

Abstract
Objective
The 5-lipoxygenase (5-LO) enzymatic pathway is widely distributed within the central nervous system, and is up-regulated in Alzheimer's disease. However, the mechanism whereby it may influence the disease pathogenesis remains elusive.
Methods
We evaluated the molecular mechanism by which 5-LO regulates Amyloid β (Aβ) formation in vitro and in vivo by pharmacological and genetic approaches.
Results
Here we show that 5-LO regulates the formation of Aβ by activating the cAMP-response element binding protein (CREB), which in turn increases transcription of the γ-secretase complex. Preventing CREB activation by pharmacologic inhibition or dominant negative mutants blocks the 5-LO-dependent elevation of Aβ formation and the increase of γ-secretase mRNA and protein levels. Moreover, 5-LO targeted gene disruption or its in vivo selective pharmacological inhibition results in a significant reduction of Aβ, CREB and γ-secretase levels.
Interpretation
These data establish a novel functional role for 5-LO in regulating endogenous formation of Aβ levels in the central nervous system. Thus, 5-LO pharmacological inhibition may be beneficial in the treatment and prevention of Alzheimer's disease.
Introduction
The enzyme 5-lipoxygenase (5-LO) catalyzes the conversion of arachidonic acid to 5-hydroxy-peroxy-eicosatetraenoic acid (5-HPETE) and subsequently to 5hydroxyeicosatetraenoic acid (5-HETE), which can be then metabolized into different leukotrienes 1. Since these metabolic products have potent inflammatory actions, this enzymatic pathway has been implicated in the pathophysiology of several chronic inflammatory diseases.
For instance, 5-LO is considered a major contributor to atherosclerosis and cardiovascular diseases 2. In addition to its presence in the cardiovascular system, this enzyme is widely expressed in the brain where significant increase in its level and activity has been associated with aging 3,4. However, while the molecular mechanisms of 5-LO-mediated atherosclerosis have been extensively studied, less is known about the molecular actions of 5-LO in the brain. Since aging is one of the strongest risk factors for developing neurodegenerative diseases, it has been suggested that this pathway may be involved in their pathogenesis 5. To this end, we recently showed that Alzheimer's disease (AD) brains had higher 5-LO protein levels than healthy controls 6. Further, we investigated the effect of 5-LO-targeted gene disruption on the amyloid β (Aβ) pathology of Tg2576 mice, a model of amyloidosis, by crossing them with 5-LO-deficient mice. In that study, we found that the genetic absence of 5-LO resulted in a significant reduction in their Aβ levels 6. However, the molecular mechanism underlying the biological effect of 5-LO on the Aβ metabolic pathway remains elusive.
In the current paper we show that over-expression or pharmacological blockade of 5-LO results in an elevation or reduction of Aβ formation, respectively, which associates with similar changes in the four protein components of the γ-secretase complex. Cell exposure to the main 5-LO metabolic product, i.e. 5-HETE, results in a significant elevation of Aβ, γ-secretase complex, and cAMP-response element binding protein (CREB) levels and activity. Pharmacological blockade of CREB, or its dominant negative mutants suppress these effects. Finally, in vivo 5-LO pharmacological inhibition or its genetic absence results in a significant reduction of CREB, γ-secretase complex and endogenous Aβ levels.
Taken together, these data provide the first experimental evidence for a novel biological role of 5-LO as an endogenous modulator of Aβ formation through a mechanism that involves transcription of the γ-secretase complex via activation of CREB.
Materials and Methods
Materials
5-HETE, 8-HETE, 13-HODE were obtained from Cayman Chemical Company; AA-861 was obtained from Biomol International; α-tocopherol (vitamin E) was from Sigma-Aldrich; L685,458 was purchased from Calbiochem; DMEM, fetal bovine serum, penicillin and streptomycin were obtained from Invitrogen.
Cell culture and transfection
N2A (neuro-2 A neuroblastoma) cells stably expressing human APP carrying the K670 N, M671 L Swedish mutation (APP swe) were grown as previously described 7. For transfection, cells were grown to 70% confluence and transfected with 0.5 μg of empty vector (pcDNA3.1) or human 5LO pcDNA3.1 (Dr. Colin Funk, Queen's University, Kingston, Canada) by using Lipofectamine reagent (Invitrogen) according to the manufacturer's instructions. After 48 h transfection, supernatants were collected, and cells harvested in lytic buffer for biochemistry analyses.
Immunoblot analyses
Cells were extracted and protein concentration was determined as previously described8,9. Samples were electrophoretically separated using 10% Bis–Tris gels or 3-8% Tris– acetate gel (Bio-Rad), according to the molecular weight of the target molecule, and then transferred onto nitrocellulose membranes (Bio-Rad). They were blocked with Odyssey blocking buffer, then incubated with primary antibodies overnight at 4 °C 8,9. After three washing cycles with T-TBS, membranes were incubated with IRDye 800CW or IRDye 680CW-labeled secondary antibodies (LI-COR) at 22°C for 1 h. Signals were developed with Odyssey Infrared Imaging Systems (LI-COR). Actin was always used as an internal loading control. Antibodies and dilutions were as follows: anti-APP N-terminal raised against amino acids 66-81 for total APP (22C11; 1:1500; Chemicon Int.), anti-BACE-1 (1:200; IBL), anti-ADAM-10 (1: 500 dilution; Chemicon Int.), anti-PS1 (1: 200; Cell Signaling), anti-nicastrin (1: 200; Cell Signaling), anti-Pen2 (1: 200; Invitrogen), anti-APH-1 (1: 200; Millipore); anti-CREB (1:200, Cell Signaling), anti-p-CREB (1:200, Cell Signaling); anti-NICD (1: 200; Cell Signaling); anti-apoE (1:100; Santa Cruz Biotech.), anti-neprilysin (1:150; Santa Cruz Biotech.), anti-IDE N-terminal (1:1000; EMD Biosciences), anti-5-LO (clone 33;1:500; BD Bioscience), anti-β actin (1;4000) 8,9.
