and tumor growth, wound healing involves interactions between cells and their microenvironment, of which the extracellular matrix (ECM) is the primary component.1–3 It is largely through these interactions that cells are directed to differentiate or dedifferentiate, proliferate or remain quiescent, and assume the architecture and function of the skin versus that of some other organ.1, 4
More than 25 years ago, it was noted that interactions between cells and the ECM occur both ways – that is, they are reciprocal.5, 6 Moreover, it was noted that these interactions were dynamic, continuously changing in response to cues from the microenvironment.5, 6 These observations were collectively termed “dynamic reciprocity” indicating the ongoing, bidirectional interactions between cells and the ECM (Figure 1).
Figure 1.

DR between cells and ECM. Cells synthesize ECM components, and also degrade and remodel ECM, the latter events occurring through the production and regulation of matrix metalloproteases (MMPs) and other enzymes. The ECM regulates cellular tension and polarity, differentiation, migration, proliferation, and survival. The ECM consists of collagen, elastin, multidomain glycoproteins (eg, fibronectin), and proteoglycans and glycosaminoglycans; the exact composition of the ECM varies by tissue and by state of the tissue (eg, intact adult tissue, healing wound, cancer, etc.)
DR conceptually encompasses many types of cell-ECM interactions, which embrace a number of fields of basic and clinical study, which include developmental biology and the pathobiology of human disease. Indeed, DR is likely to be relevant during tissue development, reparative and regenerative processes, and human pathogenesis, including embryogenesis, angiogenesis, lactogenesis, cancer, and wound healing. Within each of these basic and clinical research domains, there have been numerous reports that reflect upon the biochemical and biomechanical effects that cells and their interacting surrounding microenvironments or ECM have on tissue or organ-specific responses during human development or disease. Reciprocally responsive biochemical and mechanochemical interactions between cells and ECM during wound healing and angiogenesis have been demonstrated and discussed by a number of authors.7–17 These interactions represent integral features of DR,9, 10 which link a deepened understanding or awareness of how cell-matrix interactions modulate cellular responses to injury and wound healing in vivo.
One goal of this review is to explore the relevance of DR as it relates to the wound microenvironment. First, we briefly review the history of DR and then turn to a consideration of specific examples of DR during each stage of wound healing. It is our contention that the concept of DR also provides a framework within which to understand the processes that are disrupted in chronic wounds and how these influence subsequent interactions in the wound microenvironment. Therefore, a second goal of this review is to re-consider some hypotheses that attempt to explain chronic wound pathophysiology and how these might be understood within the framework of DR. We end with a consideration of the implications of DR for clinical wound healing and future therapeutic development.
History of Dynamic Reciprocity
The phrase “dynamic reciprocity” was coined by Bornstein and colleagues in 1982 to describe the effects of the ECM on endothelial cell function.6 They noted that endothelial cells secreted macromolecules that continually modulated cellular behavior and, presumably, this in turn influenced the identity and quantity of secreted macromolecules.6 Although, at the time, it was not known how the ECM influenced cells, they hypothesized that these macromolecules may interact with the internal cytoskeleton “to regulate cellular shape and movement and affect metabolic function by, for example, modulating ionic fluxes or protein phosphorylation/dephosphorylation.”6
In the same year, Bissell and colleagues published a review elaborating on the concept of DR and presenting a model by which the ECM could affect gene expression.5 In this model, the ECM was believed to influence cells via transmembrane receptors that interacted with the cytoskeleton to eventually alter the pattern of gene expression.5 This paper also outlined evidence demonstrating the importance of the ECM for cell shape and function, as well as maintenance of the differentiated state.5 The emphasis in Bissell’s model was that “the influence of ECM on the cell, both during the developmental process and in established tissues, appears to evolve continually.” Thus, the ECM was believed to affect the cell, which in turn altered the composition and structure of the ECM through synthesis or degradation of ECM components that then influenced the cell, and so on.5
The model that Bissell and colleagues proposed in 1982 was supported by the discovery and characterization of integrins several years later.18 Today, integrins are recognized as multi-domain receptors whose extracellular portion interacts with ECM molecules and whose intracellular portion interacts with signaling proteins (e.g., kinases) and adaptor proteins that link to the cytoskeleton (Figure 2).2, 19 These interactions in part regulate this DR through gene expression, protein synthesis, actin organization, cell polarity, differentiation, proliferation, migration, and survival. Integrin binding also modulates the signaling by several other receptor mechanisms, including syndecans, growth factors, and cytokines.20 Cells and the ECM also interact via additional mechanisms such as discoidin domain receptors, hyaluronan receptors, and cell surface proteoglycans, although the integrin pathways are the best characterized to date. Conversely, cells regulate the distribution and affinity of integrins for their matrix ligands. Interactions between cells and the ECM via integrins have been described as inside-out and outside-in, which describe their mutual influence.
Figure 2.

Dynamic and reciprocal signaling through the integrin- and growth factor receptor-rich plasma membrane. In this stylized representation, integrins (membrane-spanning proteins shown in green) bind to extracellular matrix components such as fibronectin (red “v”s) and collagen (yellow striped rods). The cytoplasmic tails of the integrin receptor directly interacts with the cytoskeleton via talin (yellow), vinculin (purple), and filamentous actin, blue). Through these dynamic protein-protein interactions, mechano-chemical signaling cascades are initiated and propagated, which modulate cell adhesion, shape, polarity, cell proliferation and migration. These reciprocally-regulated interactions can influence gene expression via effector and adaptor pathways. Molecular components, here, include members of the focal adhesion complex, including paxillin (shown in red), Crk, Cas, and the focal adhesion kinase, FAK. FAK and src can signal ‘downstream’ via linked effector pathways (e.g. shown as green, blue, and purple shapes). Integrins can also laterally interact with growth factor receptors (membrane-spanning protein shown in pink) via the MEK1 pathway (shown as purple stacked cylinders).
The concept of DR is well established in cell biology, where it continues to provide a basis for research into the mechanisms of ECM-cell interactions.1–3 This research has been instrumental in establishing the ECM as a signaling entity rather than simply an inert scaffold and support structure for cells.1, 21, 22 These non-structural properties of the ECM are best demonstrated by the functions of matricellular proteins, such as the thrombospondins, SPARC and hevin, tenascins and periostin, which influence cell function in numerous ways.3 Over the years, DR has been expanded to encompass interactions between cells and their entire microenvironment, which in addition to the ECM, includes adhesive signals, paracrine signals such as growth factors derived from neighboring cells, and systemic cues.1
The parallels between wound healing and other types of tissue development – both normal and pathological – are numerous. In particular, angiogenesis is integral to all of these processes and has been suggested as an organizing principle underlying wound healing, tumor formation, diabetic retinopathy, and selected other conditions.23 Angiogenic abnormalities are characteristic of both tumors24 and chronic wounds.25 Additionally, similar cytoskeletal mechanisms mediate epithelial cell migration in embryogenesis and normal, acute wound healing26 and both processes require the coordinated activities of cytokines and growth factors, cell-cell interactions, and cell-ECM interactions.
Dynamic Reciprocity During Normal Wound Healing
Given the parallels between wound healing and other types of tissue development, it is reasoned that wound healing is also subject to the principles of DR.5 In the following sections, we consider examples of DR that occur across the wound healing stages. Rather than providing a comprehensive review of wound healing biology, this section uses examples in an attempt to establish that cells and ECM are changed by their interactions with one another, and the orderly procession of these changes, which occur through a multitude of different signaling pathways, results in wound healing. In a subsequent section, we consider how alteration in the sequence, magnitude, or timing of these changes may contribute to stalled wound healing and/or the formation of aberrant tissue (e.g., fibrin cuffs).
Hemostasis
Hemostasis begins after the disruption of blood vessels, which leads to a series of events designed to halt blood loss. Events in this phase of healing include vasoconstriction, formation of a platelet plug, and coagulation, and involve cells responding to changes in the ECM and vice-versa.
Vascular damage leads to the extravasation of blood components and the exposure of ECM proteins such as collagen, fibronectin, laminin, and the matricellular protein thrombospondin-1.27 Platelets bind to exposed collagen via integrins of the β1 and β3 families,28 as well as to the glycoprotein (GP) VI immunoglobulin superfamily.27 This binding initiates an intracellular signaling cascade that activates platelets, causing them to degranulate and release a multitude of chemokines and other soluble mediators that increase intracellular calcium, promote reorganization of the cytoskeleton, and activate αIIbβ3 integrins via cytoskeletal proteins (Table 1; Figure 3).27
Table 1.
Selected examples of dynamic reciprocity between ECM and cells during wound healing
| Cells → | ECM → | Cells → | ECM → | Cells → | |
|---|---|---|---|---|---|
|
Hemostasis | |||||
| Platelets | Bind to exposed collagen via vWF | Platelets degranulate, release growth factors and bioactive lipids, and shift to functional fibrinogen receptors 128 (αIIbβ3 integrins) | Bind to fibrinogen | Platelet aggregation | |
|
Inflammation | |||||
| Neutrophils and monocytes bind to endothelium (selectins), integrins change from low to high affinity state, tight binding to endothelial cells, release of chemokines | Chemokines bind to heparan sulfate proteoglycans on endothelial cells | Bound chemokines stimulate expression of β1 and/or β2 integrins by neutrophils and monocytes 129 | Bind to fibronectin, collagen | Facilitates chemotaxis along cytokine gradient | |
| Monocytes | Bind to fibronectin | Increases phagocytic capacity 34 | Increased degradation of ECM components and other debris | Transition from an inflammatory phenotype (M1) to constructive phenotype (M2) | |
|
Migration and Proliferation | |||||
| Fibroblasts | Bind to fibronectin | Stimulates production of MMPs 53 | MMPs degrade certain ECM components | Enhances cell migration | |
| Endothelial cells | Bind to ECM via integrins | Integrins connected to cytoskeleton exert tension, maintain cell shape | Disruptions in ECM (e.g., degradation, increased density) alter tension and cell shape | Differentiation, proliferation, or migration (in presence of growth factors) 75 | |
| Keratinocytes | MMPs dissolve hemidesmosomes (ECM attachments), cells detach from basement membrane | Cell shape changes, migration initiated, expression of specific integrins, 109, 130 phagocytosis of ECM debris, release of MMPs 130, 131 | Deposition of fibronectin, tenascin-C, laminin-5; cells bind via integrins 130 | Guide cell migration | |
|
Contraction and Maturation | |||||
| Fibroblasts | Bind to fibronectin via αvβ5 orαvβ3 integrins 79 | Differentiate into myofibroblasts in presence of TGF-β1 (possibly from self-generated ECM) 80 | Interact with collagen via β1 integrins to contract and remodel the wound; Collagen and MMP expression modulated by mechanical tension | Myofibroblast and fibroblast levels decline in response to declining levels of growth factors and induction of CXCL2 cytokines132 | |
ECM=extracellular matrix, MMPs=matrix metalloproteases, vWF=von Willebrand factor
Figure 3.

