

Abstract
BACKGROUND AND PURPOSE
Cytochrome c when released from mitochondria into cytosol triggers assembly of the apoptosome resulting in caspase activation. Recent evidence suggests that reduced cytochrome c is unable to activate the caspase cascade. In this study, we investigated whether a chemical reductant of cytochrome c, N,N,N′,N′-tetramethylphenylene-1,4-diamine (TMPD), which we have previously shown to block cytochrome c-induced caspase activation, could prevent ischaemia-induced apoptosis in the rat perfused heart.
EXPERIMENTAL APPROACH
The Langendorff-perfused rat hearts were pretreated with TMPD and subjected to stop-flow ischaemia or ischaemia/reperfusion. The activation of caspases (measured as DEVD-p-nitroanilide-cleaving activity), nuclear apoptosis of cardiomyocytes (measured by dUTP nick end labelling assay), mitochondrial and cytosolic levels of cytochrome c (measured spectrophotometrically and by elisa), and reperfusion-induced necrosis (measured as the activity of creatine kinase released into perfusate) were assessed.
KEY RESULTS
We found that perfusion of the hearts with TMPD strongly inhibited ischaemia- or ischaemia/reperfusion-induced activation of caspases and partially prevented nuclear apoptosis in cardiomyocytes. TMPD did not prevent ischaemia- or ischaemia/reperfusion-induced release of cytochrome c from mitochondria into cytosol. TMPD also inhibited ischaemia/reperfusion-induced necrosis.
CONCLUSIONS AND IMPLICATIONS
These results suggest that TMPD or related molecules might be used to protect the heart against damage induced by ischaemia/reperfusion. The mechanism of this protective effect of TMPD probably involves electron reduction of cytochrome c (without decreasing its release) which then inhibits the activation of caspases.
Keywords: ischaemia/reperfusion, cytochrome c, mitochondria, caspases, tetramethylphenylenediamine, apoptosis, necrosis
Introduction
Heart ischaemia causes myocardial infarction and heart failure, which, according to the World Health Organization, are among the most common causes of human death in the world. Heart ischaemia or heart ischaemia plus reperfusion can induce necrosis and/or apoptosis of cardiomyocytes (Ganote et al., 1975; Freude et al., 1998; Lemasters et al., 1999).
In general, it is found that relatively short periods of ischaemia induce apoptosis, longer periods of ischaemia induce necrosis, while reperfusion induces necrosis with additional apoptosis (Schumer et al., 1992; Fliss and Gattinger, 1996; Kametsu et al., 2003), but the relation between these events remains unclear. We and others have shown that ischaemia induces rapid cytochrome c release from mitochondria in rat perfused hearts, followed by caspase activation and nuclear apoptosis (De Moissac et al., 2000; Borutaite et al., 2001), and these events are prevented by inhibiting the mitochondrial permeability transition pore (Borutaite et al., 2003). Cytochrome c release activates apoptosis by binding to a cytosolic adaptor protein, APAF-1, which then recruits and activates procaspase-9 within the apoptosome (Li et al., 1997). Recently, we and others have shown that the reduced form of cytochrome c (c2+) has little or no ability to activate the caspases, and that factors that keep the cytosolic cytochrome c reduced during apoptosis induction prevent caspase activation and death of cultured cell lines (Pan et al., 1999; Suto et al., 2005; Borutaite and Brown, 2007). One such factor is N,N,N′,N′-tetramethylphenylene-1,4-diamine (TMPD), which readily crosses cell membranes and rapidly reduces cytochrome c, and can be re-reduced by a number of cellular reductases or reductants (Sarti et al., 1992). Because we had previously found that TMPD was effective in preventing apoptosis and death of cultured cells, in this study we investigated the effectiveness of TMPD at preventing apoptosis and/or necrosis of ischaemic or ischaemic/reperfused hearts, and by what mechanism any effects were produced.
Methods
Animal model
All experiments were performed on hearts from 2–4 months old male Wistar rats. The procedures used in this study are approved by the European Convention for the protection of vertebrate animals used for experimental and other purposes and according to the Republic of Lithuania law on the care, keeping and use of animals (License of Lithuanian State Veterinary Service for Working with Laboratory Animals no. 0155). Rats were killed by increasing the concentration of CO2 in the air followed by cervical dislocation.