mNotch ΔE transfection and Notch cleavage
N2A-APPSwe cells were transiently transfected to overexpress mNotchΔE, a N-terminal truncated Notch-1 lacking most of the Notch extracellular domain with a C-terminal hemagglutinin tag (a generous gift of Dr. D'Adamio, A. Eistein College of Medicine, New York). Cultures were incubated with 5-HETE, AA-861, or L-685,458, a specific γ-secretase inhibitor 10, overnight, then cell lysates were used for immunoblot analysis for full length mNotchΔE and its γ-secretase dependent cleavage product NICD 11.
Immunofluorescence microscopy
Cell immunostaining was carried out as previously described 7. Briefly, N2A-APPswe cells were plated on glass cover slips and the following day fixed in 4% paraformaldehyde in PBS for 15 min at 22°C. After several rinses, cells were incubated in blocking solution (5% normal serum/0.4% TX100) for 1 h at 22°C and then with the primary antibody against p-CREB (1:200; Cell Signaling) overnight at 4°C. After washing with PBS, cells were incubated for 1 h with a secondary Alexa546- conjugated antibody (1:800; Invitrogen), and analyzed with an Olympus BX60 fluorescent microscope. Fluorescence emission was collected at 425–475 nm for 4’, 6-diamidino-2-phenylindole and 555–655 nm for Alexa546. Control cover slips were processed as described above except that no primary antibody was added to the solution.
Quantitative real time RT-PCR
RNA was extracted and purified using the RNeasy mini-kit (Qiagen), as previously described 4,7. Briefly, 1μg of total RNA was used to synthesize cDNA in a 20 μl reaction using the RT2 First Strand Kit for reverse transcriptase-PCR (Super Array Bio.). Mouse PS1, nicastrin, APH-1, Pen-2 and BACE-1 genes were amplified by using the corresponding primers obtained from Super Array Bioscience. β-actin was used as an internal control gene to normalize for the amount of RNA. Quantitative real-time RT-PCR was performed by using Eppendorf® ep realplex thermal cyclers. Two μl of cDNA was added to 10μl of SYBR Green PCR Master Mix (Applied Biosystems). Each sample was run in duplicate, and analysis of relative gene expression was done by using the 2-ΔΔCt method 12.
Luciferase assay
N2A-APPswe cell were transfected with Lipofectamin 2000 (Invitrogen) according to the manufacturer's instruction. Each transfection contained CREB-responsive luciferase reporter plasmid (SuperArray Biosc.) or a combination of CREB-responsive luciferase reporter plasmid and dominant-negative mutant vectors (pCMV-CREB133, pCMV-KCREB). In another set of experiments, cells were also transfected with Sp1-responsive luciferase reporter plasmid (SuperArray Biosc.). After 12 hours transfection, cells were treated by 5-HETE (10μM). Cell lysates were collected, luciferase and renilla luciferase activities were measured sequentially using a dual-luciferase reporter assay system (Promega), and a POLARstar OPTIMA Luminometer (Imgen). Firefly luciferase activity was divided by Renilla's one to normalize luciferase activity toward transfection efficiency. Data reported are means of triplicates. Each experiment was repeated at least 3 times. The solvent, ethanol, alone had no effect on basal luciferase activity of any of the luciferase reporter constructs used.
Dominant-negative mutant vector
CREB dominant-negative mutant vectors pCMV-CREB133 and pCMV-KCREB were from BD Biosciences Clontech. These vectors were transfected into N2A-APPswe cells by using Lipofectamin 2000 (Invitrogen) in accordance with the manufacturer's protocol. Following the transfection, cells were treated by 5-HETE (10μM) for 12 hours, then harvested and lysates used for immunoblot analyses.
Animals
Studies were approved by the Institutional Animal Care and Usage Committee, in accordance with the NIH guidelines. Tg2576, 5-LO-/-, Tg2576/5-LO-/- mice and their wild type littermates were anesthetizes and euthanized following procedures recommended by the American Veterinarian Medical Association. All mice used in the present study were females. Groups of 7month-old Tg2576 mice were randomized to receive AA-861 (80mg/Kg) or vehicle in their regular rodent chow diet, which was prepared by a commercial vendor (Arlan Teklad). Diets were always matched for kilocalories. During the study animals had free access to food and water, and fresh diet was changed every other day. After 5 weeks on the diet, mice were sacrificed. Brain was removed, gently rinsed in cold 0.9% PBS and immediately stored at -80°C until analysis.