Key cellular examples of DR at each wound healing stage. The left-most column summarizes a general mechanism that invokes DR, beginning with the binding of cell types to ECM components. This binding (adhesion) or reduced/altered adhesion then leads to changes in the cells, which in turn leads to changes in the ECM. Examples of DR are provided at each wound healing stage, using one or two cell types for the purposes of illustration.
Blood-derived fibrinogen and von Willebrand factor (VWF) then bind to activated αIIbβ3 integrins, promoting connections between platelets and the formation of a platelet plug (thrombus).27 Fibrinogen is concurrently converted into fibrin by thrombin as the final step in the coagulation cascade. Fibrin then polymerizes to form a clot that is further stabilized by factor XIII. The fibrin clot minimizes blood loss and, together with bound fibronectin, serves as a provisional matrix that incorporates adherence sites for cells, modulates cell function, and serves as a reservoir for growth factors, proteases and protease inhibitors.29
Many of the proteins released by platelets are chemoattractants and/or mitogens for neutrophils, fibroblasts, monocytes/macrophages, and smooth muscle cells (Figure 3). Among these is platelet-derived growth factor (PDGF), of which several isoforms bind to heparan sulfate proteoglycan (HSPG), heparin, and other glycosaminoglycans present on cell surfaces and in the ECM.30, 31 Heparin has been found to potentiate PDGF-α receptor phosphorylation, to increase the PDGF-induced activation of mitogen-activated protein (MAP) kinase, and to enhance chemotaxis of Chinese hamster ovary cells.32 Platelets also store and release transforming growth factor (TGF)-α and TGF -β, which affect proliferation and matrix metabolism, respectively.
Inflammation
The next phase in healing is the inflammatory phase, which is characterized by the sequential influx of immune cells that, among their other activities, remove bacteria, debris, and devitalized tissue. Platelet-derived cytokines are chemotactic for neutrophils, which in addition to their proteolytic and oxidative functions, initially upregulate factors necessary for the extravasation and chemotaxis of other immune cells and inflammatory responses.33 Monocytes bind to adhesion molecules on the luminal surface of activated endothelium and then utilize integrins to migrate from the blood into the wound and bind to ECM proteins (Table 1). ECM binding also enhances their phagocytic capacity34, resulting in increased degradation of ECM fragments (Table 1; Figure 3). Monocyte binding to ECM proteins also induces their differentiation into macrophages and upregulates the production of growth factors such as PDGF-B and TGF-α.35, 36 Activated monocytes and macrophages produce and release thrombospondin-1, which is chemotactic for macrophages37, and this effect correlates with the activated angiogenesis demonstrated by these cells.38 As this phase progresses, it is critical that macrophages remove apoptotic neutrophils, as failure to do so results in reduced release of active TGF-β1 and a subsequent reduction in myofibroblast differentiation and wound contraction.39 This function of macrophages is dependent on the binding of β2 integrin to ECM proteins.39 TGF-β also stimulates fibroblasts to synthesize collagen, fibronectin, hyaluronic acid, thrombospondins 1 and 2, tenascin-C, and other proteins,40–42, in addition to increasing the expression of integrins that bind collagen, fibronectin, and vitronectin43, 44.
Among the circulating, marrow-derived mononuclear cells that are recruited during inflammation, there are several populations of cells that express characteristics of granulation tissue such as collagen expression or endothelial growth factor receptors. The early wound infiltrate includes cells of the hematopoietic lineage (CD45+; fibrocytes) and the mesenchymal lineage (CD45−, CD44+; mesenchymal progenitor cells) that can transiently contribute to ECM production within the provisional matrix.45–47 Unlike resident (skin) fibroblasts, these cells also have significant effector functions.
Migration and Proliferation
The proliferative phase is characterized by the formation of granulation tissue – new blood vessels, macrophages, fibroblasts, and loose connective tissue10 – as well as early wound contraction and re-epithelialization.
The provisional fibrin matrix supports the ingrowth of cells by incorporating ECM proteins such as fibronectin and vitronectin, which interact with migratory cells via β1, β3, and β5 integrins. Adhesive interactions permit fibroblasts and endothelial cells to migrate toward growth factors such as PDGF that are localized in the early wound environment.29, 48 PDGF induces fibroblast proliferation and the expression of proteoglycans by fibroblasts, which, in conjunction with integrins, are required for fibroblast migration and binding to the provisional matrix.49 Furthermore, bioactive lipids modulate cellular injury and reparative responses, including fibroblast and vascular cell proliferation. Indeed, lysophospholipid-stimulated cytoskeletal responses impact cellular adhesion, migration, and contraction.50 In turn, lysophosphatidic acid interaction with ECM proteins have further been demonstrated to influence fibroblast migration rates in a laminin- and fibronectin-dependent manner. Indeed, the binding of fibroblasts to fibronectin stimulates the production of collagen, proteoglycans, and hyaluronic acid (Table 1).51,52 In addition to modifying the mechanical environment, these molecules serve a host of functions, including cell and growth factor attachment, increased cellular motility (hyaluronic acid), and interactions with one another (e.g., proteoglycans may facilitate collagen fibrillogenesis).10 Chemoattractant signals in the provisional matrix, including PDGF and TGF-β, activate cognate receptors on fibroblasts and other adherent cells. Sphingosine-1 phosphate interacts with PDGF to regulate vascular cell migration and with TGF-β to regulate MMP expression.50 This growth factor signaling requires the cooperative engagement of integrins. Hyaluronan, an early addition to the provisional matrix, is recognized by CD44 receptors on migrating cells. Fibroblast binding to newly-deposited collagen via the β1 integrin receptors facilitates migration and also stimulates the production of MMPs, which, by degrading matrix components, permit cell migration.53
Disruption of the epidermis and its association with the ECM of the basement membrane activates migration and proliferation of keratinocytes. With the MMP-driven breakdown of the hemidesmosomes, the anchor that held keratinocytes in place at basement membrane is disrupted, allowing keratinocytes to migrate.54 Unlike mesenchymal and endothelial cells that migrate through a three-dimensional matrix, polarized epithelial cells migrate as sheets from the wound edge or remnant skin appendages. In order to migrate, the leading edge of the sheet detaches from the underlying basal lamina, where hemidesmosomes bind to laminin and type IV collagen through β4 and β1 integrins, respectively.55, 56 The migrating keratinocyte relocates collagen-binding β1 integrins from the lateral membrane surface to the basal surface to permit migration out over the newly-formed granulation tissue bed. Keratinocytes do not bind to fibrin-fibrinogen because they lack αvβ3 integrins; thus fibrin is anti-adhesive for these cells.57 Instead, keratinocytes express integrin subtypes that bind to collagen I, fibronectin, tenascin-C, and vitronectin,58 which guide their migration over granulation tissue. To resupply the advancing epidermal sheet, these cells rapidly proliferate just distal to the wound margin under the influence of growth factors.