The hearts were perfused on a Langendorff perfusion system with Krebs-Henseleit solution (11 mM glucose, 118 mM NaCl, 25 mM NaHCO3, 4.8 mM KCl, 1.2 mM KH2PO4, 1.2 mM CaCl2, 1.7 mM MgSO4 and 0.7 mM Na pyruvate, saturated with 95% O2–5% CO2, pH 7.4 at 37°C) at a pressure of 80 cmH2O. After a 15 min equilibration period, freshly prepared 50 µM TMPD (Sigma, St Louis, MO, USA) was added to the perfusate and hearts were perfused for another 15 min. Control hearts were perfused for the same time but without TMPD. After perfusion with/without TMPD, hearts were subjected to 60 min stop-flow global ischaemia. In reperfusion experiments, hearts were subjected to 30 min stop-flow global ischaemia followed by 30 min reperfusion. In some experiments (where indicated), 40 min stop-flow global ischaemia followed by 30 min reperfusion was used.
Preparation of cytosolic and mitochondrial fractions
After control or ischaemic perfusions, the hearts were cooled in 0.9% KCl, then cut into small pieces and homogenized with a Teflon-glass homogenizer in the isolation buffer (10 mL·g−1 of tissue) containing 180 mM KCl, 20 mM Tris HCl, 1 mM EGTA, pH 7.3 at 4°C temperature. Cytosolic and mitochondrial fractions were separated by differential centrifugation (5 min at 750×g, 10 min at 6800×g). The post-mitochondrial supernatant was additionally centrifuged for 30 min at 10 000×g and the resulting supernatant (S10) was used for the determination of cytochrome c content in cytosol. Total cytosolic and mitochondrial protein was measured by a Biuret method.
Measurement of caspase activity
For measurement of caspase-3 activity, 1 mg·mL−1 of total cytosolic protein was incubated for 30 min in buffer containing 10% sucrose, 50 mM HEPES, 1 mM MgCl2 (pH 7.4, 37°C) and 0.1 mM z-DEVD-p-nitroanilide (Alexis, San Diego, CA, USA). The hydrolysis of caspase substrate was followed spectrophotometrically at 405 nm and was calibrated with p-nitroanilide. DEVD-cleaving activity was completely suppressed by 0.02 mM DEVD-CHO (Alexis), a reversible inhibitor of caspase-3.
Measurement of apoptosis
The number of apoptotic cells was quantified by dUTP nick end labelling (TUNEL) using CardioTACS in situ apoptosis detection kit (R&D Systems, Minneapolis, MN, USA). Hearts were fixed in 10% formalin for 24 h. The tissue of ventricles was cut and embedded in molten paraffin and frozen. Other procedures were performed according to the manufacturer's instructions. The number of TUNEL-positive cardiomyocytes was counted in at least 10 different microscopic fields on each section containing approximately 100 cardiomyocytes in each. Only cells with clear myocytic morphology were counted.
Measurement of mitochondrial and cytosolic cytochrome c content
For measurement of cytochrome c content, mitochondria were solubilized with 1% Triton X-100 (w/v). Sodium hydrosulphite-reduced minus hydrogen peroxide-oxidized absorption spectra difference was recorded with a Hitachi-557 spectrophotometer (Hitachi Ltd., Tokyo, Japan). Cytochrome c+c1 content was estimated by using the absorption difference at the wavelength pair 550/535 nm and ε= 14.5 mM−1cm−1 as described in Rieske (1967).
Cytochrome c content in cytosolic fractions was detected using Quantikinine M rat/mouse Immunoassay elisa kit (R&D Systems). Cytosolic fraction proteins were dissolved in 0.5% Triton X-100 and further procedures were performed according to the manufacturer's instructions.
Measurement of mitochondrial respiration
Mitochondrial respiration was measured with a Clarke-type oxygen electrode in the buffer containing 110 mM KCl, 2.24 mM MgCl2, 10 mM Tris HCl, 5 mM KH2PO4, 50 mM creatine (pH 7.2 at 37°C), and 1 mM pyruvate plus 1 mM malate were used as respiratory substrates. Mitochondrial state 3 respiration rate was achieved by adding 1 mM ADP.