Biochemical analyses
Supernatants from cell culture experiments were collected at the end of each incubation time and assayed for human Aβ1-40 levels by a specific and sensitive ELISA kit (IBL America, Minneapolis, MN) as previously described 7. Aliquots of brain samples were homogenized in 0.2% diethylamine (DEA)/50 mM NaCl at 1:10 W/v., then centrifuged for 1 hr at 100,000 × g at 4°C, and supernatants neutralized to pH 8.0 with 1:10 v/v of 0.5 Tris-HCl/pH 6.8. DEA extracts were assayed for endogenous murine Aβ40 and Aβ42 levels by a sandwich ELISA kit 8,9 (Wako Chem.). Aliquots of brain homogenates underwent total lipids extraction with ice-cold Folch solution, then base hydrolysis, and finally assayed for total 5(S)-HETE by HPLC. Briefly, 5(S)-HETE standard was purchased from Cayman Chem. Co. Reverse-phase liquid chromatography was carried out on a C18 column. The isocratic mobile phase consisted of methanol-10mM ammonium acetate-1M acetic acid (70:30:0.1; v:v:v), and the flow rate was 1.0mL/min. The 5(S)-HETE peak was identified and quantitated by UV absorbance at 240-270 nm, respectively (13). LTB4, PGE2, and 12-HETE were assayed by standardized EIAs following the manufacturer's instructions (Assay Designs) and as previously described4,7,14.
Immunohistochemistry
Immunohistochemistry analysis was performed as previously described 8,9. Briefly, brain sections were deparaffinized, hydrated, and after blocking with 2% serum were incubated with primary antibody against 5-LO overnight at 4°C. After three washings, sections were incubated with secondary antibody and finally developed using the avidin-biotin complex method (Vector Lab.) with 3’,3’-diaminobenzidine as chromogen. As controls, sections underwent the above described treatment except that no primary antibody was used.
Data analysis
Data analyses were performed using SigmaStat for Windows version 3.00. Values in all figures and table represent mean ± S.E.M. Significance was set at p < 0.05.
Results
5-LO increases Aβ formation via a γ-secretase-dependent mechanism
N2A-APPswe cells transiently transfected with human 5-LO cDNA or empty vector. After 48 hr, compared with controls, supernatants from cells receiving the 5-LO cDNA showed a significant increase in the amount of Aβ1-40 (Fig 1A). Compared with cells transfected with empty vector, the other ones had a significant increase in the expression levels of 5-LO, but not for total APP, BACE-1, or ADAM-10 levels (Fig 1B, C). By contrast, up-regulation of 5-LO resulted in a significant increase in the steady state levels of all four components of the γ-secretase complex, PS1, nicastrin, APH-1 and Pen-2 (Fig 1B, C).
Figure 1.

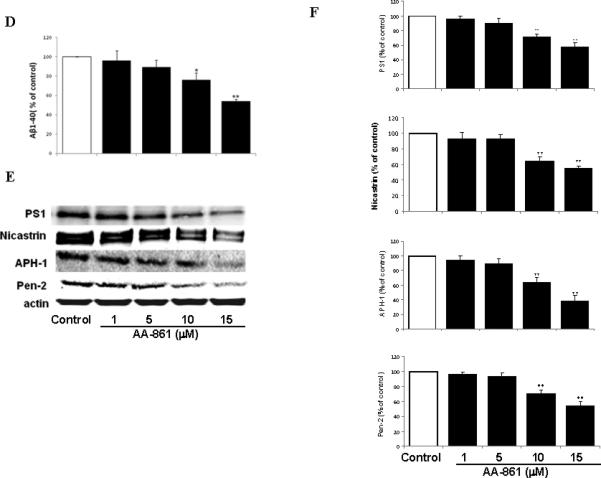
5-LO modulates Aβ formation in a γ-secretase-dependent manner. N2A-APPswe cells were transiently transfected with human 5-LO cDNA and supernatant and cells lysates were collected after 48hr. A. Aβ 1-40 levels in the supernatant. B. Representative immunoblots for 5-LO, APP, BACE-1, ADAM-10, PS1, nicastrin, APH-1 and Pen-2 from cell lysates. C. Densitometric analysis of the immunoblots data after normalization with actin. D. Cells were incubated with increasing concentration of AA-861 overnight and supernatants collected and assayed for Aβ 1-40. E. Representative immunoblots for PS1, nicastrin, APH-1 and Pen-2 in cell lysates harvested from the same experiments. F. Densitometric analysis of the western blots after normalization with actin levels. Values are mean ± s.e.m. of 3 independent experiments repeated in triplicate (*p<0.05; **p<0.01).
Pharmacologic blockade of 5LO activation reduces Aβ formation
Incubation of N2A-APPswe with AA-861, a selective and specific inhibitor of 5-LO (15), dose-dependently decreased Aβ 1-40 (Figure 1D), and LTB4 levels (Suppl. Fig 1). This effect was not associated with significant changes in total APP, BACE-1 or ADAM-10 levels (Suppl. Fig 2). By contrast, it resulted in a significant reduction in the steady state levels of PS1, nicastrin, APH-1 and Pen-2 (Fig 1 E,F). Since 5-LO has been also implicated in oxidative stress responses, we incubated the same cells with vitamin E (50μM), a potent anti-oxidant. Under this condition, no effect was observed on Aβ formation (not shown).
