

Abstract
BACKGROUND AND PURPOSE
Dietary intake of ω-3 polyunsaturated fatty acids (ω-3 PUFAs) like eicosapentaenoic acid (EPA) decreases cancer risk, while arachidonic acid and other ω-6 PUFAs increase risk, but the underlying mechanisms are unclear. Cytochrome P450 (CYP)-derived epoxides contribute to enhanced tumourigenesis due to ω-6 PUFA intake. Thus, ω-6 arachidonic acid epoxides (EETs) inhibit apoptosis and stimulate proliferation by up-regulating cyclin D1 expression in cells. The present study evaluated the corresponding ω-3 PUFA epoxides and assessed their role in the regulation of cell proliferation.
EXPERIMENTAL APPROACH
Four chemically stable EPA epoxides (formed at the 8,9-, 11,12-, 14,15- and 17,18-olefinic bonds) were synthesized and tested against growth-related signalling pathways in brain microvascular endothelial bEND.3 cells. Cell cycle distribution was determined by flow cytometry and cyclin gene expression by immunoblotting and real-time PCR. The role of the p38 mitogen-activated protein (MAP) kinase in cyclin D1 dysregulation was assessed using specific inhibitors and dominant-negative expression plasmids.
KEY RESULTS
The ω-3 17,18-epoxide of EPA decreased cell proliferation, interrupted the cell cycle in S-phase and down-regulated the cyclin D1/cyclin-dependent kinase (CDK)-4 complex, whereas the 8,9-, 11,12- and 14,15-epoxides were either inactive or enhanced proliferation. Cyclin D1 down-regulation by 17,18-epoxy-EPA was mediated by activation of the growth-suppressing p38 MAP kinase, but the alternate EPA-epoxides were inactive.
CONCLUSIONS AND IMPLICATIONS
The present findings suggest that the epoxide formed by CYP enzymes at the ω-3 olefinic bond may contribute to the beneficial effects of ω-3 PUFA by down-regulating cyclin D1 and suppressing cell proliferation.
Keywords: epoxy fatty acids, cell proliferation, ω-3 polyunsaturated fatty acid, cyclin-dependent kinases, p38 MAP kinase, bEND.3 cell
Introduction
Epidemiological studies suggest that there is an inverse relationship between cancer rates in humans and intake of ω-3 polyunsaturated fatty acids (PUFAs), such as eicosapentaenoic acid (EPA; 20:5ω-3) (Bougnoux, 1999). In contrast, intake of ω-6 PUFAs, such as arachidonic acid (AA, 20:4ω-6), increases the risk of tumourigenesis (Maillard et al., 2002). Although the mechanisms by which PUFAs influence tumour progression have not been fully elaborated, findings from several laboratories, including our own, suggest that biotransformation to eicosanoid products may be important (Tapiero et al., 2002; Szymczak et al., 2008).
Recent evidence indicates that epoxyeicosatrienoic acids (EETs) formed by the action of cytochromes P450 (CYPs) on AA stimulate tumour cell growth. AA-derived EETs promote the neoplastic phenotype of carcinoma cells and increase the proliferation of tumour cells (Chen et al., 2001; Jiang et al., 2005). Furthermore, EETs may also enhance tumour growth by stimulating angiogenesis (Fleming, 2007), which increases delivery of nutrients and oxygen to tumours and promotes metastatic disease.
Cell growth is highly controlled, involving the coordinated activation of numerous regulatory proteins including cyclins, catalytic cyclin-dependent kinase (CDK) subunits and CDK inhibitors (CDKi) (Walker and Assoian, 2005). Cyclin D1 is a pivotal component that is activated in response to mitogens at the point of entry to the cell cycle. The cyclin D1/CDK4 complex regulates the transition to S-phase in cells and acts in concert with cyclin E/CDK2 to phosphorylate the retinoblastoma protein and activate genes that enable the progression of the cell cycle towards mitogenesis (Stacey, 2003). In previous studies the treatment of cells with EETs, or over-expression of CYP epoxygenases, activated cyclin D1 expression and accelerated cell cycle progression (Potente et al., 2002). In coculture systems, astrocytes, which generate and release EETs, stimulated mitogenesis and angiogenesis in cerebral microvascular endothelial cells (Zhang and Harder, 2002). These actions of EETs may be elicited via modulation of the activity of several receptor-mediated signalling pathways, including those for ligands such as thromboxane, peroxisome proliferators and epidermal growth factor (Chen et al., 1999; Michaelis et al., 2003; Ng et al., 2007; Behm et al., 2009); however, a clear consensus regarding potential mechanisms has not yet emerged. Importantly, in the context of tumour cell proliferation, it has been demonstrated that 14,15-EET inhibits apoptosis and promotes cell survival (Potente et al., 2003). An important target for the promitogenic activity of EETs appears to be the cell cycle regulatory gene cyclin D1 (Potente et al., 2002; Michaelis et al., 2003).
The antitumour effects of ω-3 PUFAs have been tested using a range of experimental approaches, including cultured cells, animal models and clinical studies (Rose and Connolly, 1993; Grammatikos et al., 1994), but their mode of action has not yet been established. In view of the wide tissue distribution of CYP PUFA epoxygenases, the objective of the present work was to address the possibility that epoxides may contribute to the beneficial actions of ω-3 PUFA in the regulation of cell proliferation. The present study offers evidence that 17,18-epoxy-EPA, which possesses an epoxide at the ω-3 double bond of EPA, impairs cellular proliferation. The principal finding to emerge was that 17,18-epoxy-EPA selectively decreased the expression of cyclin D1 and its functional partner CDK4, and paused the cell cycle in S-phase. Furthermore, 17,18-epoxy-EPA selectively activated the growth-suppressing p38 mitogen-activated protein (MAP) kinase (Ensembl ID ENSG00000112062) pathway to down-regulate cyclin D1. This effect was specific for the 17,18-epoxide of EPA because the alternate epoxy-EPA regioisomers were inactive. The p38 MAP kinase has been found to be a target for ω-3 PUFA in vascular cells (Diep et al., 2000; Abedin et al., 2006); the present findings suggest that these actions may be mediated, at least in part, by ω-3 PUFA epoxides.
Methods
Materials
The reagents used in this work were obtained as follows: 8,9-EET, 11,12-EET, 14,15-EET and the extracellular signal-regulated kinase (ERK) inhibitor PD98059 (Cayman Chemical, Ann Arbor, MI, USA); the p38 and c-Jun-N-terminal kinase (JNK) MAP kinase inhibitors SB203580 and SP600125 respectively (Alexis Biochemicals, Farmingdale, NY, USA); dimethyl sulphoxide (DMSO), RNase A, propidium iodide, 3-(4,5-dimethylthiazol-2-yl)-2,5-biphenyl tetrazolium bromide (MTT), horseradish peroxidase-conjugated anti-rabbit and anti-mouse IgGs, and etoposide (Sigma Chemical, St. Louis, MO, USA); trypan blue and Lipofectamine™ 2000 (Invitrogen, Mount Waverley, Vic., Australia); Ponceau S red stain (ICN biomedical, Solon, OH, USA); Dulbecco's modified eagle medium (DMEM), fetal bovine serum, L-glutamine, trypsin/EDTA, penicillin and streptomycin (Thermo Fisher Scientific, Waltham, MA, USA); phosphate-buffered saline (PBS) (Amresco, Solon, OH, USA); anti-cyclin A, anti-cyclin B1, anti-cyclin E, anti-cyclin D1, anti-CDK4, anti-CDK6 and anti-β-actin antibodies (Santa Cruz biotechnology, Santa Cruz, CA, USA); rabbit antibodies against p38, ERK and JNK MAP kinases, and phospho-p38, phospho-ERK and phospho-JNK MAP kinases (Cell Signalling Technology, Arundel, Qld, Australia) and anti-p27kip1 and anti-p21cip1 antibodies (BD Biosciences, San Jose, CA, USA); colorimetric caspase-3 activity assay kit (Promega, Annandale, NSW, Australia); bicinchoninic acid protein assay kit (Pierce Biotechnology, Woburn, MA, USA); enhanced chemiluminescence (ECL) detection reagents and Hyperfilm ECL autoradiography film (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK).
Synthesis of 17,18-, 14,15-, 11,12- and 8,9-epoxy-EPA isomers
The methyl ester of EPA was synthesized as described previously (Cui et al., 2009). m-Chloroperoxybenzoic acid (3 mM) in dichloromethane was added to the EPA methyl ester, excess acid was neutralized with aqueous sodium hydrogen carbonate (5%), and the organic phase was dried with anhydrous sodium sulphate. The isomeric monoepoxides were partially resolved by normal-phase short-column vacuum chromatography (4 cm × 5 cm i.d.) using ethyl acetate : hexane (2:98). An Alltech Econosil Silica column (10 µm; 250 mm × 10 mm i.d.) was used to purify the monoexpoxides on a Beckman System Gold 126P system (Global Medical-Instrumentation, Ramsey, MN, USA) with a mobile phase of ethyl acetate : hexane (3:97). Structures were assigned using 1D- and 2D-NMR and LC-MS/MS.