MMPs and other enzymes are important in generating active ligands. For instance, heparin-binding epidermal growth factor (Hb-EGF) is initially bound to the membranes of monocytes/macrophages, T cells, and keratinocytes, where it is released by ectodomain shedding in response to MMPs.59 Hb-EGF is abundant in acute porcine wound fluid 1–3 days after injury, where it binds to heparin or heparan sulfate and is mitogenic for fibroblasts, smooth muscle cells, and epithelial cells.59, 60 The ectodomain shedding of Hb-EGF bound to keratinocyte membranes is required for keratinocyte migration.61
In humans, capillary sprouts begin to migrate into the wound by day 4 post injury. This process depends on the degradation of existing basement membrane by MMPs and other enzymes,62 as well as on the presence of fibrin, vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF)-2, and other factors.63 Capillary sprouts initially express αvβ3 integrins that allow them to bind to fibrin-fibrinogen.63 As endothelial cells migrate into the wound, integrin expression is spatiotemporally controlled, with ECM adhesions being assembled and disassembled at various times and places.64 By analogy with epidermal migration, endothelial sprouts advancing through the newly-forming ECM switch the types of integrins they express, leading to differences in adhesion dynamics, cytoskeletal organization, and signaling pathway activation over the course of angiogenesis.64
FGF, a key player in the regulation of angiogenesis, is an example of a growth factor that must be bound to the ECM to exert its effects. FGF is made by multiple types of cells (e.g., macrophages during the inflammatory phase and fibroblasts and endothelial cells during the proliferative phase) during wound healing, but FGF interacts with heparin-like moieties in the ECM and on the plasma membrane in order to stimulate target cells throughout the phases of wound healing. Heparanase, an enzyme that cleaves heparan sulfate to liberate bound growth factors from the ECM, has been found to accelerate wound healing primarily through an enhanced angiogenic response.65 FGF molecules, either secreted from cells or liberated from the ECM by heparanase, are stabilized by heparan sulfate fragments and escorted to the FGF receptors by cell surface HSPG to form a tetrameric complex leading to receptor dimerization and signaling (Figure 3).66 Prevention of FGF-2 binding to heparan sulfate prevents its ability to support fibroblast growth and reduces binding to its cell-surface receptors.67, 68
MMPs also liberate angiogenic factors from the ECM proteins to which they are bound, and they unmask cryptic sites in ECM components or generate bioactive ECM fragments that can bind to cells via growth factor or other receptors and initiate signaling pathways.62, 69–71 Endothelial cells express αvβ3 integrin, which binds to fibrin, fibronectin, vitronectin, and vWF, and mediates endothelial cell adhesion and migration.63 Matrix components regulate the temporal sequence of endothelial cell proliferation, with laminin inducing a higher proliferation rate than collagen IV.72 PDGF-B is also mitogenic and chemotactic for pericytes – cells that surround capillary tubes where they promote new capillary growth and uniform structure. As angiogenesis progresses, endothelial cells form tubes, which involves the recruitment of pericytes in response to endothelial cell-derived FGF-2 and PDGF.73 Thrombospondins are potent modulators of angiogenesis.74
Dynamic and reciprocal interactions between cells and ECM are evident not only in the biochemistry of the wound microenvironment, but also in the biomechanical interactions that occur at the cell-ECM interface.75 Cell shape is mediated by the tension generated when cells anchor to ECM via integrins, which are connected to the cytoskeleton. Cell tension and matrix elasticity interact to regulate cell differentiation. In combination with growth factor gradients, cell shape and tension generated through cell-ECM adhesions are critical determinants of cell migration.76 Under homeostatic conditions in adults, the dynamic interactions between cell traction forces and resistance to these forces by the ECM strike a balance that maintains cell shape.77 However, during tissue development, including wound repair, degradation of ECM disrupts mechanical tension and consequently cell shape, influencing cellular proliferation and migration.75 The altered cell shape caused by changes in the ECM then feeds back to modify cell behaviors such as growth, differentiation, motility, etc.75
Contraction and Remodeling
During the proliferative phase, fibroblasts that are initially bound to fibronectin via αvβ5 or αvβ3 integrins migrate and proliferate in response to PDGF, producing an ECM that is relatively sparse but enriched in hyaluronan and with relatively higher levels of type III collagen.10 Under the influence of TGF-β and connective tissue growth factor (CTGF), collagen I becomes the predominant fibrous protein. In the continued presence of TGF-β1, a fraction of the maturing granulation tissue further differentiates into myofibroblasts (Table 1).78, 79 It has been proposed that integrin-mediated myofibroblast contraction directly activates latent TGF-β1 from self-generated ECM, thereby restricting the progression of fibrosis to the area of mechanical stress.80 Lysophosophatidic acid also enhances wound contraction via activation of G-protein-linked receptors and activation of the Rho and ROK pathways, eventually leading to increased phosphorylation of myosin light chain by impacting myosin phosphatase activity and actomyosin interactions.50, 81
One example of ECM-growth factor binding is the interaction between TGF-β and the protein components of decorin and betaglycan. TGF- β1 induces the synthesis of decorin and biglycan.82 Binding of TGF-β1 to decorin, betaglycan, and biglycan inhibits its activity, suggesting a negative feedback loop 83. In addition, the latent TGF-β binding proteins are closely related to the fibrillins, both of which can affect TGF-β bioavailability. TGF-β1 and 2 are involved in scar formation and TGF- β type II receptors are important in wound contraction.84
Myofibroblasts interact with collagen bundles and growth factors to contract the wound.85 Macrophages, endothelial cells, and epidermal cells release MMPs that remodel the early matrix, while myofibroblasts replace it with the stronger collagen type I (Figure 3).86 This newly deposited collagen is organized along the stress lines of the skin and shows increased tensile strength. Fibroblasts and myofibroblasts appear to continue to accumulate collagen until the compliance of the ECM reaches mechanical equilibrium with surrounding tissue. In hypertrophic scarring, there is an inappropriate response to tensional forces, leading to excess matrix deposition. The slow remodeling of collagen, including the formation of bundles and cross-links, progresses over a period of months to form a scar.
What Are the Problems In Chronic Wounds and How Can They Be Understood Within the Framework of Dynamic Reciprocity?
While most wounds heal in a timely and orderly pattern, the process can be stalled or halted in individuals with a variety of diseases or conditions, including diabetes mellitus, venous or arterial insufficiency, and immunosuppression, or following a period of immobility that leads to prolonged pressure. Chronic wounds may develop in these cases, potentially leading to pain, immobility, hospitalization, amputation, or even death.87, 88
Viewed in the context of DR, non-healing wounds fail to exhibit the normal sequence of actions and reactions between cells and ECM that characterizes acute wound healing. This is not to say that DR does not apply to chronic wounds; on the contrary, the highly organized nature of the ECM and cells evident in fibrin cuffs of venous stasis ulcers has led to the suggestion that this tissue is actively synthesized,89 but perhaps inappropriately so. Additionally, the normal, sequential pattern of these interactions does not occur, and the disruption of these interactions – potentially at a variety of different points – leads to downstream effects on other cell-ECM interactions that ultimately delay or preclude healing.
In the following sections, we consider some of the disruptions in chronic wounds that may relate to non-healing and examine how they may fit into the framework of DR.
Disruptions due to disease state
No single hypothesis has emerged as a comprehensive explanation for why chronic wounds such as diabetic foot or venous leg ulcers fail to heal in a timely fashion. In fact, it has been suggested that more than 100 physiologic factors contribute to wound healing deficits in individuals with diabetes.90 Diabetes-induced alterations in progenitor cell recruitment and homing or those that result from excessive nonenzymatic glycation of matrix proteins, would be expected to alter the dynamic and reciprocal interactions between cells and ECM that are necessary for wound healing. These changes also would be expected to have reverberating downstream effects that would interfere with the normal sequential interactions that characterize wound healing, thereby preventing timely wound closure. While many processes lead to the occurrence of chronic wounds, such wounds seem to share a number of characteristics regardless of cause. These include changes in cellular responsiveness, elevated proteolytic environments, and microvascular abnormalities. Table 2 lists some of the disease-related abnormalities observed in individuals with diabetes or venous insufficiency and/or animal models of these conditions, any or all of which may contribute to delayed wound healing.
Table 2.
Selected Disease-Related Abnormalities That May Contribute to Delayed Healing of Diabetic and Venous Ulcers
| Disease | Documented Abnormalities |
|---|---|
| Diabetes |
|
| Venous Insufficiency |
|
ECM=extracellular matrix, IL=interleukin
Elevated proteases
Following observations of elevated levels of various MMPs in chronic wound fluid, it was hypothesized that these enzymes could be causing excessive degradation of ECM proteins and chronic tissue turnover that prevented the wounds from healing. In their 1993 study, Wysocki and colleagues showed that levels of MMP-2 and MMP-9 were increased by 5- to 10-fold in acute wounds, but were increased by another 5- to 10-fold in chronic wounds.91 Both activated enzymes and proenzyme species were present. Numerous subsequent studies have documented the presence of elevated levels of various MMPs and decreased levels of tissue inhibitors of metalloproteases (TIMPs) in chronic wounds, or an imbalance between the levels of MMPs and TIMPs.92–95 Other studies have found a correlation between elevated MMP levels and non-healing.95–98
In addition to degrading ECM, elevated levels of proteases would also be expected to degrade growth factors. The evidence indicates that chronic but not acute wound fluid rapidly degrades PDGF and TGF-β1 – effects that appear to be due to the activity of neutrophil elastase.99
The process of DR would predict that excessive degradation of the ECM would deprive cells of attachment sites and signals required for migration, differentiation, and proliferation. These consequences would interfere with the cell’s response to the normal ECM interactions (e.g., increased or decreased protein synthesis), which would, in turn, prevent changes in the matrix composition needed for wound healing to progress. The degradation of growth factors by excess MMPs would also deprive the cell of critical signals. Finally, high levels of MMPs may be expected to cleave proteins with cryptic signaling sequences or shed ectodomains to generate active forms of some membrane-bound precursor proteins such as Hb-EGF. The flooding of these signaling components into the wound microenvironment, theoretically followed by their rapid degradation, may further interfere with the carefully orchestrated interactions of cells with their microenvironment that normally causes wound healing to progress.
Biofilm formation
A biofilm is defined as a structured community of bacterial cells enclosed in a self-produced polymeric matrix that is adherent to an inert or living surface.100 As a result of their structured community, biofilms are largely resistant to innate immune mechanisms and antimicrobial agents.101 Furthermore, biofilms provide a continuous source of inflammation101, 102—one of the hallmarks of chronic wounds. Few studies have evaluated the extent of biofilm formation in chronic wounds. In one small study using molecular sequence analyses, 30 of 50 (60%) of chronic venous ulcers contained biofilm (although 40% did not); in contrast, only 1 of 16 (6%) acute wounds contained biofilm.103
From the perspective of the process of DR, the formation of wound biofilm may be expected to interfere with the effects of leukocytes and their subsequent interactions with the ECM that would normally lead to differentiation and release of chemokines and growth factors. For instance, because neutrophils cannot engulf and digest bacteria within the biofilm, they release large quantities of proinflammatory cytokines that lead to chronic inflammation.101 Biofilm also inhibits the chemotaxis and degranulation of neutrophils104 and phagocytosis by macrophages.105 Inhibition of these processes and massive release of inflammatory mediators would be expected to interfere with the sequential series of events that characterize normal wound healing, resulting in aberrant leukocyte-ECM interactions.
Over the past few years, increasing attention has focused on the hypothesis that chronic wounds may fail to heal because they have developed biofilms,101, 102 a concept that is supported by preclinical research.106 However, evidence for a causal relationship between biofilm formation and delayed wound healing in humans is either inconclusive107 or requires further analysis. For example, it will be important for wound care practitioners and wound repair scientists to clarify whether biofilms actually cause non-healing, chronic wounds or whether the chronic wound microenvironment renders all of these wounds or a select fraction thereof to become highly susceptible or conducive for biofilm development. Clearly, more work will be required before any causal relationships between bacterial biofilms and chronic wound are established.