5-LO increases Aβ formation by promoting transcription of the γ-secretase complex
5-LO is an enzyme which inserts molecular oxygen at C5 of the arachidonic acid resulting in the formation of 5-hydroxyl-peroxy-eicosatetraenoic acid (5-HPETE), which is immediately converted into the more stable 5-hydroxy-eicosatetraenoic acid (5-HETE). To further support a direct role of 5-LO in the Aβ metabolism, we embarked in a series of experiments using 5-HETE. Incubation of N2A-APPswe with this metabolite induced a significant increase in Aβ (Fig 2A), which was not associated with changes in APP, BACE-1 or ADAM-10 levels (Suppl. Fig 3). By contrast, under these conditions we observed a significant increase in the steady state levels of PS1, nicastrin, APH-1 and Pen-2 (Fig 2B,C). Quantitative real time RT-PCR showed that the same treatment also resulted in a significant increase in their mRNAs (Suppl. Fig 4). Finally, to confirm the specificity of this effect we incubated the same cells with 8-HETE (10μM) and 13-HODE (10μM), products of two distinct LOs (i.e., 8-LO and 15-LO respectively). In both cases no significant effect on Aβ formation was observed (not shown).
Figure 2.
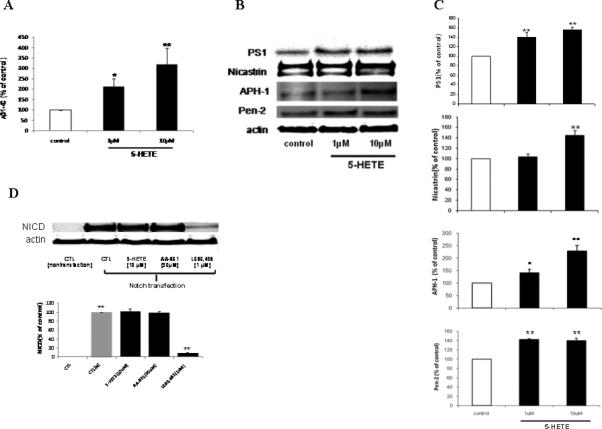
5-HETE increases Aβ formation, PS1, nicastrin, APH1 and Pen-2 levels, but has no effect on Notch processing. N2A-APPswe cells were incubated with 5-HETE (1 and 10 μM) overnight and supernatants and cell lysates collected. A. Aβ 1-40 levels in supernatant. B. Representative immunoblots for PS1, nicastrin, APH-1 and Pen-2. C. Densitometric analysis of the immunoblots after normalization with actin levels. D. Representative immunoblot and densitometric analysis for NICD. Values are mean ± s.e.m. of 3 independent experiments repeated in triplicate (*p<0.05; **p<0.01).
5-LO effect on the γ-secretase does not influence Notch processing
Since the data so far support an effect of 5-LO on the γ-secretase complex, which is also involved in the processing of the Notch receptor, next we wanted to see whether this enzymatic pathway influenced the release of Notch intracellular domain (NICD) 16. Cells were transiently transfected with an N-terminally truncated Notch-1 plasmid (mNotchΔE) and then incubated with 5-HETE, or the 5-LO selective blocker AA-861. As a positive control, cells were treated with L685,458, a potent γ-secretase inhibitor 16. As shown in Figure 2D, while L685,458 reduced the levels of NICD, both 5-HETE and AA-861 were without effect, suggesting that 5-LO does not interfere with Notch cleavage.
5-LO-dependent transcription of the γ-secretase complex is regulated by CREB activation
Previous studies have shown that HETEs activates the cAMP-response element binding protein (CREB), a transcriptional factor, which by translocating into the nucleus regulates gene expression 17,18. For this reason, we wanted to test if this was also the case in our system. Cells incubated with 5-HETE showed a significant increase in the steady state levels of total CREB and its phosphorylated form at Ser133 (Fig 3A). Activation of CREB by 5-HETE was also documented by immuno-fluorescence studies, where we observed that while non-stimulated cells expressed most of the p-CREB in their cytoplasm, addition of 5-HETE resulted in the translocation of p-CREB immuno-reactivity into the nuclear region (Fig 3B). Next we sought to further demonstrate that 5-HETE directly activates CREB by using a luciferase reporter system. As shown in Figure 3C, we found that in cells treated with 5-HETE this transcription factor was significantly activated already at 1 hr with a maximum increase at 3hr. By contrast, no significant changes were observed in the activation state of another transcription factor, which has been implicated in regulating the γ-secretase complex, i.e. Sp1 (Fig 3C). Under the same conditions we also investigated mRNAs changes for the four components of the γ-secretase complex. Compared with control, APH-1 mRNA showed the fastest (3hr) and strongest (6 hr) increase, which by 24hr went back almost to control values. By contrast, the other 3 mRNAs showed a slower increase, which became significant at 6hr for all, and then plateau for Pen-2 and nicastrin at 12 and 24 hr, but continued to increase for PS1 up to 24 hr (Fig 3 D). By contrast, 5-HETE had no effect on BACE-1 mRNA levels (Fig 3D). Since protein kinase A (PKA) has been implicated in the activation of CREB 9, in the next sets of experiments we used selective pharmacological inhibitors of this kinase to prevent 5-HETE-dependent effects. Incubation of cells with two distinct PKA specific inhibitors, H89 and Rp, completely prevented the 5-HETEdependent Aβ and p-CREB increase (Fig 3E,F), and induced a significant reduction in the steady state levels of PS1, nicastrin, APH-1 and Pen-2 (Fig 3G) To further support the role of CREB in the 5-LO mediated effect on the transcription of the γ-secretase complex, next we used CREB dominant negative mutant vectors. First, we identified the concentrations of CREB dominant negative mutants, CREB133 and KCREB, which prevented the 5-HETE-dependent increase in CREB activity (Fig 4A). Then, by using this amount of negative dominants, we tested the 5-HETE-dependent effect on Aβ formation, γ-secretase complex and CREB levels. As shown in Figure 4 B to D, we found that indeed by blocking CREB activation with these negative mutants we could fully prevent these effects.