Individual epoxy-EPA methyl esters were hydrolysed to the corresponding epoxy-acids with NaOH at 50°C. After acidification with 1 M HCl, epoxy-acids were extracted with dichloromethane and analysed by 300 MHz 1H-NMR before further purification using normal-phase short-column vacuum chromatography. Structures were confirmed by LC-MS and LC-MS/MS, using negative ion electrospray ionization as described previously (Cui et al., 2009).
Cell culture and cell treatments
Astrocytes have been shown to generate EETs that regulate proliferation and differentiation in adjacent microvascular endothelial cells in a paracrine fashion (Zhang and Harder, 2002). Thus, endothelial cells are an important target cell for CYP-derived PUFA epoxides. In the present study, the polyoma virus middle T antigen-transformed bEND.3 microvascular endothelial cell line (generously provided by Prof. N. H. Hunt, Department of Pathology, University of Sydney, NSW, Australia) was used to study the actions of individual epoxy-EPA and EET isomers. Cells were maintained in monolayers at 37°C in DMEM containing penicillin and streptomycin, L-glutamine and 10% fetal bovine serum in an atmosphere of 95% air and 5% CO2. Cells were seeded in 24-well plates (7 × 104 cells·mL−1 per well) and media was replaced after 24 h with serum-free DMEM to synchronize cells in G0-phase. Test compounds were added 24 h later in fresh serum-free medium. In experiments with MAP kinase inhibitors, cells were treated with SB203580 (p38 MAP kinase inhibitor, 25 µM), SP600125 (JNK MAP kinase inhibitor, 25 µM) or PD98059 (ERK MAP kinase inhibitor, 10 µM), respectively, 2 h before the addition of 17,18-epoxy-EPA (10 µM in 0.03% DMSO); solvent alone was added to control cells.
Transient transfection of bEND.3 cells with a dominant-negative mutant of p38 MAP kinase
Plasmid DNA (2 µg) encoding a dominant-negative mutant of the p38 MAP kinase (generously provided by Dr R. J. Davis, Howard Hughes Medical Institute, University of Massachusetts Medical School, Worcester, MA, USA) was transfected into bEND.3 cells using Lipofectamine™ 2000 in serum-free DMEM according to the manufacturer's protocol. Twenty-four hours later, transfection medium was replaced with serum-free DMEM containing 17,18-epoxy-EPA (10 µM) or DMSO (0.03%, control) for 6 or 16 h.
Cell morphology, proliferation and apoptosis
17,18-epoxy-EPA-treated and untreated cells were washed with PBS and morphological changes were observed using an OlympuS-phase-contrast microscope (Melville, NY, USA). Cells were then harvested with trypsin/EDTA and viable cells were counted by trypan blue exclusion.
Effects of EETs and epoxy-EPA derivatives on cell proliferation were quantified by the reduction of MTT at 540 nm in a PerkinElmer multilabel counter (Waltham, MA, USA); the topoisomerase II inhibitor etoposide (10 µM) was used as a positive control. In further experiments, cells were harvested with trypsin/EDTA, resuspended in serum-free medium, and reconstituted Caspase-Glo3/7 reagent (Promega) was added for the assessment of apoptosis. Luminescence was measured using a PerkinElmer multilabel counter.
Cell cycle analysis
Treated cells were fixed overnight at −20°C in 80% ethanol, centrifuged at 660× g for 15 min at 4°C and 0.1 M PBS containing 0.1% NP40, and 0.1 mg·mL−1 RNAse A was added. After being stained with propidium iodide (50 µg·mL−1), cells were incubated on ice for 1 h and subjected to flow cytometry on a FACS-Calibur instrument (BD Biosciences) using CellQuest software (BD Biosciences, North Ryde, NSW, Australia). The cell cycle distribution was determined using Modfit version 1.2 (Verity Software House, Topsham, ME, USA).
Western blot analysis
Treated cells were harvested in trypsin/EDTA and lysed in buffer containing 1% Igepal, 0.5% sodium deoxycholate, 0.1% sodium dodecylsulphate, 0.1 mM phenylmethylsulphonyl fluoride, 5 µg·mL−1 aprotinin and 5 µg·mL−1 leupeptin. Washed pellets were isolated by centrifugation and the protein contents of lysates were determined by the bicinchoninic acid method.
Lysate protein (20 µg) was electrophoresed on 12% sodium dodecylsulphate-polyacrylamide gels and transferred to nitrocellulose membranes (Whatman, Dassel, Germany) essentially as described previously (Murray et al., 2005). Membranes were probed overnight at 4°C with the primary antibodies followed by washing and incubation with horseradish peroxidase-labelled anti-mouse IgG or anti-rabbit IgG. Detection was performed by ECL and autoradiography. X-ray films were analysed using a Bio-Rad GS-800 Calibrated Densitometer (Hercules, CA, USA). Membranes were re-probed for β-actin to normalize protein expression.
RNA extraction and real-time RT-PCR
Total RNA was extracted using Tri Reagent (Astral Scientific, Gymea, NSW, Australia) according to the manufacturer's protocol and was quantified spectrophotometrically. RNA samples were DNAse treated (RQ1 kit, Promega) and the first cDNA strands for cyclin D1 and RPL-13a were generated using iScript (Bio-Rad). PCR products were cloned into the pGEM-T vector (Promega) and plasmid DNA was used to generate the standard curves for cyclin D1 and RPL-13a. Previously reported gene-specific primers for cyclin D1 (Floyd et al., 2006) and the internal control RPL-13a (Ball et al., 2007) were used in gene amplification. Cycling conditions (Rotor-Gene™ 6000; Corbett Research, Sydney, NSW, Australia) were as follows: 95°C (15 min); 40 cycles of denaturation (95°C, 30 s); annealing (59°C, 1 min); elongation (72°C, 1 min).
Real-time RT-PCR was performed using the QuantiTect SYBR Green RT-PCR kit (Qiagen, Doncaster, Vic., Australia), and the purity of RT-PCR products was assessed by melting curve analysis. For each gene, three or four independent experiments were performed with triplicate aliquots of each RNA sample and the cyclin D1 copy number was normalized to RPL-13a. Gene expression of cyclins A and E (Schmetsdorf et al., 2007) was quantified by delta, delta CT analysis by co-amplification with RPL-13a: relative expression = 2−ΔΔCt, where ΔΔCt = (ΔCttarget–ΔCtRPL-13a)treated– (ΔCttarget–ΔCtRPL-13a)control (Livak and Schmittgen, 2001).
Statistical analysis
All data are expressed as means ± SEM. After one-way anova, the Fisher's protected least significant difference test was used to detect differences between multiple treatments. The number of replicate experiments is indicated in figure legends.
Results
Synthesis and characterization of epoxy-EPA isomers
In order to investigate the effects of the individual monoepoxides of EPA, the isomers were synthesized as described in the Methods section. Because the potential biological activity of contaminants could confound experimental outcomes, the purity of the products was assessed by LC-MS and found to be at least 90%, 95%, 95%, 95% and 87% for 17,18-, 14,15-, 11,12-, 8,9- and 5,6-epoxy-EPA respectively; retention times on reversed-phase C18 column chromatography were 11.8, 12.4, 12.6, 12.9 and 13.8 min respectively (I–V; Figure 1A). Structures (Figure 1B) were further characterized by MS/MS of the negative molecular ions ([M-H]-; m/z, 317), which were observed in the spectra of each compound (Figure 1C). The unique fragmentation patterns show major peaks arising from cleavage of epoxides and α- to the epoxy ring (Figure 1B) as proposed previously (Cui et al., 2009). These characteristic fragments, arising principally from [M-H]- and [M-H-COOH]-, respectively, are indicated as structures a and b in the panels showing the 17,18- and 14,15-epoxy-EPA isomers (I and II respectively; Figure 1B). However, 5,6-epoxy-EPA was chemically unstable and was not included in subsequent experiments undertaken in cells.
Figure 1.