Integrin switching
Over the course of wound healing, cells are stimulated to change the integrins they express, allowing them to bind to different ECM components or different parts of a single component that variously foster migration, differentiation, polarity, survival, and other properties (Figure 4).108 As in development and morphogenesis, this process is clearly illustrated during wound healing, when successive interactions between matrix receptors and matrix ligands exert a critical influence on cell shape and movement.109 In the resting epidermis, basal keratinocytes are anchored to laminin332 in the underlying basal lamina through α6β4 integrin. Basal keratinocytes maintain lateral associations, in part, through α2β1 and α3β1 integrins, which interact with E-cadherin. After injury, the area beyond the intact epithelial sheet has no basal lamina. It is initially covered by fibrin and plasma fibronectin, and subsequently by collagen and cellular fibronectin, as the provisional matrix matures into granulation tissue.
Figure 4.

Integrin switching helps mediate keratinocyte migration across wounded skin. In this graphic representation, keratinocytes are depicted as ovals containing the major integrin subunits they express, and the extracellular matrix is depicted as elongated brown cylinders. Intact keratinocytes bound to basement membrane are shown on the left and migrating keratinocytes at the wound edge are shown on the right. MMPs enable migration by breaking down the underlying basal lamina at the leading edge of the keratinocyte sheet, where the cells assume a flattened shape and express an array of integrins that permits migration across the newly-formed granulation tissue. The leading epithelial cells rearrange their distribution of β1 integrins to engage with type I collagen below the damaged/absent basement membrane.
As basal cells at the wound margin assume a migratory phenotype, several key processes occur. The β1 integrins shift from the lateral to the basal surface of the cell, where interactions with collagen occur. There is an increase in β1 integrin and αv integrins that can interact with fibronectin and other adhesive proteins.109 Furthermore, there is an induction of the filopodial expression of αvβ6, which also participates in the activation of TGF-β.110, 111 Keratinocytes at the leading edge of the wound beyond the limits of the basal lamina activate the expression of MMP-9 and MMP-10 (stromelysin 2), whereas the proximal proliferating population that supplies the leading edge expresses MMP-3 (stromelysin 1) and a more distal, intact population expresses MMP-28/epilysin.112, 113 These findings indicate that different MMPs are required for mobilization from the basal lamina versus advancement across the granulation tissue.112, 113 Keratinocytes appear to require the ability to cleave collagen with MMP-1/MMP-13 at their trailing membrane edge in order to migrate across this substrate.114
As basal lamina components reappear under the advancing epithelial front, αvβ1 and αvβ6 expression disappears. Adherence to laminin resumes, and β1 integrins resume a lateral location. Concurrently, underlying granulation tissue expresses MT1-MMP, an important activator of other MMPs and TIMPs, which modulate MMP activity.54
During angiogenesis, endothelial mobilization similarly requires disengagement of α6β4integrin from laminin in the endothelial basement membrane. The engagement of the αvβ5 and α5β1 integrins by their insoluble arginine-glycine-aspartic acid (RGD) ligands is required for the execution of the angiogenic signals from VEGF and FGF-2, respectively.
Integrin switching is also evident in fibroblasts. For instance, fibroblasts expressing α5β1 or αvβ1 integrins both adhere to fibronectin, which leads to their rapid movement.64 However, those expressing the former integrin migrate in a random and non-persistent fashion, whereas those expressing the latter integrin migrate in a highly persistent fashion.64
Deficits in integrin switching would be expected to interfere dramatically with wound healing,108 and may be a reason that chronic wounds fail to heal. Several studies have found altered integrin expression in chronic wounds.115, 116 In the mouse, deletion of α1 or α2 integrins produces subtle effects on wound healing. This may reflect the redundancy of the collagen-binding integrins. However, there are numerous factors that could lead to defects in integrin switching in chronic wounds, and thus this hypothesis may be consistent with a variety of other potential explanations.
Clearly, deficits in integrin switching would interfere with the DR that characterizes the process of wound healing. Lack of appropriate spatial and temporal integrin presentation and switching would interfere with cell-ECM interactions that guide cell migration and other processes. Changes in the integrin repertoire can lead to changes in the ECM,19 which would be expected to further influence the dynamic and reciprocal interactions in the wound.
Alterations in specific proteins (other than proteases)
Alterations in many other proteins have been noted in chronic wounds. These include decreased levels of intact fibronectin, an increase in fibronectin degradation products,117 a loss of type II TGF-β receptors on fibroblasts,118 reduced levels of PDGF,119 and dramatic down regulation of keratins in epithelial cells.120 Chronic wounds also show upregulation of β6 integrins, with overexpression in mice leading to spontaneous chronic wounds in approximately 20% of animals.116 Venous or diabetic ulcers that heal slowly show decreased levels of intact fibronectin (Figure 5),89 decreased levels of type II TGF-β receptors,118 increased levels of inflammatory cytokines, and decreased levels of PDGF.119
Figure 5.
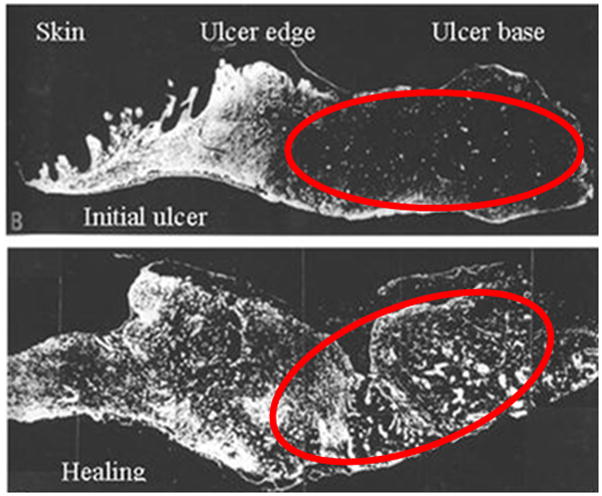
Degradation of fibronectin in base of chronic venous ulcer (top photo) reverses with initiation of healing (bottom photo). Reprinted from Am J Pathol 1992, 141:1085–109589 with permission from the American Society for Investigative Pathology.
In most of these cases, it is not known whether the alterations cause or are caused by changes in the wound microenvironment. Also in most cases, the alterations in these proteins may be compatible with one or more hypotheses as to why chronic wounds occur and/or why they fail to heal. The alterations in individual proteins described in this section may be a glimpse into disruptions that occur at one slice of time in the chronic wound – a piece of the overall disrupted sequence of DR in wound healing. Because alterations in each of these proteins may be expected to have downstream effects, it is unclear whether replacement of any single protein would be sufficient to remedy the slowed healing in chronic wounds. That is, we may be unable to restore the normal pattern of DR with the addition or deletion of any single component that is altered in cells or extracellular fluid of chronic wounds. This hypothesis is supported by the variable effects of growth factors on chronic wounds.
Clinical Manifestations
Given the above data suggesting that normal DR is disrupted in many chronic wounds, various attempts have been made to re-set the “normal” processes. For example, some efforts to remove all or part of the ulcer through debridement to restore normality have been suggested,121 although the quality of the data that supports this approach has been questioned.122 Recent work proposes that intrinsic factors create a geometry of the wound as exemplified by biopsies taken from the edge of venous ulcers that heal back to their original position.123 Cytokines, tissue, and cell therapies have been successful in improving healing in various wound types,124–127 but, overall, results have been moderate.
What Are the Implications of Dynamic Reciprocity for Chronic Wound Healing and Future Therapeutic Development?
Multiple biochemical and structural abnormalities have been documented in chronic wounds.89–91, 117 DR may help us understand how these abnormalities fit together and how disruptions in one part of the wound healing process may lead to disruptions in subsequent interactions that ultimately prevent chronic wounds from healing. That is, understanding how the sequential changes in the ECM lead to specific changes in cells that then lead to alterations in the ECM and so forth, forces us to think about how defects in the early stages of wound healing may have downstream effects that eventually preclude wound closure.
Viewed this way, many of the biochemical alterations noted in chronic wounds may be responses to lack of adhesion to (or detachment from) an ECM of specific structure and composition at the right time in the wound healing sequence. Of course, this may be due to the inability of cells to generate the ECM or ECM attachment sites (integrins or other receptors) appropriate to the particular point in the healing process. Even the over-production of MMPs in chronic wounds could be viewed as a reaction to inadequate attachment of cells to the specific variant of the ECM needed at that point, which would naturally maintain appropriate levels of enzymes. Alternately, the excess MMPs could be an attempt by cells to degrade ECM that is not matched to the expressed cellular adhesion sites. Ultimately, a better understanding of the dynamic, reciprocal processes that take place during wound healing has the potential to influence the development of improved diagnostics, as well as therapeutics.
In this article, we have used the phrase “dynamic reciprocity” as a point of departure to explain the pathophysiology of chronic wounds. However, it is important to recognize that DR does not, in itself, explain the pathophysiology of the complex phenomena and factors that result in delayed healing. Instead, we view this concept as a working construct that provides a framework within which to interpret data. It brings together discrete observations from different fields and will hopefully stimulate ideas as to why chronic wounds don’t heal.
An example of the data that may be integrated under the rubric of DR is the interface between biochemical and biomechanical cell-ECM interactions. In addition to the widely appreciated biochemical interactions between cells and ECM, biomechanical interactions are increasingly recognized as key determinants of cell shape, polarity, and tissue architecture, ultimately influencing cell differentiation, proliferation, motility, and survival.75 An understanding of the combined influence of biomechanical and biochemical factors may be a cornerstone to the development of future tissue replacement products.
Acknowledgments
The authors acknowledge the professional writing assistance of Mary Ann Chapman, PhD, in the preparation of this article; the development of graphics by Mike Austin; and the financial support provided by Healthpoint, Ltd. Grant support for the authors is as follows: GSS: NIH EY05587; JMD: NIH AG06528, AR056138, DK065656, and the Department of Veterans Affairs; IMH: NIH NEI EY15125 and NIH NEI EY19533.