Determination of creatine kinase activity in the perfusates
For evaluation of necrosis, perfusate from the hearts was collected over the reperfusion time and creatine kinase activity was measured. In preliminary experiments, it was found that TMPD interferes with the NADH-based assays for measurement of lactate dehydrogenase or creatine kinase activity. Therefore, we used a fluorimetric procedure for the measurement of creatine kinase activity based on chemical determination of creatine formed from phosphocreatine in the reaction of the enzyme (Rokos et al., 1972). This assay was insensitive to TMPD. Briefly, the procedure included the following steps: 10 µL of perfusate was added into 100 µL of 0.1 M Tris – Mg acetate buffer (pH 6.9) containing 25 mM creatine phosphate, 15 mM ADP, 0.015% mercaptoethanol. The blank contained all the same chemicals except ADP. After 15 min incubation at 37°C, the reaction was stopped by placing the tubes on ice. Then 1 mL of 56 mM ninhydrin and 2 mL of 1.7 M KOH solution in ethanol were added. After 6 min incubation at room temperature, the fluorescence of the sample and the blank was measured (excitation 400 nm, emission 495 nm). The concentration of creatine formed in the reaction was calculated using a calibration curve and the activity of creatine kinase was expressed as U·L−1 of sample.
Statistical analysis
Data are expressed as means ± SE of at least three separate experiments. Statistical comparisons between experimental groups were performed by anova followed by Tukey's or LSD tests. A value of P < 0.05 was considered statistically significant.
Results
TMPD prevents caspase activation induced by ischaemia or ischaemia/reperfusion
In this study, we used two models of heart ischaemia: 30 min stop-flow ischaemia followed by 30 min reperfusion or 60 min stop-flow ischaemia only. As can be seen in Figure 1, in both the ischaemia and ischaemia/reperfusion models, there was four- to fivefold increase in caspase-3 activity compared with untreated, control hearts. However, when hearts were pre-loaded with TMPD, the ischaemia-induced caspase activation was significantly decreased: in the presence of TMPD, caspase-3 activity was lower by 50–75% compared with the respective ischaemic group without TMPD (Figure 1). Perfusion of the non-ischaemic control hearts with TMPD had no effect on caspase activity (data not shown).
Figure 1.
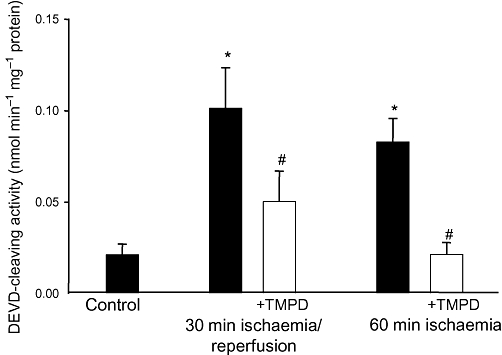
Perfusion of hearts with N,N,N′,N′-tetramethylphenylene-1,4-diamine (TMPD) prevents ischaemia-induced caspase activation. Hearts were pre-perfused for 15 min with 50 µM TMPD prior to induction of stop-flow ischaemia. Cytosolic fractions of the hearts were used for determination of caspase activity using z-DEVD-p-nitroanilide, a caspase-3-substrate. * Statistically significant effect (P < 0.01) if compared with control. # Statistically significant effect of TMPD (P < 0.01) if compared with ischaemia or ischaemia/reperfusion group. Means ± SE of 4–9 experiments on separate hearts are presented.