Figure 3.


5-HETE by activating CREB induces PS1, nicastrin, APH-1 and Pen-2 mRNA. N2A-APPswe cells were incubated with 5-HETE overnight and cell lysates collected. A. Representative immunoblots and densitometric analysis for CREB and phosphorylated CREB at Ser 133. B. Representative pictures of immunoflourescence analysis for cellular compartimentalization of p-CREB in cells incubated with vehicle or 5-HETE (a-c) negative control: no primary antibody; (d-f) vehicle; (g-i) 5-HETE. C. Time-dependent activation of CREB and Sp1 after challenge with 5-HETE. D. Time-dependent changes for PS1, nicastrin, APH-1, Pen-2 and BACE-1 mRNAs levels after challenge with 5-HETE. E-G. Cells were pre-incubated with H89 and Rp, two specific PKA inhibitors, then challenged with 5-HETE overnight, supernatant collected for Aβ levels (E), cell lysates for immunoblot analysis of p-CREB (F), PS1, nicastrin, APH-1 and Pen-2 protein levels (G). Values are mean ± s.e.m. of 3 independent experiments repeated in triplicate (*p<0.05; **p<0.01).
Figure 4.
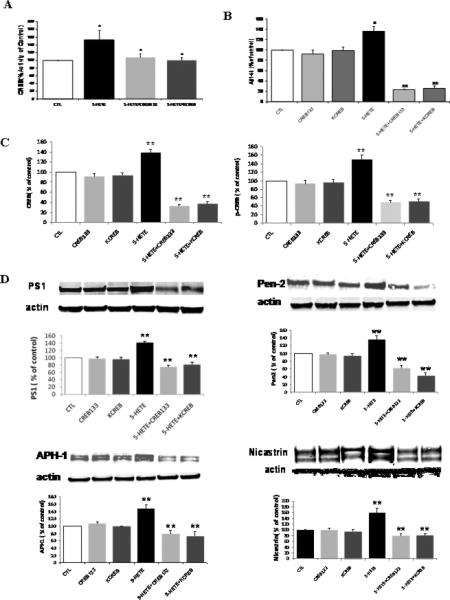
Blockade of CREB activation suppresses 5-LO-dependent Aβ elevation and γ-secretase complex up-regulation. A. Transfection of cells with two mutant CREB negative dominant vectors prevents the 5-HETE-dependent CREB activation, (B) and Aβ formation. C. Densitometric analyses for total CREB and p-CREB after transfection with CREB negative dominant vectors and 5-HETE challenge. D. Representative immunoblot and densitometric analyses for PS1, nicastrin, APH-1 and Pen-2 after transfection with CREB negative dominant vectors and 5-HETE challenge. Values are mean ± s.e.m. of 3 independent experiments repeated in triplicate (*p<0.05; **p<0.01).
In vivo modulation of CREB by 5-LO
Immunohistochemistry analysis of brains from 7-month-old Tg2576 showed a significant increase in 5-LO immunoreactivity compared with nontransgenic wild type (WT) littermates (Fig 5A). Further, quantitation of 5(S)-HETE in the same brain regions confirmed a significant increase of its levels in Tg2576 mice when compared with WT (63±7 vs 29±4 pg/mg, p<0.01). Importantly, the levels of this 5-LO specific metabolic product were similar to the ones we have been used in our in vitro experiments (low micromolar). To further support the involvement of CREB in the 5-LO-mediated effect on Aβ formation, 7-month-oldTg2576 mice were orally dosed with the selective 5-LO inhibitor AA-861, or vehicle (n=6 each group) and brain levels of total SDS-soluble Aβ 1-40 and 1-42 were measured by ELISA. Inhibition of 5-LO was monitored by measuring levels of brain 5(S)-HETE, which at the end of the treatment were significantly reduced (>70%). Under these conditions we observed that compared with vehicle, mice receiving the active drug had a significant decrease in Aβ levels (Aβ40: 24±2 vs 13±1.2 pmol/mg; Aβ42: 10.5± 0.9 vs 5.4±0.6 pmol/mg; p<0.01 for both), and this effect was associated with a significant reduction in p-CREB protein levels (Fig 5B). A similar pattern was observed when we compared brains form Tg2576 with Tg2576/5-LO knock-out (5-LO-/-) mice (Figure 5C).
Figure 5.