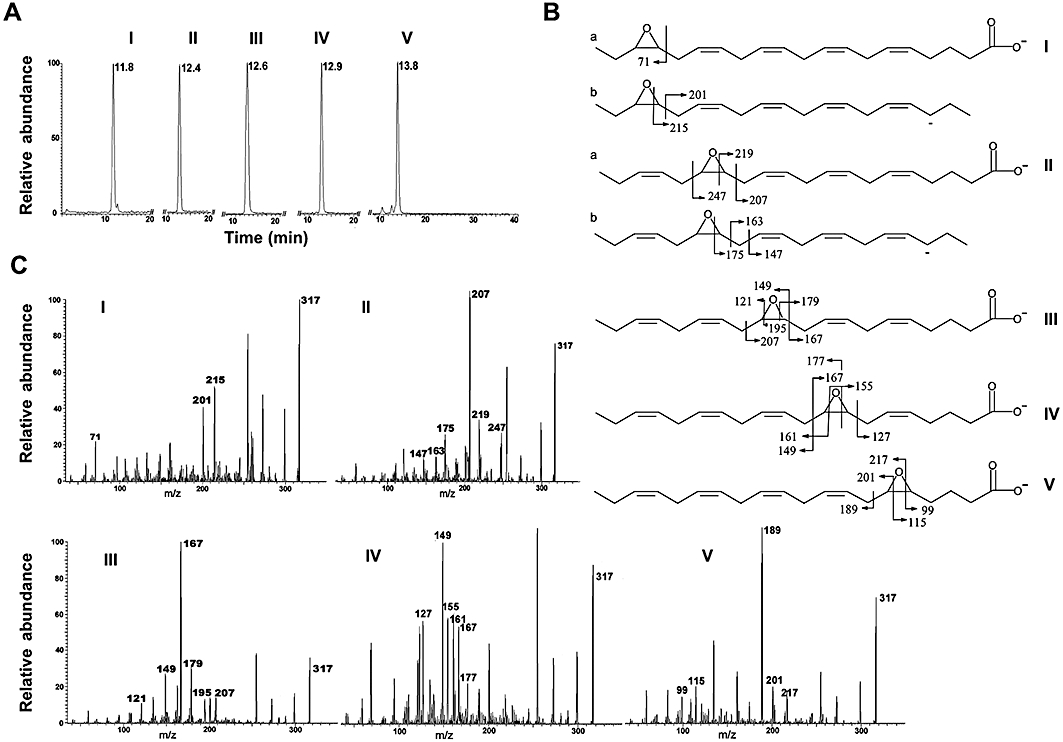
LC-MS and MS/MS analysis of eicosapentaenoic acid (EPA) monoepoxides. (A) Retention times of isomeric EPA monoepoxides on reverse-phase LC-MS: 17,18-epoxide (I), 14,15-epoxide (II), 11,12-epoxide (III), 8,9-epoxide (IV) and 5,6-epoxide (V). (B) Structures and predicted fragments arising from the epoxide ring systems in isomeric EPA monoepoxides (I–V) on MS/MS analysis. (C) MS/MS spectra showing major diagnostic ions for EPA monoepoxide isomers (I–V).
17,18-epoxy-EPA, but not other epoxy-EPA isomers, inhibits cell proliferation
MTT reduction was used to assess the effects of the four stable epoxides of EPA (17,18-, 14,15-, 11,12- and 8,9-epoxy-EPA) on the proliferation of cells that had been synchronized by removal of serum. Proliferation was decreased by 17,18-epoxy-EPA (10 µM, P < 0.01) to 61 ± 6% of control after 24 h of treatment, whereas the 8,9- and 11,12-isomers (10 µM) elicited slight increases in MTT reduction by 22 ± 8% and 20 ± 3% over control, respectively (P < 0.05), and 14,15-epoxy-EPA was without significant effect (Figure 2A). The actions of 17,18-epoxy-EPA were apparently maximal at 10 µM and were not increased further at 50 µM (Figure 2B). Similarly, when tested at a concentration of 50 µM, the effects of 8,9-, 11,12- and 14,15-epoxy-EPA on MTT reduction (124 ± 6, 114 ± 10 and 104 ± 13% of control respectively) were similar to those observed at 10 µM. By comparison, epoxides formed from ω-6 AA (8,9-EET, 11,12-EET and 14,15-EET; 10 µM) increased bEND.3 cell proliferation by 45 ± 14%, 42 ± 5% (both P < 0.05) and 71 ± 36% (P < 0.01) over control respectively (Figure 2A).
Figure 2.
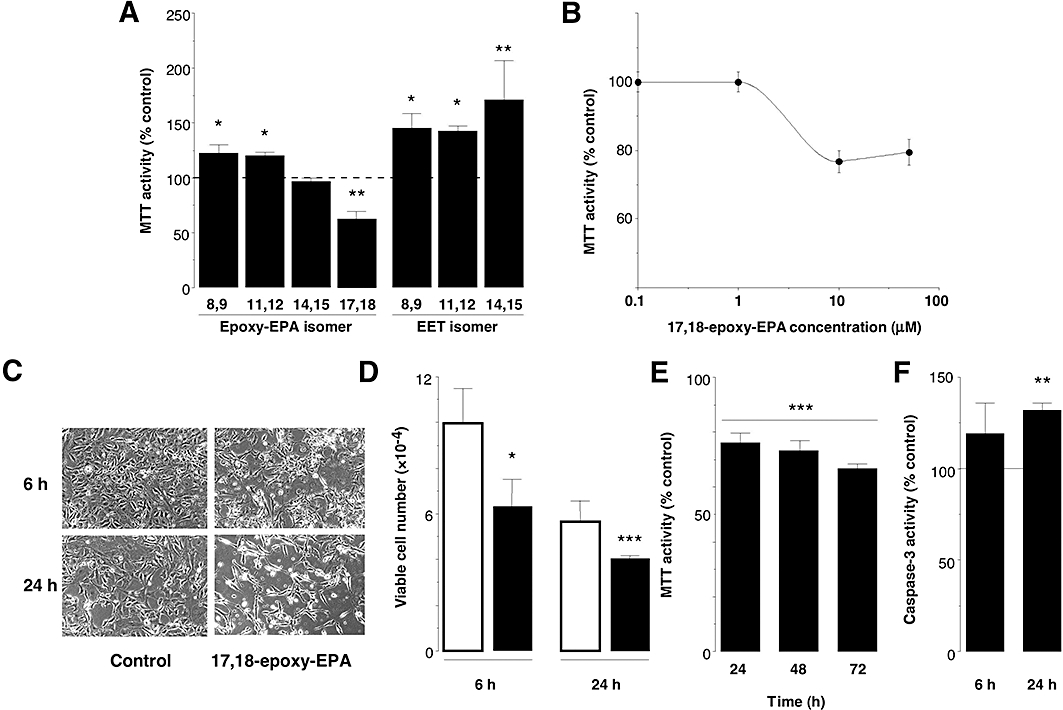
Effects of eicosapentaenoic acid (EPA) monoepoxides on cell proliferation. (A) Effects of alternate EPA monoepoxides and epoxyeicosatrienoic acid (EET) isomers (10 µM) on cell proliferation. (B) Concentration-dependent effects of 17,18-epoxy-EPA on MTT reduction in bEND.3 cells over 24 h. (C) Appearance of cultured cells 6 and 24 h after treatment with 17,18-epoxy-EPA (10 µM), showing decreased confluence and increased non-viable unattached cells; magnification: ×20. (D) Trypan blue exclusion assay showing decreases in number of viable cells after 6 and 24 h of treatment with 17,18-epoxy-EPA (10 µM; solid columns) relative to control (open columns). (E) Time-dependent decreases in MTT reduction in bEND.3 cells treated with 17,18-epoxy-EPA (10 µM) at 24 h intervals. (F) Caspase-3 activity in 17,18-epoxy-EPA-treated (10 µM) bEND.3 cells after 6 and 24 h. Data are means ± SEM of three separate experiments. Different from corresponding time-matched control: *P < 0.05, **P < 0.01, ***P < 0.001.
Short-term treatment with 17,18-epoxy-EPA for 6 and 24 h produced morphological changes in bEND.3 cells that were consistent with decreased cell growth and proliferation. Compared to control cells, the viable cell density was decreased by treatment with 17,18-epoxy-EPA (10 µM for 6 and 24 h) and there were increased numbers of unattached non-viable cells (Figure 2C); similar morphological effects were observed up to 72 h of treatment (not shown). A single application of 17,18-epoxy-EPA to cells decreased cell viability (as determined by trypan blue exclusion assay) by 37 ± 12% and 31 ± 3% relative to time-matched control, after 6 and 24 h of treatment respectively (Figure 2D). By comparison, the 11,12-isomer increased cell viability slightly after 24 h of treatment to 112% of DMSO-treated control (P < 0.05), but the other isomers were inactive. To test whether degradation may limit the effects of a single application, 17,18-epoxy-EPA (10 µM) was added at 24 h intervals (Figure 2E). Cell proliferation (MTT reduction) in synchronized cells decreased progressively by 23 ± 4% (24 h), 27 ± 2% (48 h) and 34 ± 3% (72 h) compared with corresponding time-matched controls. These data suggest that the effects of the epoxide on cell proliferation may be relatively short-term.
We tested the additional possibility that the decreased proliferation of bEND.3 cells elicited by 17,18-epoxy-EPA might be due to increased apoptosis. Because of the central role of caspase-3 in apoptotic pathways, the effect of 17,18-epoxy-EPA on the activity of the enzyme was assessed. Although caspase-3 activity was not significantly different from control after 6 h of treatment with 17,18-epoxy-EPA (10 µM), an increase to 131 ± 6% of control was evident by 24 h (P < 0.01; Figure 2F). By comparison, the cytotoxic agent etoposide (100 µM, 24 h) was used as a positive control and strongly increased caspase-3 activity in bEND.3 cell lysates to 200 ± 32% of control (P < 0.05, data not shown). These findings suggest that decreased proliferation of bEND.3 cells elicited by 17,18-epoxy-EPA may be due to initial cytostatic effects followed by an increase in apoptosis.