List of abbreviations
- CTGF
connective tissue growth factor
- ECM
extracellular matrix
- FGF
fibroblast growth factor
- GP
glycoprotein
- Hb-EGF
heparin-binding epidermal growth factor
- HSPG
heparan sulfate proteoglycan
- IL
interleukin
- MAP
mitogen-activated protein
- MMP
matrix metalloprotease
- PDGF
platelet derived growth factor
- RGD
arginine-glycine-aspartic acid
- TGF
transforming growth factor
- TIMP
tissue inhibitor of metalloprotease
- VEGF
vascular endothelial growth factor
- VWF
von Willebrand factor
References
- 1.Nelson CM, Bissell MJ. Of extracellular matrix, scaffolds, and signaling: tissue architecture regulates development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2006;22:287–309. doi: 10.1146/annurev.cellbio.22.010305.104315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–9. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bornstein P, Sage EH. Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol. 2002;14:608–16. doi: 10.1016/s0955-0674(02)00361-7. [DOI] [PubMed] [Google Scholar]
- 4.Xu R, Boudreau A, Bissell MJ. Tissue architecture and function: dynamic reciprocity via extra- and intra-cellular matrices. Cancer Metastasis Rev. 2009;28:167–76. doi: 10.1007/s10555-008-9178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bissell MJ, Hall HG, Parry G. How does the extracellular matrix direct gene expression? J Theor Biol. 1982;99:31–68. doi: 10.1016/0022-5193(82)90388-5. [DOI] [PubMed] [Google Scholar]
- 6.Bornstein P, McPherson J, Sage H. Synthesis and secretion of structural macromolecules by endothelial cells in culture. In: Nossel HL, Vogel HJ, editors. Pathobiology of the Endothelial Cell. New York: Academic Press; 1982. pp. 215–28. [Google Scholar]
- 7.Sage EH, Bornstein P. Extracellular proteins that modulate cell-matrix interactions. SPARC, tenascin, and thrombospondin. J Biol Chem. 1991;266:14831–4. [PubMed] [Google Scholar]
- 8.Raghow R. The role of extracellular matrix in postinflammatory wound healing and fibrosis. Faseb J. 1994;8:823–31. doi: 10.1096/fasebj.8.11.8070631. [DOI] [PubMed] [Google Scholar]
- 9.Clark RA. Basics of cutaneous wound repair. J Dermatol Surg Oncol. 1993;19:693–706. doi: 10.1111/j.1524-4725.1993.tb00413.x. [DOI] [PubMed] [Google Scholar]
- 10.Clark RA. Biology of dermal wound repair. Dermatol Clin. 1993;11:647–66. [PubMed] [Google Scholar]
- 11.Grinnell F. Fibroblast biology in three-dimensional collagen matrices. Trends Cell Biol. 2003;13:264–9. doi: 10.1016/s0962-8924(03)00057-6. [DOI] [PubMed] [Google Scholar]
- 12.Eckes B, Nischt R, Krieg T. Cell-matrix interactions in dermal repair and scarring. Fibrogenesis Tissue Repair. 2010;3:4. doi: 10.1186/1755-1536-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–63. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 14.Herman IM. Extracellular matrix-cytoskeletal interactions in vascular cells. Tissue Cell. 1987;19:1–19. doi: 10.1016/0040-8166(87)90052-8. [DOI] [PubMed] [Google Scholar]
- 15.Young WC, Herman IM. Extracellular matrix modulation of endothelial cell shape and motility following injury in vitro. J Cell Sci. 1985;73:19–32. doi: 10.1242/jcs.73.1.19. [DOI] [PubMed] [Google Scholar]
- 16.Lee S, Zeiger A, Maloney JM, Kotecki M, Van Vliet KJ, Herman IM. Pericyte actomyosin-mediated contraction at the cell–material interface can modulate the microvascular niche. J Physics: Condensed Matter. 2010;22:1941–15. doi: 10.1088/0953-8984/22/19/194115. [DOI] [PubMed] [Google Scholar]
- 17.Kotecki M, Zeiger AS, Van Vliet KJ, Herman IM. Calpain- and talin-dependent control of microvascular pericyte contractility and cellular stiffness. Microvasc Res. 2010;80:339–48. doi: 10.1016/j.mvr.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamkun JW, DeSimone DW, Fonda D, Patel RS, Buck C, Horwitz AF, Hynes RO. Structure of integrin, a glycoprotein involved in the transmembrane linkage between fibronectin and actin. Cell. 1986;46:271–82. doi: 10.1016/0092-8674(86)90744-0. [DOI] [PubMed] [Google Scholar]
- 19.Berrier AL, Yamada KM. Cell-matrix adhesion. J Cell Physiol. 2007;213:565–73. doi: 10.1002/jcp.21237. [DOI] [PubMed] [Google Scholar]
- 20.Streuli CH, Akhtar N. Signal co-operation between integrins and other receptor systems. Biochem J. 2009;418:491–506. doi: 10.1042/BJ20081948. [DOI] [PubMed] [Google Scholar]
- 21.MacNeil S. What role does the extracellular matrix serve in skin grafting and wound healing? Burns. 1994;20 (Suppl 1):S67–70. doi: 10.1016/0305-4179(94)90094-9. [DOI] [PubMed] [Google Scholar]
- 22.Nelson CM, Bissell MJ, Of S. What role does the extracellular matrix, scaffolds, serve in skin grafting and signaling: tissue architecture regulates development, homeostasis, and cancerwound healing? Annu Rev Cell Dev BiolBurns 2006. 1994;2220(Suppl 1):287-309S67–70. doi: 10.1146/annurev.cellbio.22.010305.104315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273–86. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 24.Papetti M, Herman IM. Mechanisms of normal and tumor-derived angiogenesis. Am J Physiol Cell Physiol. 2002;282:C947–70. doi: 10.1152/ajpcell.00389.2001. [DOI] [PubMed] [Google Scholar]
- 25.Falanga V. Wound healing and its impairment in the diabetic foot. Lancet. 2005;366:1736–43. doi: 10.1016/S0140-6736(05)67700-8. [DOI] [PubMed] [Google Scholar]
- 26.Martin P, Parkhurst SM. Parallels between tissue repair and embryo morphogenesis. Development. 2004;131:3021–34. doi: 10.1242/dev.01253. [DOI] [PubMed] [Google Scholar]
- 27.Rivera J, Lozano ML, Navarro-Nunez L, Vicente V. Platelet receptors and signaling in the dynamics of thrombus formation. Haematologica. 2009;94:700–11. doi: 10.3324/haematol.2008.003178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nieswandt B, Varga-Szabo D, Elvers M. Integrins in platelet activation. J Thromb Haemost. 2009;7 (Suppl 1):206–9. doi: 10.1111/j.1538-7836.2009.03370.x. [DOI] [PubMed] [Google Scholar]
- 29.Clark RA. Fibrin is a many splendored thing. J Invest Dermatol. 2003;121:xxi–xxii. doi: 10.1046/j.1523-1747.2003.12575.x. [DOI] [PubMed] [Google Scholar]
- 30.Garcia-Olivas R, Hoebeke J, Castel S, Reina M, Fager G, Lustig F, Vilaró S. Differential binding of platelet-derived growth factor isoforms to glycosaminoglycans. Histochem Cell Biol. 2003;120:371–82. doi: 10.1007/s00418-003-0576-6. [DOI] [PubMed] [Google Scholar]
- 31.Zafiropoulos A, Fthenou E, Chatzinikolaou G, Tzanakakis GN. Glycosaminoglycans and PDGF signaling in mesenchymal cells. Connect Tissue Res. 2008;49:153–6. doi: 10.1080/03008200802148702. [DOI] [PubMed] [Google Scholar]
- 32.Rolny C, Spillmann D, Lindahl U, Claesson-Welsh L. Heparin amplifies platelet-derived growth factor (PDGF)- BB-induced PDGF alpha -receptor but not PDGF beta -receptor tyrosine phosphorylation in heparan sulfate-deficient cells. Effects on signal transduction and biological responses. J Biol Chem. 2002;277:19315–21. doi: 10.1074/jbc.M111805200. [DOI] [PubMed] [Google Scholar]
- 33.Theilgaard-Monch K, Knudsen S, Follin P, Borregaard N. The transcriptional activation program of human neutrophils in skin lesions supports their important role in wound healing. J Immunol. 2004;172:7684–93. doi: 10.4049/jimmunol.172.12.7684. [DOI] [PubMed] [Google Scholar]
- 34.Brown EJ, Goodwin JL. Fibronectin receptors of phagocytes. Characterization of the Arg-Gly-Asp binding proteins of human monocytes and polymorphonuclear leukocytes. J Exp Med. 1988;167:777–93. doi: 10.1084/jem.167.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shaw RJ, Doherty DE, Ritter AG, Benedict SH, Clark RA. Adherence-dependent increase in human monocyte PDGF(B) mRNA is associated with increases in c-fos, c-jun, and EGR2 mRNA. J Cell Biol. 1990;111:2139–48. doi: 10.1083/jcb.111.5.2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haskill S, Johnson C, Eierman D, Becker S, Warren K. Adherence induces selective mRNA expression of monocyte mediators and proto-oncogenes. J Immunol. 1988;140:1690–4. [PubMed] [Google Scholar]
- 37.Agah A, Kyriakides TR, Lawler J, Bornstein P. The lack of thrombospondin-1 (TSP1) dictates the course of wound healing in double-TSP1/TSP2-null mice. Am J Pathol. 2002;161:831–9. doi: 10.1016/S0002-9440(10)64243-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DiPietro LA, Polverini PJ. Angiogenic macrophages produce the angiogenic inhibitor thrombospondin 1. Am J Pathol. 1993;143:678–84. [PMC free article] [PubMed] [Google Scholar]
- 39.Sindrilaru A, Peters T, Schymeinsky J, Oreshkova T, Wang H, Gompf A, Mannella F, Wlaschek M, Sunderkötter C, Rudolph KL, Walzog B, Bustelo XR, Fischer KD, Scharffetter-Kochanek K. Wound healing defect of Vav3−/− mice due to impaired {beta}2-integrin-dependent macrophage phagocytosis of apoptotic neutrophils. Blood. 2009;113:5266–76. doi: 10.1182/blood-2008-07-166702. [DOI] [PubMed] [Google Scholar]