TMPD has no effect on ischaemia- induced release of mitochondrial cytochrome c and inhibition of mitochondrial respiration
In principle, in order to block caspase activation, TMPD might act either upstream or downstream of cytochrome c release from mitochondria during ischaemia. To test which, we measured the content of cytochrome c in mitochondria isolated from control and ischaemic hearts pre-perfused with/without TMPD. Cytochrome c content in mitochondria isolated from hearts after 30 min ischaemia/reperfusion or 60 min ischaemia was lower by about 20–30% compared with control (non-ischaemic) mitochondria (Figure 2A). Pre-loading of hearts with TMPD had no effect on these levels: the content of cytochrome c in mitochondria from hearts treated with or without TMPD was similarly decreased after 30 min ischaemia/reperfusion or 60 min ischaemia alone (Figure 2A). In accordance with the decrease in mitochondrial cytochrome c, we observed an increase in the levels of cytochrome c in the cytosol after 60 min ischaemia, and this was not prevented when hearts were pretreated with TMPD prior to ischaemia: the cytosolic cytochrome c content in ischaemia and ischaemia plus TMPD groups were not statistically significantly different from each other but significantly higher than in non-ischaemic control cytosols (Figure 2B). Some cytochrome c was always present in cytosols from control, non-ischaemic hearts, which may be caused by the disruption of mitochondria during the homogenization of heart tissue and the mitochondrial isolation procedure. These data suggest that TMPD does not prevent ischaemia-induced loss of cytochrome c from mitochondria.
Figure 2.
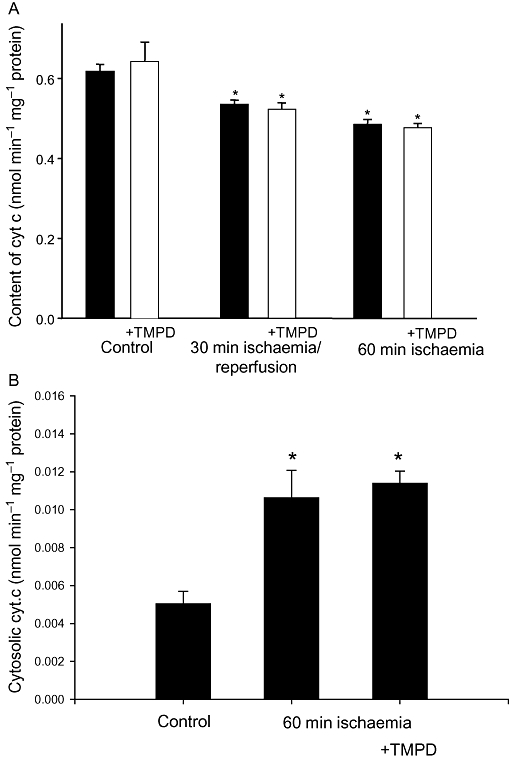
Perfusion of hearts with N,N,N′,N′-tetramethylphenylene-1,4-diamine (TMPD) has no effect on ischaemia-induced release of cytochrome c from mitochondria into cytosol. (A) Cytochrome c concentration in mitochondria; (B) cytochrome c concentration in cytosolic fractions from control and ischaemic hearts. Hearts were pre-perfused for 15 min with 50 µM TMPD prior to induction of stop-flow ischaemia. The cytochrome c concentration in isolated mitochondria was measured spectrophotometrically as described in Methods. Cytosolic concentrations of cytochrome c were measured using elisa. *Statistically significant effect if compared with control (P < 0.01). Means ± SE of 4–9 separate experiments are presented.
We have previously shown that one of the consequences of ischaemia-induced loss of cytochrome c is the inhibition of mitochondrial respiration. Figure 3 shows the effects of ischaemia and pre-ischaemic treatment of the hearts with TMPD on mitochondrial state 3 respiration rate. As can be seen, ischaemia caused an inhibition of mitochondrial respiration by 22% (30 min ischaemia/30 min reperfusion) and by 48% (60 min ischaemia) compared with control mitochondrial respiration (from non-ischaemic hearts), and this ischaemia-induced suppression of respiration was not affected by TMPD: the respiratory rates were still lower by 28–39% in mitochondria from ischaemic hearts pre-loaded with TMPD.
Figure 3.