17,18-epoxy-EPA interrupts cell cycle progression
The effects of 17,18-epoxy-EPA on cell cycle progression in unstimulated bEND.3 cells were assessed by flow cytometry. Cells were treated with 17,18-epoxy-EPA (10 µM) for 16 h, stained with propidium iodide, and populations in different phases of the cell cycle were quantified; an increase in S-phase was apparent (Figure 3A). The time dependence of the pause in cell cycle progression was assessed. Thus, 6 h of treatment with 17,18-epoxy-EPA increased the proportion of cells in S-phase by 45% over control (12.3 ± 1.0% vs. 8.5 ± 1.1% in control; P < 0.01; Figure 3B), which was more pronounced after 16 h of treatment (cells in S-phase were 70% over control; P < 0.001). However, by 24 h the proportion of cells in S-phase had essentially normalized; it is feasible that 17,18-epoxy-EPA may undergo biotransformation in cells to less active products over 24 h of incubation as PUFA epoxides are readily hydrated by soluble epoxide hydrolase (Yu et al., 2000). The relatively small decreases in cell populations in G0/G1- and G2/M-phases effected by 17,18-epoxy-EPA did not attain statistical significance. Considered together, the short-term effect of 17,18-epoxy-EPA is consistent with a pause in the progression of cells already in the cell cycle, leading to accumulation of cells in S-phase as well as an increase in apoptosis.
Figure 3.
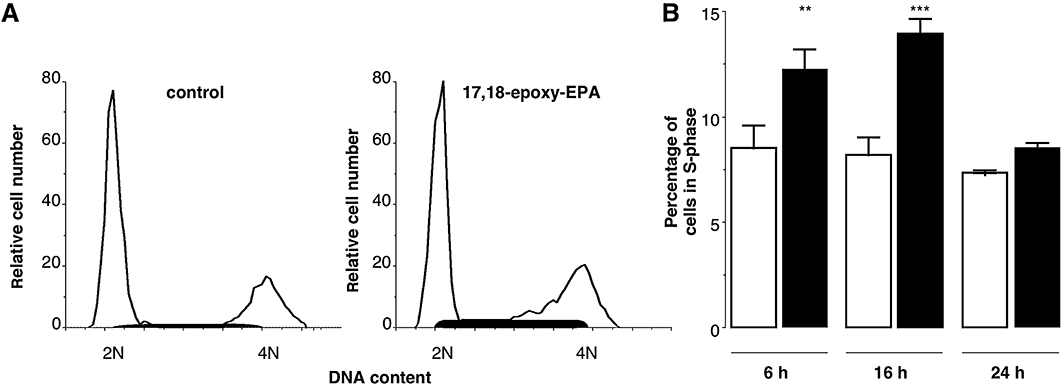
Inhibition of cell cycle progression by 17,18-epoxy-EPA (10 µM). Exponentially growing bEND.3 cells were synchronized to G1-phase by removal of serum and were treated 24 h later with either 17,18-epoxy-EPA or dimethyl sulphoxide (control). (A) Cell cycle histogram of a representative experiment after 16 h of treatment: control (left), 17,18-epoxy-EPA (right). The shaded areas indicate cells in S-phase; DNA content: 2N diploid, 4N tetraploid. (B) Percentage of bEND.3 cells in S-phase after 6, 16 or 24 h of treatment with 17,18-epoxy-EPA (open columns) relative to time-matched control (solid columns). Data are means ± SEM of three separate experiments. Different from corresponding control: **P < 0.01, ***P < 0.001.
In comparison with these findings, the positive-control etoposide (10 µM, 16 h) increased the proportion of cells in S- and G2/M-phases by 56% (P < 0.001) and 16% (P < 0.05) over control (data not shown). When tested at 100 µM, etoposide increased the distribution of bEND.3 cells in S- and G2/M-phases by 90% (P < 0.001) and 20% (P < 0.01) over control; the G0/G1 population was decreased proportionately (not shown).
Selective impairment of cyclin D1 protein and mRNA expression in 17,18-epoxy-EPA-treated cells
Because flow cytometry indicated that 17,18-epoxy-EPA interrupted the progression of the cell cycle, we evaluated the expression of cyclin regulatory proteins in bEND.3 cells. After 2, 6 and 16 h of treatment, cyclin D1 protein expression was significantly decreased in 17,18-epoxy-EPA-treated cells relative to time-matched control cells (Figure 4A); in contrast, cyclin D1 protein expression was unimpaired when cells were treated for 6 and 16 h with 8,9- and 11,12-epoxy-EPA (not shown). The effect of 17,18-epoxy-EPA on cyclin D1 mRNA expression was also investigated. In accord with findings at the protein level, 6 and 16 h of treatment of bEND.3 cells with the epoxide decreased cyclin D1 mRNA expression by 27 ± 8% (P < 0.05) and 41 ± 13% (P < 0.01) respectively (Figure 4B). However, cyclin D1 mRNA was unchanged at 2 h, thus implicating a bimodal regulatory effect of 17,18-epoxy-EPA on cyclin D1. Expression of the catalytic subunit CDK4 in control cells was also assessed. Thus, 17,18-epoxy-EPA treatment decreased CDK4 expression (by 34 ± 8% after 2 h and by 40 ± 19% after 16 h; Figure 4C).
Figure 4.
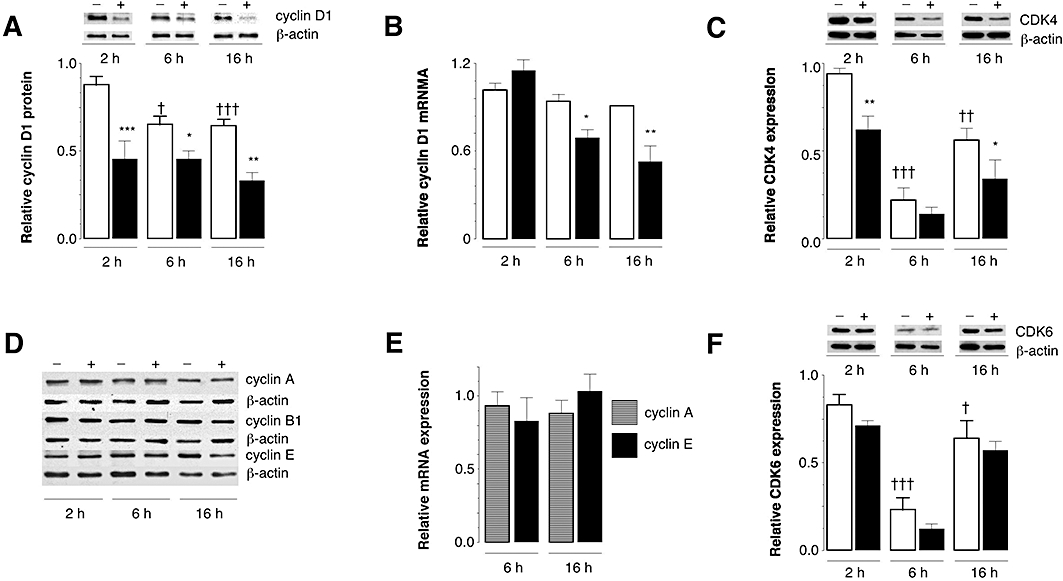
Effects of 17,18-epoxy-EPA (10 µM) on the expression of cell cycle regulatory gene expression in bEND.3 cells. (A) Down-regulation of cyclin D1 protein with 17,18-epoxy-EPA (solid columns) compared with time-matched control cells (open columns). (B) Time-dependent down-regulation of cyclin D1 mRNA with 17,18-epoxy-EPA (solid columns) compared with control (open columns). (C) Time-dependent expression of cyclin-dependent kinase (CDK)-4 protein in bEND.3 cells and down-regulation by 17,18-epoxy-EPA. (D) Lack of effect of 17,18-epoxy-EPA on expression of A, B1 and E cyclins. (E) Lack of effect of 17,18-epoxy-EPA on expression of cyclin A and cyclin E mRNA. (F) Time-dependent expression of CDK6 protein in bEND.3 cells. Open columns are controls whereas solid columns indicate treatment with 17,18-epoxy-EPA. Data are means ± SEM of three to four separate experiments, each conducted in triplicate. Different from corresponding time-matched control: *P < 0.05, **P < 0.01, ***P < 0.001. Different from control (2 h): †P < 0.05, ††P < 0.01, †††P < 0.001.
In contrast with these findings, expression of alternate cell cycle regulatory proteins was essentially unaltered by 17,18-epoxy-EPA treatment. Thus, cyclin A, cyclin B1 and cyclin E proteins were unchanged (Figure 4D), which was corroborated at the mRNA level for cyclins A and E (Figure 4E). The expression of CDK6, which also forms a catalytic complex with cyclin D1 (Brotherton et al., 1998), varied with time in culture. However, the trend towards decreased CDK6 expression after 17,18-epoxy-EPA treatment (10 µM) did not attain statistical significance at any time point (Figure 4F). Expression of the CDKi proteins p21cip1 and p27kip1 was also unaffected by 17,18-epoxy-EPA in bEND.3 cells (data not shown). Taken together, these findings demonstrate that 17,18-epoxy-EPA selectively decreased the expression of cyclin D1 and its important regulatory kinase CDK4 in bEND.3 cells.