- 40.Roberts AB, Heine UI, Flanders KC, Sporn MB. Transforming growth factor-beta. Major role in regulation of extracellular matrix. Ann N Y Acad Sci. 1990;580:225–32. doi: 10.1111/j.1749-6632.1990.tb17931.x. [DOI] [PubMed] [Google Scholar]
- 41.Ignotz RA, Massague J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 1986;261:4337–45. [PubMed] [Google Scholar]
- 42.Varga J, Rosenbloom J, Jimenez SA. Transforming growth factor beta (TGF beta) causes a persistent increase in steady-state amounts of type I and type III collagen and fibronectin mRNAs in normal human dermal fibroblasts. Biochem J. 1987;247:597–604. doi: 10.1042/bj2470597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ignotz RA, Heino J, Massague J. Regulation of cell adhesion receptors by transforming growth factor-beta. Regulation of vitronectin receptor and LFA-1. J Biol Chem. 1989;264:389–92. [PubMed] [Google Scholar]
- 44.Ignotz RA, Massague J. Cell adhesion protein receptors as targets for transforming growth factor-beta action. Cell. 1987;51:189–97. doi: 10.1016/0092-8674(87)90146-2. [DOI] [PubMed] [Google Scholar]
- 45.Chesney J, Metz C, Stavitsky AB, Bacher M, Bucala R. Regulated production of type I collagen and inflammatory cytokines by peripheral blood fibrocytes. J Immunol. 1998;160:419–25. [PubMed] [Google Scholar]
- 46.Li H, Fu X, Ouyang Y, Cai C, Wang J, Sun T. Adult bone-marrow-derived mesenchymal stem cells contribute to wound healing of skin appendages. Cell Tissue Res. 2006;326:725–36. doi: 10.1007/s00441-006-0270-9. [DOI] [PubMed] [Google Scholar]
- 47.Spaeth EL, Dembinski JL, Sasser AK, Watson K, Klopp A, Hall B, Andreeff M, Marini F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS One. 2009;4:e4992. doi: 10.1371/journal.pone.0004992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Greiling D, Clark RA. Fibronectin provides a conduit for fibroblast transmigration from collagenous stroma into fibrin clot provisional matrix. J Cell Sci. 1997;110 (Pt 7):861–70. doi: 10.1242/jcs.110.7.861. [DOI] [PubMed] [Google Scholar]
- 49.Clark RA, Lin F, Greiling D, An J, Couchman JR. Fibroblast invasive migration into fibronectin/fibrin gels requires a previously uncharacterized dermatan sulfate-CD44 proteoglycan. J Invest Dermatol. 2004;122:266–77. doi: 10.1046/j.0022-202X.2004.22205.x. [DOI] [PubMed] [Google Scholar]
- 50.Watterson KR, Lanning DA, Diegelmann RF, Spiegel S. Regulation of fibroblast functions by lysophospholipid mediators: potential roles in wound healing. Wound Repair Regen. 2007;15:607–16. doi: 10.1111/j.1524-475X.2007.00292.x. [DOI] [PubMed] [Google Scholar]
- 51.McDonald JA, Kelley DG, Broekelmann TJ. Role of fibronectin in collagen deposition: Fab’ to the gelatin-binding domain of fibronectin inhibits both fibronectin and collagen organization in fibroblast extracellular matrix. J Cell Biol. 1982;92:485–92. doi: 10.1083/jcb.92.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McDonald JA. Fibronectin: a primitive matrix. In: Clark RAF, Henson PM, editors. The Molecular and Cellular Biology of Wound Repair. New York: Plenum Press; 1988. pp. 405–26. [Google Scholar]
- 53.Steffensen B, Hakkinen L, Larjava H. Proteolytic events of wound-healing--coordinated interactions among matrix metalloproteinases (MMPs), integrins, and extracellular matrix molecules. Crit Rev Oral Biol Med. 2001;12:373–98. doi: 10.1177/10454411010120050201. [DOI] [PubMed] [Google Scholar]
- 54.Chen P, Parks WC. Role of matrix metalloproteinases in epithelial migration. J Cell Biochem. 2009;108:1233–43. doi: 10.1002/jcb.22363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Russell AJ, Fincher EF, Millman L, Smith R, Vela V, Waterman EA, Dey CN, Guide S, Weaver VM, Marinkovich MP. Alpha 6 beta 4 integrin regulates keratinocyte chemotaxis through differential GTPase activation and antagonism of alpha 3 beta 1 integrin. J Cell Sci. 2003;116:3543–56. doi: 10.1242/jcs.00663. [DOI] [PubMed] [Google Scholar]
- 56.Zhang ZG, Bothe I, Hirche F, Zweers M, Gullberg D, Pfitzer G, Krieg T, Eckes B, Aumailley M. Interactions of primary fibroblasts and keratinocytes with extracellular matrix proteins: contribution of alpha2beta1 integrin. J Cell Sci. 2006;119:1886–95. doi: 10.1242/jcs.02921. [DOI] [PubMed] [Google Scholar]
- 57.Kubo M, Van de Water L, Plantefaber LC, Mosesson MW, Simon M, Tonnesen MG, Taichman L, Clark RA. Fibrinogen and fibrin are anti-adhesive for keratinocytes: a mechanism for fibrin eschar slough during wound repair. J Invest Dermatol. 2001;117:1369–81. doi: 10.1046/j.0022-202x.2001.01551.x. [DOI] [PubMed] [Google Scholar]
- 58.Clark RA, Ashcroft GS, Spencer MJ, Larjava H, Ferguson MW. Re-epithelialization of normal human excisional wounds is associated with a switch from alpha v beta 5 to alpha v beta 6 integrins. Br J Dermatol. 1996;135:46–51. [PubMed] [Google Scholar]
- 59.Raab G, Klagsbrun M. Heparin-binding EGF-like growth factor. Biochim Biophys Acta. 1997;1333:F179–99. doi: 10.1016/s0304-419x(97)00024-3. [DOI] [PubMed] [Google Scholar]
- 60.Marikovsky M, Breuing K, Liu PY, Eriksson E, Higashiyama S, Farber P, Abraham J, Klagsbrun M. Appearance of heparin-binding EGF-like growth factor in wound fluid as a response to injury. Proc Natl Acad Sci U S A. 1993;90:3889–93. doi: 10.1073/pnas.90.9.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tokumaru S, Higashiyama S, Endo T, Nakagawa T, Miyagawa JI, Yamamori K, Hanakawa Y, Ohmoto H, Yoshino K, Shirakata Y, Matsuzawa Y, Hashimoto K, Taniguchi N. Ectodomain shedding of epidermal growth factor receptor ligands is required for keratinocyte migration in cutaneous wound healing. J Cell Biol. 2000;151:209–20. doi: 10.1083/jcb.151.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lafleur MA, Handsley MM, Edwards DR. Metalloproteinases and their inhibitors in angiogenesis. Expert Rev Mol Med. 2003;5:1–39. doi: 10.1017/S1462399403006628. [DOI] [PubMed] [Google Scholar]
- 63.Tonnesen MG, Feng X, Clark RA. Angiogenesis in wound healing. J Investig Dermatol Symp Proc. 2000;5:40–6. doi: 10.1046/j.1087-0024.2000.00014.x. [DOI] [PubMed] [Google Scholar]
- 64.Truong H, Danen EH. Integrin switching modulates adhesion dynamics and cell migration. Cell Adh Migr. 2009;3:179–81. doi: 10.4161/cam.3.2.8036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zcharia E, Zilka R, Yaar A, Yacoby-Zeevi O, Zetser A, Metzger S, Sarid R, Naggi A, Casu B, Ilan N, Vlodavsky I, Abramovitch R. Heparanase accelerates wound angiogenesis and wound healing in mouse and rat models. FASEB J. 2005;19:211–21. doi: 10.1096/fj.04-1970com. [DOI] [PubMed] [Google Scholar]
- 66.Schlessinger J, Plotnikov AN, Ibrahimi OA, Eliseenkova AV, Yeh BK, Yayon A, Linhardt RJ, Mohammadi M. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol Cell. 2000;6:743–50. doi: 10.1016/s1097-2765(00)00073-3. [DOI] [PubMed] [Google Scholar]
- 67.Rapraeger AC, Krufka A, Olwin BB. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science. 1991;252:1705–8. doi: 10.1126/science.1646484. [DOI] [PubMed] [Google Scholar]
- 68.Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell. 1991;64:841–8. doi: 10.1016/0092-8674(91)90512-w. [DOI] [PubMed] [Google Scholar]
- 69.Tran KT, Griffith L, Wells A. Extracellular matrix signaling through growth factor receptors during wound healing. Wound Repair Regen. 2004;12:262–8. doi: 10.1111/j.1067-1927.2004.012302.x. [DOI] [PubMed] [Google Scholar]
- 70.Tran KT, Lamb P, Deng JS. Matrikines and matricryptins: Implications for cutaneous cancers and skin repair. J Dermatol Sci. 2005;40:11–20. doi: 10.1016/j.jdermsci.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 71.Demidova-Rice TN, Geevarghese A, Herman IM. Bioactive peptides derived from vascular endothelial cell extracellular matrices promote microvascular morphogenesis and wound healing in vitro. Wound Repair Regen; (In Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Form DM, Pratt BM, Madri JA. Endothelial cell proliferation during angiogenesis. In vitro modulation by basement membrane components. Lab Invest. 1986;55:521–30. [PubMed] [Google Scholar]
- 73.Kutcher ME, Herman IM. The pericyte: cellular regulator of microvascular blood flow. Microvasc Res. 2009;77:235–46. doi: 10.1016/j.mvr.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bornstein P. Thrombospondins function as regulators of angiogenesis. J Cell Commun Signal. 2009;3:189–200. doi: 10.1007/s12079-009-0060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ghosh K, Ingber DE. Micromechanical control of cell and tissue development: implications for tissue engineering. Adv Drug Deliv Rev. 2007;59:1306–18. doi: 10.1016/j.addr.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 76.Parker KK, Brock AL, Brangwynne C, Mannix RJ, Wang N, Ostuni E, Geisse NA, Adams JC, Whitesides GM, Ingber DE. Directional control of lamellipodia extension by constraining cell shape and orienting cell tractional forces. FASEB J. 2002;16:1195–204. doi: 10.1096/fj.02-0038com. [DOI] [PubMed] [Google Scholar]