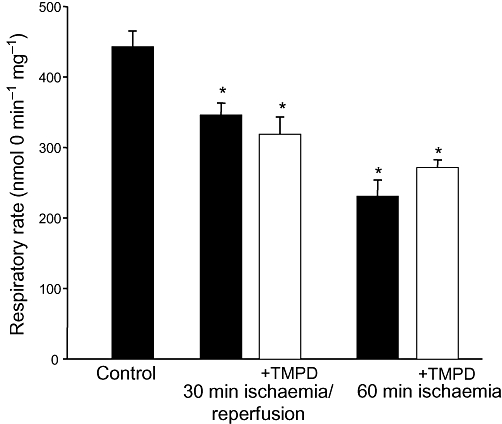
Perfusion of hearts with N,N,N′,N′-tetramethylphenylene-1,4-diamine (TMPD) did not prevent ischaemia-induced inhibition of mitochondrial respiration. Hearts were pre-perfused for 15 min with 50 µM TMPD prior to induction of stop-flow ischaemia. Mitochondrial respiration was measured using 1 mM pyruvate plus 1 mM malate as substrates. State 3 respiration rate was obtained by the addition of 1 mM ADP. *Statistically significant effect if compared with control (P < 0.01). Means ± SE of 4–9 separate experiments are presented.
In addition, we tested the direct effect of TMPD on respiration of isolated mitochondria. In these experiments, TMPD was added to control or ischaemia-damaged mitochondria respiring in state 3 with succinate as substrate. As can be seen from data presented in Table 1, TMPD had no significant effect on the steady-state rate of mitochondrial respiration in control or ischaemia-damaged mitochondria if compared with the state 3 respiratory rate in the absense of TMPD.
Table 1.
Effect of TMPD on the respiration of isolated mitochondria
| VADP (nmol·mg−1 protein) | VADP+TMPD (nmol·mg−1 protein) | |
|---|---|---|
| Control | 382 ± 31 | 356 ± 10 |
| 60 min ischaemia | 186 ± 21* | 200 ± 22* |
Mitochondria were isolated from normal rat hearts (control) or those exposed to 60 min stop-flow ischaemia. Mitochondrial respiration was measured as described in Methods, using 10 mM succinate as a respiratory substrate (plus 1 µM rotenone).
VADP– state 3 respiration rate (after addition of 1 mM ADP); VADP+TMPD– state 3 respiration rate in the presence of 50 µM TMPD.
Means ± SE of data from three hearts in each condition (control and ischaemic) are presented.
Statistically significant difference (P < 0.01) if compared with control group.
Altogether, these results provide further evidence that the protective effect of TMPD against ischaemia-induced caspase activation is induced downstream of cytochrome c release from mitochondria; we had previously shown that the ischaemia-induced respiratory inhibition is largely due to the release of cytochrome c (Borutaite et al., 1996).
TMPD prevents ischaemia-induced nuclear apoptosis
Pre-perfusion of the hearts with TMPD not only suppressed ischaemia-induced caspase activation but it also prevented ischaemia-induced nuclear DNA fragmentation, which is an indicator of apoptotic cell death. We only counted cells with clear myocytic morphology and we found that the number of TUNEL-positive (DNA-fragmented) cardiomyocytes increased from <1% up to 6% (of all cardiomyocytes) after 60 min ischaemia (Figure 4). However, when hearts were pre-loaded with TMPD before the induction of ischaemia, there was a statistically significant (∼ 45%) decrease in the number of TUNEL-positive cardiomyocytes (Figure 4), indicating that perfusion of the hearts with TMPD decreases ischaemia-induced nuclear apoptosis of cardiac myocytes.
Figure 4.
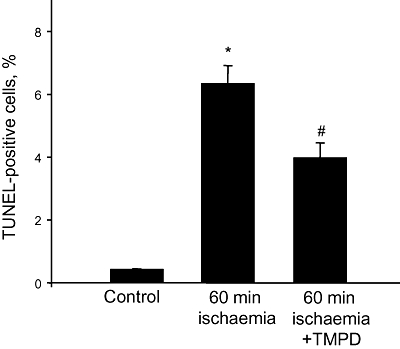
Perfusion of the hearts with N,N,N′,N′-tetramethylphenylene-1,4-diamine (TMPD) prevents ischaemia-induced apoptosis. Hearts were pre-perfused for 15 min with 50 µM TMPD prior to induction of 60 min stop-flow ischaemia. After ischaemia, hearts were fixed and stained using the Cardiotask kit for determination of cardiomyocytes with fragmented DNA. Number of TUNEL-positive cells is expressed as the percentage of total number of cardiomyocytes in tissue sections investigated. *Statistically significant effect (P < 0.01) if compared with control. #Statistically significant effect of TMPD (P < 0.01) if compared with ischaemia alone. Means ± SE of data from three hearts in each condition are presented. TUNEL, dUTP nick end labelling.