Selective activation of the p38 MAP kinase pathway mediates cyclin D1 suppression by 17,18-epoxy-EPA
Previous studies have implicated MAP kinase signalling in the regulation of cyclin D1 and cell proliferation by ω-6 EETs (Potente et al., 2002). It has also been shown that the p38 MAP kinase negatively regulates cyclin D1 (Lavoie et al., 1996). In this study we tested the possibility that 17,18-epoxy-EPA may down-regulate cyclin D1 expression and decrease cell viability by activation of specific MAP kinases. As shown in Figure 5A, the relative accumulation of phosphorylated p38 MAP kinase was increased by short-term treatment of cells with 17,18-epoxy-EPA (10 µM), but at longer treatment times up to 24 h phospho-p38 MAP kinase expression was not different from control (not shown). In contrast, the apparent increase in phospho-JNK at 30 min normalized rapidly and there was no evidence for ERK activation (Figure 5A). The selective activation of the p38 MAP kinase by 17,18-epoxy-EPA was not observed after treatment of cells with the 8,9- and 11,12-epoxy-EPA isomers (10 µM; Figure 5B). In follow-up studies the relative activation of the p38 and ERK MAP kinases by PUFA epoxides was studied further. Lower concentrations of 17,18-epoxy-EPA (1 and 5 µM) also increased phospho-p38 MAP kinase expression but ω-6 EETs were inactive (Figure 6). In accord with previous reports, phospho-ERK accumulation was increased in cells after treatment with the isomeric EETs (Chen et al., 1999; Yang et al., 2007), but not by 17,18-epoxy-EPA (Figure 6).
Figure 5.
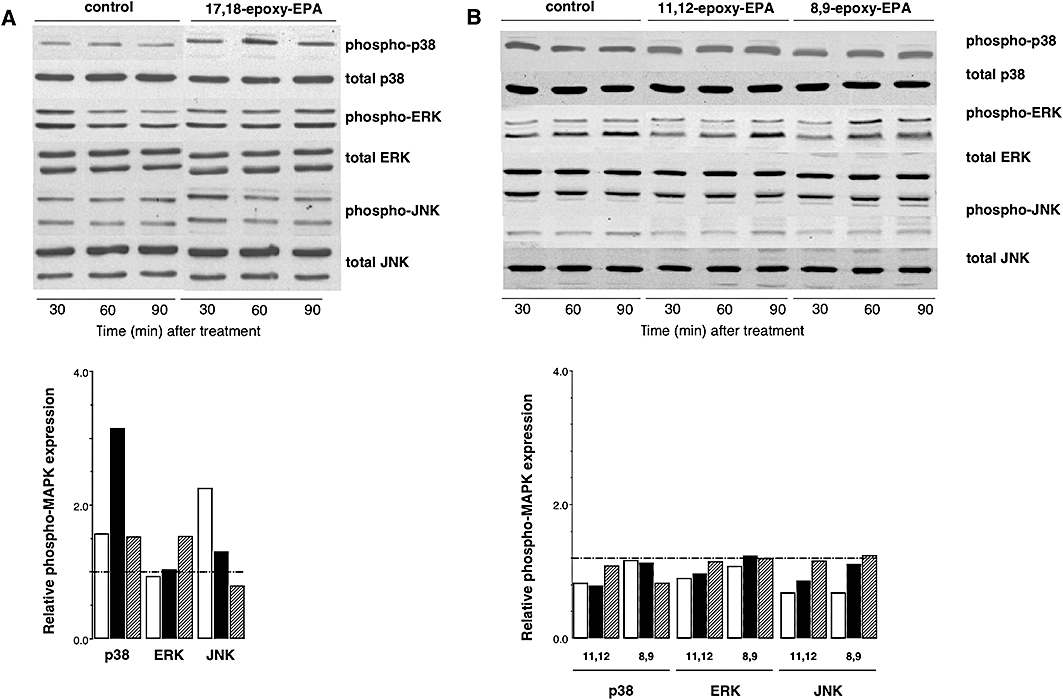
Selective increase in phospho-p38 mitogen-activated protein (MAP) kinase expression in lysates of endothelial cells treated with (A) 17,18-epoxy-EPA (10 µM), but not (B) 8,9- or 11,12-epoxy-EPA (10 µM). Phosphorylated/total MAP kinase ratios were determined relative to control (dimethyl sulphoxide-treated) cells. Densitometric analyses of representative experiments from three separate experiments are shown: open columns, 30 min; solid columns, 60 min; and hatched columns, 90 min of treatment.
Figure 6.
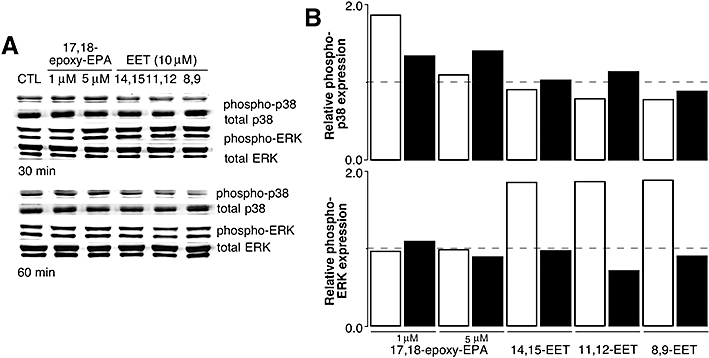
(A) Phospho- and total p38 and extracellular signal-regulated kinase (ERK) mitogen-activated protein (MAP) kinase expression in lysates from bEND.3 cells treated with lower concentrations of 17,18-epoxy-EPA (1 and 5 µM) or isomeric epoxyeicosatrienoic acids (EETs) (10 µM) for 30 or 60 min. (B) Phosphorylated/total MAP kinase ratios were determined relative to control (dimethyl sulphoxide-treated) cells. Densitometric analyses of single representative experiments from three separate experiments are shown: open columns, 30 min, and solid columns, 60 min of treatment.
The inhibitory effect of 17,18-epoxy-EPA on cyclin D1 mRNA expression after 6 and 16 h treatment was suppressed by the p38 MAP kinase inhibitor SB203580 (25 µM, Figure 7A), but inhibitors of the ERK and JNK pathways (10 µM PD98059 and 25 µM SP600125 respectively) were without significant effect (Figure 7B). Moreover, the lack of effect of SB203580 on cyclin A mRNA expression (Figure 7C) is consistent with the selective decrease in cyclin D1 expression following 17,18-epoxy-EPA treatment and the role of the p38 MAP kinase in the pathway (Figure 7A). To corroborate the apparent involvement of the p38 MAP kinase in the down-regulation of cyclin D1 mRNA expression by 17,18-epoxy-EPA, bEND.3 cells were transiently transfected with a dominant-negative p38 MAP kinase (DN-p38) mutant and then treated with 17,18-epoxy-EPA for 6 and 16 h (Figure 7D). This also prevented the down-regulatory effect of 17,18-epoxy-EPA on cyclin D1 expression. The functional significance of p38 MAP kinase inhibition was supported by the findings that treatment with SB203580 (25 µM) prevented the decrease in MTT reduction (Figure 7E) and cyclin D1 protein expression (Figure 7F) in b.END3 cells elicited by 17,18-epoxy-EPA.
Figure 7.
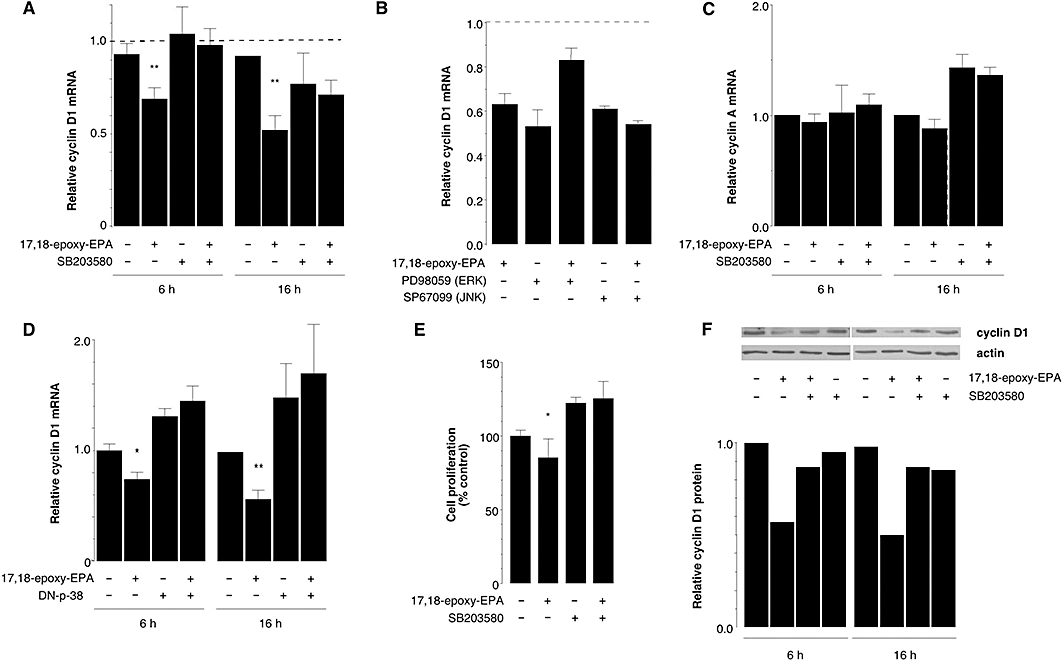
Effects of p38 mitogen-activated protein (MAP) kinase inhibition on cyclin D1 expression and cell proliferation in bEND.3 cells treated with 17,18-epoxy-EPA (10 µM). (A) Down-regulation of cyclin D1 mRNA expression by 17,18-epoxy-EPA (6 and 16 h) was prevented by SB203580 (25 µM). (B) Effect of c-Jun-N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK) inhibitors on cyclin D1 mRNA expression in the presence and absence of 17,18-epoxy-EPA. (C) Lack of effect of p38 MAP kinase inhibition on cyclin A mRNA expression. (D) Down-regulation of cyclin D1 mRNA by 17,18-epoxy-EPA was prevented by transfection with a dominant-negative-p38 MAP kinase expression plasmid. (E) Decreased proliferation induced by 17,18-epoxy-EPA treatment was prevented by application of SB203580. (F) Down-regulation of cyclin D1 protein expression by 17,18-epoxy-EPA (6 and 16 h) was prevented by SB203580 (25 µM). Cyclin D1 expression relative to actin was determined densitometrically; data from a representative of duplicate experiments are shown. In (A-E) data are means ± SEM of three to four separate experiments, each conducted in triplicate. Different from corresponding time-matched control: *P < 0.05, **P < 0.01.