- 77.Ingber DE. Cellular tensegrity: defining new rules of biological design that govern the cytoskeleton. J Cell Sci. 1993;104 (Pt 3):613–27. doi: 10.1242/jcs.104.3.613. [DOI] [PubMed] [Google Scholar]
- 78.Jester JV, Huang J, Petroll WM, Cavanagh HD. TGFbeta induced myofibroblast differentiation of rabbit keratocytes requires synergistic TGFbeta, PDGF and integrin signaling. Exp Eye Res. 2002;75:645–57. doi: 10.1006/exer.2002.2066. [DOI] [PubMed] [Google Scholar]
- 79.Lygoe KA, Norman JT, Marshall JF, Lewis MP. AlphaV integrins play an important role in myofibroblast differentiation. Wound Repair Regen. 2004;12:461–70. doi: 10.1111/j.1067-1927.2004.12402.x. [DOI] [PubMed] [Google Scholar]
- 80.Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. 2007;179:1311–23. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Parizi M, Howard EW, Tomasek JJ. Regulation of LPA-promoted myofibroblast contraction: role of Rho, myosin light chain kinase, and myosin light chain phosphatase. Exp Cell Res. 2000;254:210–20. doi: 10.1006/excr.1999.4754. [DOI] [PubMed] [Google Scholar]
- 82.Okuda S, Languino LR, Ruoslahti E, Border WA. Elevated expression of transforming growth factor-beta and proteoglycan production in experimental glomerulonephritis. Possible role in expansion of the mesangial extracellular matrix. J Clin Invest. 1990;86:453–62. doi: 10.1172/JCI114731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yamaguchi Y, Mann DM, Ruoslahti E. Negative regulation of transforming growth factor-beta by the proteoglycan decorin. Nature. 1990;346:281–4. doi: 10.1038/346281a0. [DOI] [PubMed] [Google Scholar]
- 84.Martinez-Ferrer M, Afshar-Sherif AR, Uwamariya C, de Crombrugghe B, Davidson JM, Bhowmick NA. Dermal transforming growth factor-beta responsiveness mediates wound contraction and epithelial closure. Am J Pathol. 176:98–107. doi: 10.2353/ajpath.2010.090283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341:738–46. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- 86.Witte MB, Barbul A. General principles of wound healing. Surg Clin North Am. 1997;77:509–28. doi: 10.1016/s0039-6109(05)70566-1. [DOI] [PubMed] [Google Scholar]
- 87.Brem H, Kirsner RS, Falanga V. Protocol for the successful treatment of venous ulcers. Am J Surg. 2004;188:1–8. doi: 10.1016/S0002-9610(03)00284-8. [DOI] [PubMed] [Google Scholar]
- 88.Brem H, Sheehan P, Rosenberg HJ, Schneider JS, Boulton AJ. Evidence-based protocol for diabetic foot ulcers. Plast Reconstr Surg. 2006;117:193S–209S. doi: 10.1097/01.prs.0000225459.93750.29. discussion 10S–11S. [DOI] [PubMed] [Google Scholar]
- 89.Herrick SE, Sloan P, McGurk M, Freak L, McCollum CN, Ferguson MW. Sequential changes in histologic pattern and extracellular matrix deposition during the healing of chronic venous ulcers. Am J Pathol. 1992;141:1085–95. [PMC free article] [PubMed] [Google Scholar]
- 90.Brem H, Tomic-Canic M. Cellular and molecular basis of wound healing in diabetes. J Clin Invest. 2007;117:1219–22. doi: 10.1172/JCI32169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wysocki AB, Staiano-Coico L, Grinnell F. Wound fluid from chronic leg ulcers contains elevated levels of metalloproteinases MMP-2 and MMP-9. J Invest Dermatol. 1993;101:64–8. doi: 10.1111/1523-1747.ep12359590. [DOI] [PubMed] [Google Scholar]
- 92.Lobmann R, Ambrosch A, Schultz G, Waldmann K, Schiweck S, Lehnert H. Expression of matrix-metalloproteinases and their inhibitors in the wounds of diabetic and non-diabetic patients. Diabetologia. 2002;45:1011–6. doi: 10.1007/s00125-002-0868-8. [DOI] [PubMed] [Google Scholar]
- 93.Subramaniam K, Pech CM, Stacey MC, Wallace HJ. Induction of MMP-1, MMP-3 and TIMP-1 in normal dermal fibroblasts by chronic venous leg ulcer wound fluid*. Int Wound J. 2008;5:79–86. doi: 10.1111/j.1742-481X.2007.00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Saito S, Trovato MJ, You R, Lal BK, Fasehun F, Padberg FT, Jr, Hobson RW, 2nd, Durán WN, Pappas PJ. Role of matrix metalloproteinases 1, 2, and 9 and tissue inhibitor of matrix metalloproteinase-1 in chronic venous insufficiency. J Vasc Surg. 2001;34:930–8. doi: 10.1067/mva.2001.119503. [DOI] [PubMed] [Google Scholar]
- 95.Beidler SK, Douillet CD, Berndt DF, Keagy BA, Rich PB, Marston WA. Multiplexed analysis of matrix metalloproteinases in leg ulcer tissue of patients with chronic venous insufficiency before and after compression therapy. Wound Repair Regen. 2008;16:642–8. doi: 10.1111/j.1524-475X.2008.00415.x. [DOI] [PubMed] [Google Scholar]
- 96.Liu Y, Min D, Bolton T, Nubé V, Twigg SM, Yue DK, McLennan SV. Increased matrix metalloproteinase-9 predicts poor wound healing in diabetic foot ulcers. Diabetes Care. 2009;32:117–9. doi: 10.2337/dc08-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rayment EA, Upton Z, Shooter GK. Increased matrix metalloproteinase-9 (MMP-9) activity observed in chronic wound fluid is related to the clinical severity of the ulcer. Br J Dermatol. 2008;158:951–61. doi: 10.1111/j.1365-2133.2008.08462.x. [DOI] [PubMed] [Google Scholar]
- 98.Mwaura B, Mahendran B, Hynes N, Defreitas D, Avalos G, Adegbola T, Adham M, Connolly CE, Sultan S. The impact of differential expression of extracellular matrix metalloproteinase inducer, matrix metalloproteinase-2, tissue inhibitor of matrix metalloproteinase-2 and PDGF-AA on the chronicity of venous leg ulcers. Eur J Vasc Endovasc Surg. 2006;31:306–10. doi: 10.1016/j.ejvs.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 99.Chen SM, Ward SI, Olutoye OO, Diegelmann RF, Kelman Cohen I. Ability of chronic wound fluids to degrade peptide growth factors is associated with increased levels of elastase activity and diminished levels of proteinase inhibitors. Wound Repair Regen. 1997;5:23–32. doi: 10.1046/j.1524-475X.1997.50108.x. [DOI] [PubMed] [Google Scholar]
- 100.Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284:1318–22. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 101.Percival SL, Bowler PG. Biofilms and their potential role in wound healing. Wounds. 2004;16:234–40. [Google Scholar]
- 102.Wolcott RD, Rhoads DD, Dowd SE. Biofilms and chronic wound inflammation. J Wound Care. 2008;17:333–41. doi: 10.12968/jowc.2008.17.8.30796. [DOI] [PubMed] [Google Scholar]
- 103.James GA, Swogger E, Wolcott R, Pulcini E, Secor P, Sestrich J, Costerton JW, Stewart PS. Biofilms in chronic wounds. Wound Repair Regen. 2008;16:37–44. doi: 10.1111/j.1524-475X.2007.00321.x. [DOI] [PubMed] [Google Scholar]
- 104.Johnson GM, Lee DA, Regelmann WE, Gray ED, Peters G, Quie PG. Interference with granulocyte function by Staphylococcus epidermidis slime. Infect Immun. 1986;54:13–20. doi: 10.1128/iai.54.1.13-20.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shiau AL, Wu CL. The inhibitory effect of Staphylococcus epidermidis slime on the phagocytosis of murine peritoneal macrophages is interferon-independent. Microbiol Immunol. 1998;42:33–40. doi: 10.1111/j.1348-0421.1998.tb01966.x. [DOI] [PubMed] [Google Scholar]
- 106.Schierle CF, De la Garza M, Mustoe TA, Galiano RD. Staphylococcal biofilms impair wound healing by delaying reepithelialization in a murine cutaneous wound model. Wound Repair Regen. 2009;17:354–9. doi: 10.1111/j.1524-475X.2009.00489.x. [DOI] [PubMed] [Google Scholar]
- 107.Thomson CH. Biofilms: do they affect wound healing? Int Wound J. 2010 doi: 10.1111/j.1742-481X.2010.00749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yamada KM, Clark RAF. Provisional matrix. In: Clark RAF, editor. The Molecular Biology of Wound Repair. New York: Plenum Press; 1996. pp. 51–93. [Google Scholar]
- 109.Larjava H, Salo T, Haapasalmi K, Kramer RH, Heino J. Expression of integrins and basement membrane components by wound keratinocytes. J Clin Invest. 1993;92:1425–35. doi: 10.1172/JCI116719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Annes JP, Chen Y, Munger JS, Rifkin DB. Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J Cell Biol. 2004;165:723–34. doi: 10.1083/jcb.200312172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–28. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 112.Madlener M, Parks WC, Werner S. Matrix metalloproteinases (MMPs) and their physiological inhibitors (TIMPs) are differentially expressed during excisional skin wound repair. Exp Cell Res. 1998;242:201–10. doi: 10.1006/excr.1998.4049. [DOI] [PubMed] [Google Scholar]