TMPD prevents ischaemia/reperfusion-induced necrosis
The loading of the hearts with TMPD also prevented ischaemia plus reperfusion-induced necrosis which was measured as an increase of creatine kinase activity in the perfusate. As shown in Figure 5, 30 min ischaemia plus 30 min reperfusion did not cause any detectable necrosis, but if the ischaemic period was prolonged to 40 min followed by 30 min reperfusion, there was a 2.3-fold increase in creatine kinase activity in the perfusate. When hearts were pre-perfused with TMPD prior to ischaemia, the ischaemia-induced release of creatine kinase from hearts was practically eliminated. This suggests that TMPD by blocking caspase activation may prevent secondary necrosis of myocardial cells, or that TMPD has some other effect beneficial for cell survival during ischaemia or ischaemia/reperfusion.
Figure 5.
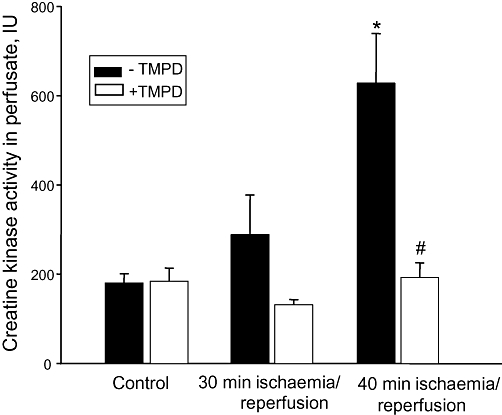
Perfusion of the hearts with N,N,N′,N′-tetramethylphenylene-1,4-diamine (TMPD) prevents ischaemia-induced necrosis. Coronary effluents during reperfusion were collected for the determination of creatine kinase activity as described in Methods. *Statistically significant effect (P < 0.01) if compared with control. #Statistically significant effect of TMPD (P < 0.01) if compared with ischaemia/reperfusion in the absence of TMPD. Means ± SE of 5–9 separate experiments are presented.
Discussion and conclusions
Both ischaemia and ischaemia plus reperfusion induce caspase activation, cytochrome c release and mitochondrial dysfunction in the rat isolated heart. We and others have previously ascribed these changes to ischaemia- and/or reperfusion-induced mitochondrial permeability transition that causes cytochrome c release, which inhibits mitochondrial respiration and activates caspases that execute apoptosis (Halestrap et al., 1998; Borutaite et al., 2003). The addition of TMPD to the hearts prior to ischaemia or ischaemia/reperfusion partially inhibited caspase activation and nuclear apoptosis without blocking cytochrome c release or the related inhibition of mitochondrial respiration. This indicates that TMPD blocks events between cytochrome c release and caspase activation. We have previously shown that TMPD and related agents can block caspase activation in cultured cells and cytosolic extracts by reducing the cytochrome c (from c3+ to c2+), which is then less able to activate the apoptosome (Brown and Borutaite, 2008). TMPD did not significantly inhibit caspases themselves after activation (Borutaite and Brown, 2007), and did not inhibit measured caspase activity in the control hearts. Our findings obtained here in the rat heart are consistent with TMPD blocking caspase activation by reducing cytochrome c (without blocking cytochrome c release), but we cannot eliminate the possibility that TMPD directly inhibited APAF-1 function.
We also showed that the addition of TMPD directly to isolated mitochondria from control and ischaemic hearts had no significant effect on the state 3 respiration rate (Table 1). This suggests that although TMPD reduces mitochondrial cytochrome c, this has little effect on the steady-state level in intact respiring mitochondria because of the relatively rapid reduction and oxidation of mitochondrial cytochrome c by the respiratory complexes; whereas TMPD may induce a more marked reduction of cytosolic cytochrome c because this pool interacts more slowly with the respiratory chain. However, we cannot rule out the possibility that TMPD may reduce other mitochondrial components without having a significant effect on respiratory function.