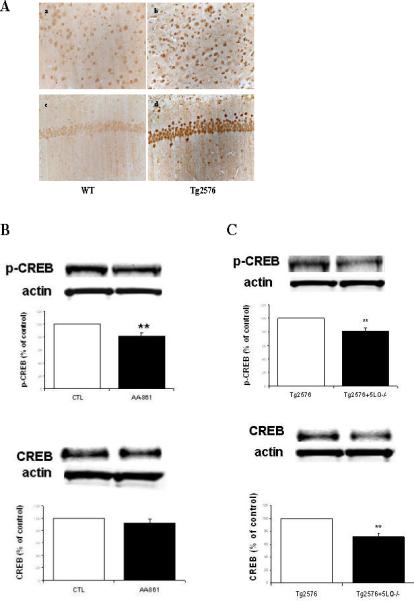
In vivo 5-LO modulation of CREB. A. Representative pictures of brain sections from Tg2576 mice and wild type littermates (WT) challenged with a specific anti-5-LO antibody. Panels a, b: entorhinal cortex; panels c,d: hippocampus CA1 region. B. Representative immunoblots and densitometric analyses for p-CREB and total CREB in brains from Tg2576 mice after AA-861 or vehicle (CTL) administration. C. Representative immunoblots and densitometric analyses for p-CREB and total CREB in Tg2576 and Tg2576/5-LO/- mice brains (n=3 per group). Values are mean ± s.e.m. (*p<0.05; **p<0.01).
Loss of 5-LO reduces endogenous Aβ, CREB and γ-secretase complex levels
If indeed 5-LO plays a physiological role in regulating CREB, which by modulating the transcription of the γ-secretase complex influences Aβ formation in vivo, then we would expect to observe reduced levels of endogenous Aβ formation, CREB and γ-secretase complex levels also in the CNS of mice, which do not carry the APP human transgene but are genetically deficient for this enzyme (i.e., 5-LO-/- mice) when compared with WT. As shown in Figure 6A, we observed that compared with WT, 5-LO-/- mice had significant lower total levels of both Aβ1-40 and 1-42 peptides. However, no significant difference between the two groups was observed for total APP, BACE-1 or ADAM-10 (Fig 6B). By contrast, we observed that steady state levels of PS1, nicastrin, APH-1 and Pen2 were significantly reduced in brain homogenates from 5-LO-/- mice (Fig 6 C). A similar pattern was observed for total CREB and p-CREB (Fig 6D). Quantitative real time RT-PCR showed that also mRNAs for all 4 components of the γ-secretase complex were significantly reduced in 5-LO-/- when compared with WT littermates (Supplemental Fig 5). Since the absence of 5-LO could result in a significant diversion of the arachidonic acid metabolism to the 12/15-LO or cyclooxygenase pathway, we measured brain levels of 12-HETE, prostaglandin E2 (PGE2), their major metabolic products, respectively. Brain homogenates from both groups did not significantly differ in the levels of those metabolites (PGE2: 1.38±0.15 vs. 1.46±0.12 ng/mg; 12-HETE: 15.5±0.20 vs. 16.1±0.22 pg/mg), suggesting that in the CNS of the 5-LO-/- mice there is not a different utilization of the same substrate, which could justify the Aβ differences between the two groups.
Figure 6.

Genetic absence of 5-LO reduces endogenous Aβ, CREB and γ-secretase complex levels in the central nervous system. A. Groups of 6-month-old 5-LO-/- mice (open symbols) and wild type (WT) littermates (closed symbols) were analyzed for total Aβ 1-40 and 1-42 levels in brain cortex homogenates. B. Representative immunoblot analysis for APP, BACE-1 and ADAM-10 in brain homogenates from these mice. C. Representative immunoblots and densitometric analysis for PS1, nicastrin, APH-1 and Pen 2 protein levels (open bars: WT, closed bars: 5-LO-/- mice). D. Representative immunoblots and densitometric analysis for CREB and p-CREB (open bars: WT, closed bars: 5-LO-/- mice). (*p<0.05; **p<0.01).
Discussion
In the present study, we demonstrate that 5-LO modulates Aβ formation in vitro and in vivo by regulating the transcription of the γ-secretase complex via activation of CREB. This finding provides the first evidence that this enzymatic pathway plays a functional role in aging-associated amyloidosis of the CNS. Previous studies have reported that 5-LO expression is increased in brain with aging, especially in the cortex and hippocampus, two regions which are known to be more susceptible to neurodenegeration. Taken together, these data support a possible biologic link between 5-LO and neurodegenerative diseases, whose strongest risk factor is aging. In line with this hypothesis, 5-LO immunoreactivity is increased in hippocampi of AD patients versus controls, and polymorphism of the 5-LO promoter can influence the age of onset of the disease 19,20. AD is a complex neurodegenerative disorder characterized by the presence of abundant extracellular and intracellular proteinaceous deposits, and results from a combination of environmental and genetic factors affecting these levels in the brain. Aβ is a major component of these deposits, and understanding of its production and clearance in the CNS is the focus of an enormous amount of work since it can help us to develop rational therapeutics to treat or prevent this disease. This peptide is generated from the sequential proteolytic processing of APP by the enzymes β- and γ-secretase. BACE-1, is a transmembrane aspartic protease which is responsible for initiating cleavage of APP. This cleavage product is then further processed by the γ-secretase, another membrane associated protease, to finally form the Aβ peptides. While it appears that BACE1 is represented by only one enzymatic protein, the γ-secretase is now known to be composed of four integral membrane proteins: PS1, nicastrin, Pen-2 and APH-1, which are both necessary and sufficient for active γ-secretase 21,22. However, despite its biological importance, studies of transcriptional regulatory mechanisms of the γ-secretase components are largely missing. Different potential transcriptional factor binding sites have been reported within the promoter regions of some of these four genes. Among them a CREB binding site has been shown