- 113.Saarialho-Kere U, Kerkela E, Jahkola T, Suomela S, Keski-Oja J, Lohi J. Epilysin (MMP-28) expression is associated with cell proliferation during epithelial repair. J Invest Dermatol. 2002;119:14–21. doi: 10.1046/j.1523-1747.2002.01790.x. [DOI] [PubMed] [Google Scholar]
- 114.Pilcher BK, Dumin JA, Sudbeck BD, Krane SM, Welgus HG, Parks WC. The activity of collagenase-1 is required for keratinocyte migration on a type I collagen matrix. J Cell Biol. 1997;137:1445–57. doi: 10.1083/jcb.137.6.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ongenae KC, Phillips TJ, Park HY. Level of fibronectin mRNA is markedly increased in human chronic wounds. Dermatol Surg. 2000;26:447–51. doi: 10.1046/j.1524-4725.2000.99281.x. [DOI] [PubMed] [Google Scholar]
- 116.Hakkinen L, Koivisto L, Gardner H, Saarialho-Kere U, Carroll JM, Lakso M, Rauvala H, Laato M, Heino J, Larjava H. Increased expression of beta6-integrin in skin leads to spontaneous development of chronic wounds. Am J Pathol. 2004;164:229–42. doi: 10.1016/s0002-9440(10)63113-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wysocki AB, Grinnell F. Fibronectin profiles in normal and chronic wound fluid. Lab Invest. 1990;63:825–31. [PubMed] [Google Scholar]
- 118.Cowin AJ, Hatzirodos N, Holding CA, Dunaiski V, Harries RH, Rayner TE, Fitridge R, Cooter RD, Schultz GS, Belford DA. Effect of healing on the expression of transforming growth factor beta(s) and their receptors in chronic venous leg ulcers. J Invest Dermatol. 2001;117:1282–9. doi: 10.1046/j.0022-202x.2001.01501.x. [DOI] [PubMed] [Google Scholar]
- 119.Pierce GF, Tarpley JE, Tseng J, Bready J, Chang D, Kenney WC, Rudolph R, Robson MC, Vande Berg J, Reid P, et al. Detection of platelet-derived growth factor (PDGF)-AA in actively healing human wounds treated with recombinant PDGF-BB and absence of PDGF in chronic nonhealing wounds. J Clin Invest. 1995;96:1336–50. doi: 10.1172/JCI118169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Charles CA, Tomic-Canic M, Vincek V, Nassiri M, Stojadinovic O, Eaglstein WH, Kirsner RS. A gene signature of nonhealing venous ulcers: potential diagnostic markers. J Am Acad Dermatol. 2008;59:758–71. doi: 10.1016/j.jaad.2008.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Brem H, Stojadinovic O, Diegelmann RF, Entero H, Lee B, Pastar I, Golinko M, Rosenberg H, Tomic-Canic M. Molecular markers in patients with chronic wounds to guide surgical debridement. Mol Med. 2007;13:30–9. doi: 10.2119/2006-00054.Brem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gottrup F. Debridement: another evidence problem in wound healing. Wound Repair Regen. 2009;17:294–5. doi: 10.1111/j.1524-475X.2009.00484.x. [DOI] [PubMed] [Google Scholar]
- 123.Panuncialman J, Hammerman S, Carson P, Falanga V. Wound edge biopsy sites in chronic wounds heal rapidly and do not result in delayed overall healing of the wounds. Vol. 18. Wound Repair Regen; pp. 21–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Marston WA. Dermagraft, a bioengineered human dermal equivalent for the treatment of chronic nonhealing diabetic foot ulcer. Expert Rev Med Devices. 2004;1:21–31. doi: 10.1586/17434440.1.1.21. [DOI] [PubMed] [Google Scholar]
- 125.Steed DL. Clinical evaluation of recombinant human platelet-derived growth factor for the treatment of lower extremity diabetic ulcers. Diabetic Ulcer Study Group. J Vasc Surg. 1995;21:71–8. doi: 10.1016/s0741-5214(95)70245-8. discussion 9–81. [DOI] [PubMed] [Google Scholar]
- 126.Veves A, Falanga V, Armstrong DG, Sabolinski ML. Graftskin, a human skin equivalent, is effective in the management of noninfected neuropathic diabetic foot ulcers: a prospective randomized multicenter clinical trial. Diabetes Care. 2001;24:290–5. doi: 10.2337/diacare.24.2.290. [DOI] [PubMed] [Google Scholar]
- 127.Mostow EN, Haraway GD, Dalsing M, Hodde JP, King D. Effectiveness of an extracellular matrix graft (OASIS Wound Matrix) in the treatment of chronic leg ulcers: a randomized clinical trial. J Vasc Surg. 2005;41:837–43. doi: 10.1016/j.jvs.2005.01.042. [DOI] [PubMed] [Google Scholar]
- 128.Bennett JS, Vilaire G. Exposure of platelet fibrinogen receptors by ADP and epinephrine. J Clin Invest. 1979;64:1393–401. doi: 10.1172/JCI109597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Muller WA. Leukocyte-endothelial cell interactions in the inflammatory response. Lab Invest. 2002;82:521–33. doi: 10.1038/labinvest.3780446. [DOI] [PubMed] [Google Scholar]
- 130.Larjava H, Koivisto L, Häkkinen L. Keratinocyte interaction with fibronectin during wound healing. In: Heino J, Kähäri V-M, editors. Cell Invasion. Landes Biosciences, Eurecah.com; Georgetown, TX: 2000. [Google Scholar]
- 131.Takashima A, Grinnell F. Human keratinocyte adhesion and phagocytosis promoted by fibronectin. J Invest Dermatol. 1984;83:352–8. doi: 10.1111/1523-1747.ep12264522. [DOI] [PubMed] [Google Scholar]
- 132.Yates CC, Krishna P, Whaley D, Bodnar R, Turner T, Wells A. Lack of CXC chemokine receptor 3 signaling leads to hypertrophic and hypercellular scarring. Am J Pathol. 2010;176:1743–55. doi: 10.2353/ajpath.2010.090564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pham HT, Economides PA, Veves A. The role of endothelial function on the foot. Microcirculation and wound healing in patients with diabetes. Clin Podiatr Med Surg. 1998;15:85–93. [PubMed] [Google Scholar]
- 134.Tilton RG, Faller AM, Burkhardt JK, Hoffmann PL, Kilo C, Williamson JR. Pericyte degeneration and acellular capillaries are increased in the feet of human diabetic patients. Diabetologia. 1985;28:895–900. doi: 10.1007/BF00703132. [DOI] [PubMed] [Google Scholar]
- 135.Tilton RG, Hoffmann PL, Kilo C, Williamson JR. Pericyte degeneration and basement membrane thickening in skeletal muscle capillaries of human diabetics. Diabetes. 1981;30:326–34. doi: 10.2337/diab.30.4.326. [DOI] [PubMed] [Google Scholar]
- 136.Ajjam ZS, Barton S, Corbett M, Owens D, Marks R. Quantitative evaluation of the dermal vasculature of diabetics. Q J Med. 1985;54:229–39. [PubMed] [Google Scholar]
- 137.Khaodhiar L, Dinh T, Schomacker KT, Panasyuk SV, Freeman JE, Lew R, Vo T, Panasyuk AA, Lima C, Giurini JM, Lyons TE, Veves A. The use of medical hyperspectral technology to evaluate microcirculatory changes in diabetic foot ulcers and to predict clinical outcomes. Diabetes Care. 2007;30:903–10. doi: 10.2337/dc06-2209. [DOI] [PubMed] [Google Scholar]
- 138.Nolan CM, Beaty HN, Bagdade JD. Further characterization of the impaired bactericidal function of granulocytes in patients with poorly controlled diabetes. Diabetes. 1978;27:889–94. doi: 10.2337/diab.27.9.889. [DOI] [PubMed] [Google Scholar]
- 139.Tarsio JF, Reger LA, Furcht LT. Decreased interaction of fibronectin, type IV collagen, and heparin due to nonenzymatic glycation. Implications for diabetes mellitus. Biochemistry. 1987;26:1014–20. doi: 10.1021/bi00378a006. [DOI] [PubMed] [Google Scholar]
- 140.McDermott AM, Xiao TL, Kern TS, Murphy CJ. Non-enzymatic glycation in corneas from normal and diabetic donors and its effects on epithelial cell attachment in vitro. Optometry. 2003;74:443–52. [PubMed] [Google Scholar]
- 141.Cohen MP, Ziyadeh FN, Hong SW, Shearman CW, Hud E, Lautenslager GT, Iglesias-de la Cruz MC, Chen S. Inhibiting albumin glycation in vivo ameliorates glomerular overexpression of TGF-beta1. Kidney Int. 2002;61:2025–32. doi: 10.1046/j.1523-1755.2002.00352.x. [DOI] [PubMed] [Google Scholar]
- 142.Fahey TJ, 3rd, Sadaty A, Jones WG, 2nd, Barber A, Smoller B, Shires GT. Diabetes impairs the late inflammatory response to wound healing. J Surg Res. 1991;50:308–13. doi: 10.1016/0022-4804(91)90196-s. [DOI] [PubMed] [Google Scholar]
- 143.Song W, Ergul A. Type-2 diabetes-induced changes in vascular extracellular matrix gene expression: relation to vessel size. Cardiovasc Diabetol. 2006;5:3. doi: 10.1186/1475-2840-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Junger M, Steins A, Hahn M, Hafner HM. Microcirculatory dysfunction in chronic venous insufficiency (CVI) Microcirculation. 2000;7:S3–12. [PubMed] [Google Scholar]
- 145.Loots MA, Lamme EN, Zeegelaar J, Mekkes JR, Bos JD, Middelkoop E. Differences in cellular infiltrate and extracellular matrix of chronic diabetic and venous ulcers versus acute wounds. J Invest Dermatol. 1998;111:850–7. doi: 10.1046/j.1523-1747.1998.00381.x. [DOI] [PubMed] [Google Scholar]
- 146.Ulrich D, Lichtenegger F, Unglaub F, Smeets R, Pallua N. Effect of chronic wound exudates and MMP-2/-9 inhibitor on angiogenesis in vitro. Plast Reconstr Surg. 2005;116:539–45. doi: 10.1097/01.prs.0000173447.81513.7a. [DOI] [PubMed] [Google Scholar]