Discussion and conclusions
The present findings indicate that the ω-3 epoxide of EPA (17,18-epoxy-EPA) inhibits the proliferation of bEND.3 microvascular endothelial cells. The observed antiproliferative effect was specific for the monoepoxide formed at the unique ω-3 olefinic bond in EPA, because isomeric EPA monoepoxides either stimulated proliferation or were inactive. Similarly, EETs formed by the action of CYPs on the ω-6 PUFA AA enhanced cellular proliferation. These alternate effects of epoxides of ω-3 and ω-6 PUFAs may differentially regulate not only the growth of endothelial cells but also their integrity, including barrier function (Alvarez et al., 2004).
Cell proliferation is regulated by the coordinated activation of CDKs that act in concert with regulatory cyclin subunits. Thus, CDK/cyclin holoenzymes phosphorylate the retinoblastoma gene product and related pocket proteins, which derepresses the E2F transcription factor and facilitates DNA synthesis during S-phase (Ekholm and Reed, 2000). Cyclin D1 expression is usually high in cells entering the cell cycle in G1-phase but is decreased in S-phase, which enables DNA synthesis. This is followed by re-establishment of high-level expression of cyclin D1 in G2-phase, which facilitates cell cycle progression and the commitment to mitogenesis (Stacey, 2003). The present findings establish that cyclin D1 is a major target for selective down-regulation by 17,18-epoxy-EPA and is consistent with decreased proliferation. Thus, impaired expression of cyclin D1 following 17,18-epoxy-EPA treatment may prevent the progression to G2-phase of cells that are already in the cell cycle. This effect was specific for cyclin D1, as expression of other cyclins (A, B1 and E) was unchanged by 17,18-epoxy-EPA treatment. 17,18-epoxy-EPA also decreased the expression of CDK4, which is an essential functional component of the cyclin D1/CDK4 complex, and which is normally activated in proliferating cells during mid-G1-phase. In contrast, expression of CDK6, which forms a complex with cyclin D1 later in the cell cycle, was relatively refractory to treatment with 17,18-epoxy-EPA.
Because the control of cell growth is complex, the independent regulation of individual cell cycle intermediates by 17,18-epoxy-EPA is perhaps not surprising (Kozar and Sicinski, 2005). Indeed, the naturally occurring vitamin A analogue lycopene has also been reported to selectively decrease the expression of cyclin D but not cyclin E (Nahum et al., 2001). Similarly, retinoids (Buckley et al., 1993) and 1,25-dihydroxyvitamin D3 (Wang et al., 1996) also decrease cyclin D1 expression and inhibit G1-phase progression. Like 17,18-epoxy-EPA, the grape polyphenol resveratrol was shown to down-regulate cyclin D1 and arrest the cell cycle in S-phase in several cancer cell lines (Joe et al., 2002). The present findings suggest that 17,18-epoxy-EPA, which is formed intracellularly by the actions of CYP enzymes on EPA (Barbosa-Sicard et al., 2005), selectively down-regulates the cyclin D1/CDK4 proliferative complex in cells.
Growth initiation is usually mediated by the intracellular activation of MAP kinase isoforms. Activation of the ERK MAP kinases occurs in response to proliferative signals and promotes cell cycle entry (Mebratu and Tesfaigzi, 2009). An initial burst of kinase activity after mitogen exposure is followed by the accumulation of cyclin D1 (Stacey, 2003). Cell proliferation is also modulated by growth suppression pathways that mediate cellular differentiation. Thus, activation of the p38 MAP kinase, which is essential for tissue maintenance and renewal by regulating stem and progenitor cell proliferation and differentiation, suppresses growth and decreases cyclin D1 expression (Conrad et al., 1999). Moreover, proliferative signals were magnified in p38α MAP kinase-null mouse lung and expression was markedly decreased in pulmonary tumours compared to normal tissue (Ventura et al., 2007). Consistent with the proposed negative role of p38 MAP kinase in growth regulation, its over-expression inhibited cell cycle progression in NIH/3T3 cells (Molnár et al., 1997) and down-regulated cyclin D1 expression in primary bovine tracheal myocytes (Page et al., 2001). In the present study the 17,18-epoxide of EPA selectively activated the p38 MAP kinase in bEND.3 cells and decreased cyclin D1 expression. Treatment with SB203580 and over-expression of a dominant-negative mutant of p38 MAP kinase both prevented the loss of cyclin D1 expression in response to 17,18-epoxy-EPA treatment.
There has been controversy surrounding the mode of action of ω-3 PUFA, with most studies to date having focused primarily on the effects of the parent fatty acids. Thus, the ω-3 PUFA docosahexaenoic acid decreased the proportion of cells in S-phase and inhibited the progression to G2/M-phase (Khan et al., 2006). MAP kinase activation was impaired and the CDKi p27Kip1 was up-regulated but cyclin D1 expression was unaltered (Khan et al., 2006). There is accumulating evidence that PUFA may regulate cell proliferation via biotransformation products, including EETs that are formed by the actions of CYPs on the ω-6 PUFA AA, which inhibit apoptosis and stimulate cell growth (Chen et al., 2001; Potente et al., 2002; 2003;). The present findings suggest that the ω-3 17,18-epoxy-EPA promotes the accumulation in S-phase of endothelial cells that are already in the cell cycle and that the p38 MAP kinase plays an essential role in this process. Activation of the p38 MAP kinase appears to exert a bimodal action on cyclin D1 expression, consisting of a rapid decline in cyclin D1 protein followed by a decline in mRNA expression. That cyclin D1 is down-regulated by p38 activation at both pre- and post-translational levels has been reported previously (Lee et al., 1994; Lavoie et al., 1996) and is consistent with the important role of this kinase as a negative regulator of proliferation. However, the signalling pathways by which these mechanisms operate are not completely clear. The p38 MAP kinase modulates the activity of a number of transcription factors, enzymes and alternate signalling cascades, some of which may regulate cyclin D1 (Zarubin and Han, 2005). The cyclin D1 gene contains regulatory elements for bZIP factors, OCT, Sp1 and other transcription factors in its 5′-flank (Albanese et al., 1995; Lavoie et al., 1996). Moreover, MAP kinases regulate several bZIP factors, including activating transcription factors, and fos and jun family proteins, which may all participate in transcriptional regulation of the cyclin D1 gene (Albanese et al., 1995; Lavoie et al., 1996). In addition, phosphorylation of cyclin D1 protein promotes degradation via the ubiquitin-proteasomal route (Diehl et al., 1997). Thus, post-translational regulation mediates the wide variation in cyclin D1 protein expression during the cell cycle (Guo et al., 2002). Certain agents, such as the polyphenolic stilbene resveratrol, activate cyclin D1 proteolysis in a range of cancer cell types (Joe et al., 2002). Such effects resemble those observed in the present study in which 17,18-epoxy-EPA, but not the other isomeric epoxides, rapidly down-regulated cyclin D1 protein in endothelial cells.