to be essential for APH-1, Pen 2 and PS1 transcriptional regulation 23. Earlier studies have also identified promoter regions of human PS1 and Pen-2, showing that their transcription can be regulated by Ets transcription factor 24,25. Our study demonstrates that 5-LO directly activates CREB, which is then responsible for the transcription of all four components of the γ-secretase complex. First, we show that 5-HETE, the specific metabolic product of 5-LO, directly activates CREB and promote its nuclear translocation. Second, that pharmacologic inhibition of CREB activation or its mutant negative dominants block the 5-HETE-dependent transcription of the γ-secretase complex and Aβ formation. The specificity of 5-LO effect on CREB is underscored by the lack of any significant effect on another transcription factor, Sp1. Interestingly, we also found that 5-LO has no effect on another important player of Aβ proteolytic pathway, BACE-1 whose mRNA levels have been previously reported to be under the control different transcription factors such as STAT3 26,27. The biological importance of 5-LO in regulating Aβ formation via the CREB pathway was also demonstrated in an animal model of AD-like amyloidosis. Thus, pharmacological blockade or genetic absence of 5-LO resulted in a reduction of both Aβ and CREB levels in Tg2576 mice brain. Besides the APP metabolism, the γ-secretase complex is also involved in multiple important physiological activities, such as the Notch signal pathway, making it a target of extensive scrutiny 27-29. Here, we demonstrate that 5-LO effect on the γ-secretase complex is independent of any effect on the Notch signaling pathway. The observed specific effect of 5-LO on γ-secretase mediated APP processing over Notch signaling is in agreement with previous observations and in line with a modulator activity of this enzymatic pathway on the secretase. It further supports the novel concept that it is possible to develop γ-secretase modulator that alter Aβ-generating activity while preserving other essential biological function of the complex 30. This fact makes any potential therapeutic application of 5-LO inhibitor(s), which could act as γ-secretase modulators, in AD-like amyloidosis, as a feasible one without the potential toxicity of the classical inhibitors of the complex.
Our observation has physiological relevance since we demonstrate that 5-LO genetic absence results in a significant reduction of endogenous Aβ levels, and the same biochemical changes also in a biological system not driven by an APP human transgene. Thus, we show that compared with WT littermates, 5-LO-deficient mice produce less murine Aβ, without any significant change in APP, BACE1 and ADAM-10 levels. By contrast, it results in a significant reduction in both protein and mRNA levels of the γ-secretase complex, which associates with lower CREB levels. Finally, since previous reports demonstrated that another LO, i.e. 12/15-LO, and the cycloxygenase, both of which use arachidonic acid as substrate, could also influence the APP metabolism 31,7, we wanted to make sure that the absence of 5-LO did not result in a diversion of the arachidonic acid towards one of these metabolic pathways. Based on our results, we can rule out this possibility since 5-LO-/- mice did not differ from WT littermates in the amount of their metabolic products. We are aware that the relationship between 5-LO and amyloidosis is clearly a complex one, our findings, however, suggest that there is a previously unrecognized link between this pathway and the APP proteolytic processing. Further studies will be required to gain better understanding of the role that 5-LO might also play in processes leading to neurodegeneration. If 5-LO is confirmed to contribute actively to human AD, inhibitors of the enzyme deserve further investigation as a potential treatment for this disease.
Acknowledgments
This study was supported by US National Institute of Health (NIH) grants AG-022152, AG-033568. Additional support was provided by the Alzheimer's Association Zenith Fellow grant, ZEN-07-59289.
References
- 1.Radmark O, Werz O, Steinhilber D, Samuelsson B. 5-Lipoxygenase: regulation of expression and enzyme activity. Trends Biochem. Sci. 2007;32:332–341. doi: 10.1016/j.tibs.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 2.Mehrabian M, Allayee H. 5-Lipoxygenase and atherosclerosis. Curr. Opin. Lipidol. 2003;14:447–457. doi: 10.1097/00041433-200310000-00005. [DOI] [PubMed] [Google Scholar]
- 3.Uz T, Pesold C, Longone P, Manev H. Age-associated up-regulation of neuronal 5-lipoxygenase expression: putative role in neuronal vulnerability. FASEB J. 1998;123:439–449. doi: 10.1096/fasebj.12.6.439. [DOI] [PubMed] [Google Scholar]
- 4.Chinnici CM, Yao Y, Praticò D. The 5-lipoxygenase enzymatic pathway in the mouse brain: young versus old. Neurobiol. Aging. 2007;28:1457–1462. doi: 10.1016/j.neurobiolaging.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 5.Praticò D. Oxidative stress hypothesis in Alzheimer's disease: a reappraisal. Trends Pharmacol. Sci. 2008;29:609–615. doi: 10.1016/j.tips.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 6.Firuzi O, Zhuo J, Chinnici CM, Wisniewski T, Praticò D. 5-Lipoxygenase gene disruption reduces amyloid-β pathology in a mouse model of Alzheimer's disease. FASEB J. 2008;22:1169–1178. doi: 10.1096/fj.07-9131.com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Succol F, Praticò D. A role for 12/15Lipoxygenase in the Amyloid β Precursor Protein metabolism. J. Neurochem. 2007;103:380–387. doi: 10.1111/j.1471-4159.2007.04742.x. [DOI] [PubMed] [Google Scholar]