TMPD also strongly inhibited ischaemia plus reperfusion-induced necrosis (measured by creatine kinase release into perfusate). It has previously been shown that caspase inhibitors can block ischaemia/reperfusion-induced necrosis of perfused heart (Yaoita et al., 1998; Holly et al., 1999; Yang et al., 2003; Bott-Flügel et al., 2005;Taki et al., 2007). Thus, the most obvious reason why TMPD protected against ischaemia/reperfusion-induced necrosis is due to the TMPD-induced inhibition of caspase activation. However, we cannot rule out other explanations, such as unknown effects of TMPD on the cell components or TMPD inducing some form of preconditioning, although we think the latter unlikely as TMPD did not prevent ischaemia-induced cytochrome c release.
The present work suggests that TMPD or related molecules might be used to protect the heart against ischaemia/reperfusion-induced damage. This protective effect of TMPD may involve electron reduction (change in the redox state) of cytochrome c and hence preventing the activation of caspase, although further work is required to elucidate the exact mechanism.
Acknowledgments
This work was supported by the Science Council of Lithuania.
Glossary
Abbreviations
- APAF-1
apoptosis activating factor 1
- TMPD
N,N,N′,N′-tetramethylphenylene-1,4-diamine
- TUNEL
dUTP nick end labelling
Conflict of interest
None.
Supporting Information
Supporting Information: Teaching Materials; Figs 1–5 as PowerPoint slide.
References
- Borutaite V, Brown GC. Mitochondrial regulation of caspase activation by cytochrome oxidase and tetramethylphenylenediamine via cytosolic cytochrome c redox state. J Biol Chem. 2007;282:31124–31130. doi: 10.1074/jbc.M700322200. [DOI] [PubMed] [Google Scholar]
- Borutaite V, Morkuniene R, Budriunaite A, Krasauskaite D, Ryselis S, Toleikis A, et al. Kinetic analysis of changes in activity of heart mitochondrial oxidative phosphorylation system induced by ischaemia. J Mol Cell Cardiol. 1996;28:2195–2201. doi: 10.1006/jmcc.1996.0211. [DOI] [PubMed] [Google Scholar]
- Borutaite V, Budriunaite A, Morkuniene R, Brown GC. Release of mitochondrial cytochrome c and activation of cytosolic caspases induced by myocardial ischemia. Biochim Biophys Acta. 2001;1537:101–109. doi: 10.1016/s0925-4439(01)00062-x. [DOI] [PubMed] [Google Scholar]
- Borutaite V, Jekabsone A, Morkuniene R, Brown GC. Inhibition of mitochondrial permeability transition prevents mitochondrial dysfunction, cytochrome c release and apoptosis induced by heart ischemia. J Mol Cell Cardiol. 2003;35:357–366. doi: 10.1016/s0022-2828(03)00005-1. [DOI] [PubMed] [Google Scholar]
- Bott-Flügel L, Weig HJ, Knödler M, Städele C, Moretti A, Laugwitz KL, et al. Gene transfer of the pancaspase inhibitor P35 reduces myocardial infarct size and improves cardiac function. J Mol Med. 2005;83:526–534. doi: 10.1007/s00109-005-0683-z. [DOI] [PubMed] [Google Scholar]
- Brown GC, Borutaite V. Regulation of apoptosis by the redox state of cytochrome c. Biochim Biophys Acta. 2008;1777:877–881. doi: 10.1016/j.bbabio.2008.03.024. [DOI] [PubMed] [Google Scholar]
- De Moissac D, Gurevich RM, Zheng H, Singal PK, Kirshenbaum LA. Caspase activation and mitochondrial cytochrome c release during hypoxia-mediated apoptosis of adult ventricular myocytes. J Mol Cell Cardiol. 2000;32:53–63. doi: 10.1006/jmcc.1999.1057. [DOI] [PubMed] [Google Scholar]
- Fliss H, Gattinger D. Apoptosis in ischaemic and reperfused rat myocardium. Circ Res. 1996;79:949–956. doi: 10.1161/01.res.79.5.949. [DOI] [PubMed] [Google Scholar]