Polyunsaturated fatty acid epoxides have been shown to activate multiple intracellular signalling pathways. Thus, ω-6 EETs activate the epidermal growth factor receptor and stimulate downstream kinases that comprise MAP kinase cascades (Chen et al., 1999). Recent studies have also implicated certain PUFAs, CYP-derived EETs and their hydration products as ligands for thromboxane and peroxisome proliferator-activated receptors, although relative potencies varied markedly between the derivatives (Fang et al., 2006; Behm et al., 2009). Reported proliferative effects of EETs in cells were observed in the concentration range 1–10 µM. In the present study, the antiproliferative effects of ω-3 17,18-epoxy-EPA were observed at similar concentrations. Why the 17,18-epoxide of EPA selectively activates the p38 MAP kinase to decrease cyclin D1 expression and inhibit cell proliferation is not completely clear but may relate to its structure. In this molecule, the epoxide ring is closer to the terminal carbon atom at C20 than is the case in the other EPA monoepoxides or in EETs formed from the ω-6 PUFA AA (Figure 1B). The interaction of these ‘non-ω-3 epoxides’ with signalling intermediates in the p38 MAP kinase pathway might be obstructed by steric hindrance from the flexible carbon chain, which would be minimized in 17,18-epoxy-EPA. However, additional studies are now required to evaluate in greater detail the interactions of epoxides with MAP kinase signalling pathways.
The possibility that the ω-3 epoxide of EPA may stimulate apoptosis in order to decrease cell proliferation was also assessed in the present study. Although addition of PUFA to cultured cells reportedly increases apoptosis (Artwohl et al., 2008), the potential significance of these observations is obscured somewhat by the use of supraphysiological concentrations of PUFA in some of these studies. High concentrations of PUFA may exceed the binding capacity of cellular proteins and undergo non-specific attack by adventitious free radicals that may activate apoptosis. In the present study we measured caspase-3 activity, a primary indicator of the induction of apoptosis in 17,18-epoxy-EPA-treated microvascular endothelial cells. Activity was elevated by 24 h of treatment by physiologically relevant concentrations of the epoxide, although not to the same extent as that elicited by the cytotoxic agent etoposide. This suggests that, in addition to interruption of cell cycle progression, 17,18-epoxy-EPA stimulates apoptosis.
Considered together, PUFA and their metabolites exert effects on multiple cell cycle control pathways. The present study indicates that the unique ω-3-epoxide of EPA activates the p38 MAP kinase and down-regulates cyclin D1 expression in microvascular endothelial cells; monoepoxides formed at alternate olefinic bonds within EPA were inactive. This finding is significant in the context of previous findings that ω-6 EETs induce cyclin D1 and suggests that this important cell cycle regulator may be a cellular switch that is modulated differentially by epoxide metabolites of different PUFA classes. Moreover, these opposing effects of ω-3 and ω-6 PUFA epoxides in cells are consistent with epidemiological studies in which dietary ω-3 PUFA appear to be beneficial in limiting the spread of certain cancers, whereas diets that are high in ω-6 PUFA may stimulate tumour growth (Bougnoux, 1999; Maillard et al., 2002). Thus, it is feasible that at least some of the beneficial effects of ω-3 PUFA in minimizing tumour growth may be due to the specific ω-3 epoxide metabolite that impairs cellular proliferation.
Acknowledgments
This work was supported by a grant from the Australian National Health and Medical Research Council. The gift of bEND.3 cells from Prof. Nick Hunt (Department of Pathology, University of Sydney) is gratefully acknowledged. The dominant-negative p38 MAP kinase mutant was generously provided by Dr Roger J. Davis (Howard Hughes Medical Institute, University of Massachusetts Medical School, Worcester, MA). The expert assistance of Dr Colin C. Duke and Mr Bruce N. Tattam in structural characterization of epoxides and LC-MS and LC-MS/MS analysis is also gratefully acknowledged.
Glossary
Abbreviations
- AA
arachidonic acid
- CDK
cyclin-dependent kinase
- CDKi
cyclin-dependent kinase inhibitor
- CYP
cytochrome P450
- DMEM
Dulbecco's modified eagle medium
- DN
dominant-negative expression plasmid
- ECL
enhanced chemiluminescence
- EET
epoxyeicosatrienoic acid
- EPA
eicosapentaenoic acid
- ERK
extracellular signal-regulated kinase
- JNK
c-Jun-N-terminal kinase
- MAP kinase
mitogen-activated protein kinase
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-biphenyl tetrazolium bromide
- PUFA
polyunsaturated fatty acid
Conflict of interest
The authors state no conflict of interest.
Supporting Information
Supporting Information: Teaching Materials; Figs 1–7 as PowerPoint slide.
References
- Abedin M, Lim J, Tang TB, Park D, Demer LL, Tintut Y. n-3 fatty acids inhibit vascular calcification via the p38-mitogen-activated protein kinase and peroxisome proliferator-activated receptor-gamma pathways. Circ Res. 2006;98:727–729. doi: 10.1161/01.RES.0000216009.68958.e6. [DOI] [PubMed] [Google Scholar]
- Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A, et al. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem. 1995;270:23589–23597. doi: 10.1074/jbc.270.40.23589. [DOI] [PubMed] [Google Scholar]
- Alvarez DF, Gjerde EB, Townsley MI. Role of EETs in regulation of endothelial permeability in rat lung. Am J Physiol Lung Cell Mol Physiol. 2004;286:L445–L451. doi: 10.1152/ajplung.00150.2003. [DOI] [PubMed] [Google Scholar]
- Artwohl M, Lindenmair A, Sexl V, Maier C, Rainer G, Freudenthaler A, et al. Different mechanisms of saturated versus polyunsaturated FFA-induced apoptosis in human endothelial cells. J Lipid Res. 2008;49:2627–2640. doi: 10.1194/jlr.M800393-JLR200. [DOI] [PubMed] [Google Scholar]
- Ball HJ, Sanchez-Pereza A, Weisera S, Austin CJD, Astelbauer F, Miu J, et al. Characterization of an indoleamine 2,3-dioxygenase-like protein found in humans and mice. Gene. 2007;396:203–213. doi: 10.1016/j.gene.2007.04.010. [DOI] [PubMed] [Google Scholar]
- Barbosa-Sicard E, Markovic M, Honeck H, Christ B, Muller DN, Schunck WH. Eicosapentaenoic acid metabolism by cytochrome P450 enzymes of the CYP2C subfamily. Biochem Biophys Res Commun. 2005;329:1275–1281. doi: 10.1016/j.bbrc.2005.02.103. [DOI] [PubMed] [Google Scholar]
- Behm DJ, Ogbonna A, Wu C, Burns-Kurtis CL, Douglas SA. Epoxyeicosatrienoic acids function as selective, endogenous antagonists of native thromboxane receptors: identification of a novel mechanism of vasodilation. J Pharmacol Exp Ther. 2009;328:231–239. doi: 10.1124/jpet.108.145102. [DOI] [PubMed] [Google Scholar]
- Bougnoux P. n-3 polyunsaturated fatty acids and cancer. Curr Opin Clin Nutr Metab Care. 1999;2:121–126. doi: 10.1097/00075197-199903000-00005. [DOI] [PubMed] [Google Scholar]
- Brotherton DH, Dhanaraj V, Wick S, Brizuela L, Domaille P, Volyanik E, et al. Crystal structure of the complex of the cyclin-D dependent kinase cdk6 bound to the cell-cycle inhibitor p19INK4d. Nature. 1998;395:244–250. doi: 10.1038/26164. [DOI] [PubMed] [Google Scholar]
- Buckley MF, Sweeney KJ, Hamilton JA, Sini RL, Manning DL, Nicholson RI, et al. Expression and amplification of cyclin genes in human breast cancer. Oncogene. 1993;8:2127–2133. [PubMed] [Google Scholar]
- Chen JK, Wang DW, Falck JR, Capdevila J, Harris RC. Transfection of an active cytochrome P450 arachidonic acid epoxygenase indicates that 14,15-epoxyeicosatrienoic acid functions as an intracellular second messenger in response to epidermal growth factor. J Biol Chem. 1999;274:4764–4769. doi: 10.1074/jbc.274.8.4764. [DOI] [PubMed] [Google Scholar]
- Chen JK, Capdevila J, Harris RC. Cytochrome P450 epoxygenase metabolism of arachidonic acid inhibits apoptosis. Mol Cell Biol. 2001;21:6322–6331. doi: 10.1128/MCB.21.18.6322-6331.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad PW, Rust RT, Han J, Millhorn DE, Beitner-Johnson D. Selective activation of p38alpha and p38gamma by hypoxia. Role in regulation of cyclin D1 by hypoxia in PC12 cells. J Biol Chem. 1999;274:23570–23576. doi: 10.1074/jbc.274.33.23570. [DOI] [PubMed] [Google Scholar]