- 8.Sung S, et al. Modulation of nuclear factor-B activity by indomethacin influences Aβ levels but not Aβ precursor protein metabolism in a model of Alzheimer's disease. Am. J. Pathol. 2004;165:2197–2206. doi: 10.1016/s0002-9440(10)63269-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shineman D, Zhang B, Leight SN, et al. Thromboxane receptor activation mediates isoprostane-induced increases in amyloid pathology in Tg2576 mice. J. Neurosci. 2008;28:4785–4794. doi: 10.1523/JNEUROSCI.0684-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Josien H. Recent advances in the development of γ-secretase inhibitors. Curr. Op. Drug Disc. Devel. 2002;5:513–525. [PubMed] [Google Scholar]
- 11.Netzer WJ, et al. Gleevec inhibits βamyloid production but not Notch cleavage. Proc. Natl. Acad. Sci. USA. 2003;100:12444–12449. doi: 10.1073/pnas.1534745100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kenneth JL, Thomas DS. Analysis of relative gene expression data using Real Time quantitative PCR and the 2-ΔΔCt method. Method. 2002;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 13.Paige M, Saprito MS, Bunyan DA, Shim YM. HPLC quantification of 5 hydroxyeicosatetraenoic acid in human lung cancer tissues. Biomed. Chromatogr. 2009;23:817–821. doi: 10.1002/bmc.1191. [DOI] [PubMed] [Google Scholar]
- 14.Praticò D, et al. 12/15Lipoxygenase is increased in Alzheimer's disease: possible involvement in brain oxidative stress. Am. J. Pathol. 2004;164:1655–1662. doi: 10.1016/S0002-9440(10)63724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Riccioni G, DiIlio C, Conti P, Theoharides TC, D'Orazio N. Advances in therapy with anti-leukotriene drugs. Ann. Clin. Lab. Sci. 2004;34:379–38. [PubMed] [Google Scholar]
- 16.Geling A, Steiner H, Willem M, Bally-Cuif L, Haas C. A gamma-secretase inhibitor blocks Notch signaling in vivo and causes a severe neurogenic phenotype in zebrafish. EMBO Rep. 2002;3:688–694. doi: 10.1093/embo-reports/kvf124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dronadula N, Rizvi F, Blaskova E, Li Q, Rao GN. Involvement of cAMPresponse element binding protein-1 in arachidonic-induced vascular smooth muscle cell motility. J. Lipid Res. 2006;47:767–777. doi: 10.1194/jlr.M500369-JLR200. [DOI] [PubMed] [Google Scholar]
- 18.Sands WA, Palmer TM. Regulating gene transcription in response to cyclic AMP elevation. Cell Signaling. 2008;20:460–466. doi: 10.1016/j.cellsig.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 19.Ikonomovic MD, Abrahamson EE, Uz T, Manev H, Dekosky ST. Increased 5-Lipooxygenase imunoreactivity in hippocampus of patients with Alzheimer’ diseases. J. Histochem. Cytochem. 2008;56:1065–1073. doi: 10.1369/jhc.2008.951855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qu T, Manev R, Manev H. 5-Lipoxygenase (5-LOX) promoter polymorphism in patients with early-onset and late-onset Alzheimer's disease. J.Neuropsychiatry Clin. Neurosci. 2001;13:304–305. doi: 10.1176/jnp.13.2.304. [DOI] [PubMed] [Google Scholar]
- 21.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 22.Wolf MS. Inhibition and modulation of gamma-secretase for Alzheimer's disease. Neurotherapeutics. 2008;5:391–398. doi: 10.1016/j.nurt.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo WJ, et al. PEN-2 and APH-1 coordinately regulate proteolytic processing of presenilin 1. J. Biol. Chem. 2003;278:7850–7854. doi: 10.1074/jbc.C200648200. [DOI] [PubMed] [Google Scholar]
- 24.Wang R, et al. Transcriptional regulation of Pen-2, a key component of the γ-secretase complex, by CREB. Mol. Cell. Biol. 2006;26:1347–1354. doi: 10.1128/MCB.26.4.1347-1354.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pastorcic M, Das HK. Alternative initiation of transcription of the human presenilin 1 gene in SH-SY5Y and SK-N-SH cells. The role of Ets factors in the regulation of presenilin 1. Eur. J. Biochem. 2004;271:4485–4494. doi: 10.1111/j.1432-1033.2004.04453.x. [DOI] [PubMed] [Google Scholar]
- 26.Wen Y, et al. Transcriptional regulation of β-secretase by p25/cdk5 leads to enhanced amyloidogenic processing. Neuron. 2008;57:680–690. doi: 10.1016/j.neuron.2008.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christensen MA, et al. Transcriptional regulation of BACE-1, the β-amyloid precursor protein β secretase, by Sp1. Mol. Cell. Biol. 2004;24:865–874. doi: 10.1128/MCB.24.2.865-874.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Strooper B, et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 29.Marambaud P, et al. A presenilin-1/γ-secreatse cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 2002;21:1948–1956. doi: 10.1093/emboj/21.8.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolfe MS. Inhibition and modulation of gamma-secretase for Alzheimer's disease. Neurotherapeutics. 2008;5:391–398. doi: 10.1016/j.nurt.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qin W, et al. Cycloxygenase (COX)-2 and COX-1 potentiate beta-amyloid peptide generation through mechanisms that involve gamma-secretase activity. J Biol Chem. 2003;278:50970–50977. doi: 10.1074/jbc.M307699200. [DOI] [PubMed] [Google Scholar]