- Freude B, Masters TN, Kostin S, Robicsek F, Schaper J. Cardiomiocyte apoptosis in acute and chronic conditions. Basic Res Cardiol. 1998;93:85–89. doi: 10.1007/s003950050066. [DOI] [PubMed] [Google Scholar]
- Ganote ChE, Gomes RS, Nayler WG, Jennings RB. Irreversible myocardial injury in anoxic perfused rat heart. Am J Pathol. 1975;80:419–438. [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Kerr PM, Javadov S, Woodfield KY. Elucidating the molecular mechanism of the permeability transition pore and its role in reperfusion injury of the heart. Biochim Biophys Acta. 1998;1366:79–94. doi: 10.1016/s0005-2728(98)00122-4. [DOI] [PubMed] [Google Scholar]
- Holly TA, Drincic A, Byun Y, Nakamura S, Harris K, Klocke FJ, et al. Caspase inhibition reduces myocyte cell death induced by myocardial ischemia and reperfusion in vivo. J Mol Cell Cardiol. 1999;31:1709–1715. doi: 10.1006/jmcc.1999.1006. [DOI] [PubMed] [Google Scholar]
- Kametsu Y, Osuga S, Hakim AM. Apoptosis occurs in the Penumbra Zone during short-duration focal ischemia in the rat. J Cereb Blood Flow Metab. 2003;23:416–422. doi: 10.1097/01.WCB.0000052281.23292.7C. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Qian T, Bradham CA, Brenner DA, Cascio WE, Trost LC, et al. Mitochondrial dysfunction in the pathogenesis of necrotic and apoptotic cell death. J Bioenerg Biomembr. 1999;31:305–319. doi: 10.1023/a:1005419617371. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/Caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Pan Z, Voehringer DW, Meyn RE. Analysis of redox regulation of cytochrome c-induced apoptosis in a cell-free system. Cell Death Differ. 1999;6:683–688. doi: 10.1038/sj.cdd.4400544. [DOI] [PubMed] [Google Scholar]
- Rieske JS. The quantitative determination of mitochondrial hemoproteins. Methods Enzymol. 1967;10:488–493. [Google Scholar]
- Rokos JA, Rosalki SB, Tarlow D. Automated fluorometric procedure for measurement of creatine phosphokinase activity. Clin Chem. 1972;18:193–198. [PubMed] [Google Scholar]
- Sarti P, Antonini G, Malatesta F, D'Itri E, Brunori M, Blanck TJ. Spectral analysis of cytochromes in rat heart myocytes: transient and steady-state photodiode array spectrophotometry measurements. Arch Biochem Biophys. 1992;299:8–14. doi: 10.1016/0003-9861(92)90237-q. [DOI] [PubMed] [Google Scholar]
- Schumer M, Colombel MC, Sawczuk IS, Gobé G, Connor J, O'Toole KM, et al. Morphologic, biochemical, and molecular evidence of apoptosis during the reperfusion phase after brief periods of renal ischemia. Am J Pathol. 1992;140:831–838. [PMC free article] [PubMed] [Google Scholar]
- Suto D, Sato K, Ohba Y, Yoshimura T, Fuji J. Suppression of the pro-apoptotic function of cytochrome c by singlet oxygen via a heme redox state-independent mechanism. Biochem J. 2005;392:399–406. doi: 10.1042/BJ20050580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taki J, Higuchi T, Kawashima A, Fukuoka M, Kayano D, Tait JF, et al. Effect of postconditioning on myocardial 99mTc-annexin-V uptake: comparison with ischemic preconditioning and caspase inhibitor treatment. J Nucl Med. 2007;48:1301–1307. doi: 10.2967/jnumed.106.037408. [DOI] [PubMed] [Google Scholar]
- Yang W, Guastella J, Huang JC, Wang Y, Zhang L, Xue D, et al. MX1013, a dipeptide caspase inhibitor with potent in vivo antiapoptotic activity. Br J Pharmacol. 2003;140:402–412. doi: 10.1038/sj.bjp.0705450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaoita H, Ogawa K, Maehara K, Maruyama Y. Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation. 1998;97:276–281. doi: 10.1161/01.cir.97.3.276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.