- Cui PH, Zhang WV, Hook J, Tattam BN, Duke CC, Murray M. Synthesis and NMR characterization of the methyl esters of eicosapentaenoic acid monoepoxides. Chem Phys Lipids. 2009;159:30–37. doi: 10.1016/j.chemphyslip.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11:957–972. doi: 10.1101/gad.11.8.957. [DOI] [PubMed] [Google Scholar]
- Diep QN, Touyz RM, Schiffrin EL. Docosahexaenoic acid, a peroxisome proliferator-activated receptor-α ligand, induces apoptosis in vascular smooth muscle cells by stimulation of p38 mitogen-activated protein kinase. Hypertension. 2000;36:851–855. doi: 10.1161/01.hyp.36.5.851. [DOI] [PubMed] [Google Scholar]
- Ekholm SV, Reed SI. Regulation of G(1) cyclin-dependent kinases in the mammalian cell cycle. Curr Opin Cell Biol. 2000;12:676–684. doi: 10.1016/s0955-0674(00)00151-4. [DOI] [PubMed] [Google Scholar]
- Fang X, Hu S, Xu B, Snyder GD, Harmon S, Yao J, et al. 14,15-Dihydroxyeicosatrienoic acid activates peroxisome proliferator-activated receptor-α. Am J Physiol Heart Circ Physiol. 2006;290:H55–H63. doi: 10.1152/ajpheart.00427.2005. [DOI] [PubMed] [Google Scholar]
- Fleming I. Epoxyeicosatrienoic acids, cell signalling and angiogenesis. Prostaglandins Other Lipid Mediat. 2007;82:60–67. doi: 10.1016/j.prostaglandins.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Floyd HS, Jennings-Gee JE, Kock ND, Miller MS. Genetic and epigenetic alterations in lung tumors from bitransgenic Ki-rasG12C expressing mice. Mol Carcinog. 2006;45:506–517. doi: 10.1002/mc.20181. [DOI] [PubMed] [Google Scholar]
- Grammatikos SI, Subbaiah PV, Victor TA, Miller WM. n-3 and n-6 fatty acid processing and growth effects in neoplastic and non-cancerous human mammary epithelial cell lines. Br J Cancer. 1994;70:219–227. doi: 10.1038/bjc.1994.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Stacey DW, Hitomi M. Post-transcriptional regulation of cyclin D1 expression during G2 phase. Oncogene. 2002;21:7545–7556. doi: 10.1038/sj.onc.1205907. [DOI] [PubMed] [Google Scholar]
- Jiang JG, Chen CL, Card JW, Yang S, Chen JX, Fu XN, et al. Cytochrome P450 2J2 promotes the neoplastic phenotype of carcinoma cells and is up-regulated in human tumors. Cancer Res. 2005;65:4707–4715. doi: 10.1158/0008-5472.CAN-04-4173. [DOI] [PubMed] [Google Scholar]
- Joe AK, Liu H, Suzui M, Vural ME, Xiao D, Weinstein IB. Resveratrol induces growth inhibition, S-phase arrest, apoptosis, and changes in biomarker expression in several human cancer cell lines. Clin Cancer Res. 2002;8:893–903. [PubMed] [Google Scholar]
- Khan NA, Nishimura K, Aires V, Yamashita T, Oaxaca-Castillo D, Kashiwagi K, et al. Docosahexaenoic acid inhibits cancer cell growth via p27Kip1, CDK2, ERK1/ERK2, and retinoblastoma phosphorylation. J Lipid Res. 2006;47:2306–2313. doi: 10.1194/jlr.M600269-JLR200. [DOI] [PubMed] [Google Scholar]
- Kozar K, Sicinski P. Cell cycle progression without cyclin D-CDK4 and cyclin D-CDK6 complexes. Cell Cycle. 2005;4:388–391. doi: 10.4161/cc.4.3.1551. [DOI] [PubMed] [Google Scholar]
- Lavoie JN, L'Allemain G, Brunet A, Muller R, Pouyssegur J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem. 1996;271:20608–20616. doi: 10.1074/jbc.271.34.20608. [DOI] [PubMed] [Google Scholar]
- Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Maillard V, Bougnoux P, Ferrari P, Jourdan ML, Pinault M, Lavillonniere F, et al. n-3 and n-6 fatty acids in breast adipose tissue and relative risk of breast cancer in a case-control study in Tours, France. Int J Cancer. 2002;98:78–83. doi: 10.1002/ijc.10130. [DOI] [PubMed] [Google Scholar]
- Mebratu Y, Tesfaigzi Y. How ERK1/2 activation controls cell proliferation and cell death: is subcellular localization the answer? Cell Cycle. 2009;8:1168–1175. doi: 10.4161/cc.8.8.8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis UR, Fisslthaler B, Medhora M, Harder D, Fleming I, Busse R. Cytochrome P450 2C9-derived epoxyeicosatrienoic acids induce angiogenesis via cross-talk with the epidermal growth factor receptor (EGFR) FASEB J. 2003;17:770–772. doi: 10.1096/fj.02-0640fje. [DOI] [PubMed] [Google Scholar]
- Molnár A, Theodoras AM, Zon LI, Kyriakis JM. Cdc42Hs, but not Rac1, inhibits serum-stimulated cell cycle progression at G1/S through a mechanism requiring p38/RK. J Biol Chem. 1997;272:13229–13235. doi: 10.1074/jbc.272.20.13229. [DOI] [PubMed] [Google Scholar]
- Murray M, Butler AM, Fiala-Beer E, Su GM. Phospho-STAT5 accumulation in nuclear fractions from vitamin A-deficient rat liver. FEBS Lett. 2005;579:3669–3673. doi: 10.1016/j.febslet.2005.05.052. [DOI] [PubMed] [Google Scholar]
- Nahum A, Hirsch K, Danilenko M, Watts CK, Prall OW, Levy J, et al. Lycopene inhibition of cell cycle progression in breast and endometrial cancer cells is associated with reduction in cyclin D levels and retention of p27(Kip1) in the cyclin E-cdk2 complexes. Oncogene. 2001;20:3428–3436. doi: 10.1038/sj.onc.1204452. [DOI] [PubMed] [Google Scholar]
- Ng VY, Huang Y, Reddy LM, Falck JR, Lin ET, Kroetz DL. Cytochrome P450 eicosanoids are activators of peroxisome proliferator-activated receptor-α. Drug Metab Dispos. 2007;35:1126–1134. doi: 10.1124/dmd.106.013839. [DOI] [PubMed] [Google Scholar]
- Page K, Li J, Hershenson MB. p38 MAP kinase negatively regulates cyclin D1 expression in airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2001;280:L955–L964. doi: 10.1152/ajplung.2001.280.5.L955. [DOI] [PubMed] [Google Scholar]
- Potente M, Michaelis UR, Fisslthaler B, Busse R, Fleming I. Cytochrome P450 2C9-induced endothelial cell proliferation involves induction of mitogen-activated protein (MAP) kinase phosphatase-1, inhibition of the c-Jun N-terminal kinase, and up-regulation of cyclin D1. J Biol Chem. 2002;277:15671–15676. doi: 10.1074/jbc.M110806200. [DOI] [PubMed] [Google Scholar]
- Potente M, Fisslthaler B, Busse R, Fleming I. 11,12-Epoxyeicosatrienoic acid-induced inhibition of FOXO factors promotes endothelial proliferation by downregulating p27Kip1. J Biol Chem. 2003;278:29619–29625. doi: 10.1074/jbc.M305385200. [DOI] [PubMed] [Google Scholar]
- Rose DP, Connolly JM. Effects of dietary omega-3 fatty acids on human breast cancer growth and metastases in nude mice. J Natl Cancer Inst. 1993;85:1743–1747. doi: 10.1093/jnci/85.21.1743. [DOI] [PubMed] [Google Scholar]
- Schmetsdorf S, Gartner U, Arendt T. Constitutive expression of functionally active cyclin-dependent kinases and their binding partners suggests noncanonical functions of cell cycle regulators in differentiated neurons. Cereb Cortex. 2007;17:1821–1829. doi: 10.1093/cercor/bhl091. [DOI] [PubMed] [Google Scholar]
- Stacey DW. Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Curr Opin Cell Biol. 2003;15:158–163. doi: 10.1016/s0955-0674(03)00008-5. [DOI] [PubMed] [Google Scholar]
- Szymczak M, Murray M, Petrovic N. Modulation of angiogenesis by omega-3 polyunsaturated fatty acids is mediated by cyclooxygenases. Blood. 2008;111:3514–3521. doi: 10.1182/blood-2007-08-109934. [DOI] [PubMed] [Google Scholar]
- Tapiero H, Ba GN, Couvreur P, Tew KD. Polyunsaturated fatty acids (PUFA) and eicosanoids in human health and pathologies. Biomed Pharmacother. 2002;56:215–222. doi: 10.1016/s0753-3322(02)00193-2. [DOI] [PubMed] [Google Scholar]
- Ventura JJ, Tenbaum S, Perdiguero E, Huth M, Guerra C, Barbacid M, et al. p38alpha MAP kinase is essential in lung stem and progenitor cell proliferation and differentiation. Nat Genet. 2007;39:750–758. doi: 10.1038/ng2037. [DOI] [PubMed] [Google Scholar]
- Walker JL, Assoian RK. Integrin-dependent signal transduction regulating cyclin D1 expression and G1-phase cell cycle progression. Cancer Metastasis Rev. 2005;24:383–393. doi: 10.1007/s10555-005-5130-7. [DOI] [PubMed] [Google Scholar]
- Wang QM, Jones JB, Studzinski GP. Cyclin-dependent kinase inhibitor p27 as a mediator of the G1-S-phase block induced by 1,25-dihydroxyvitamin D3 in HL60 cells. Cancer Res. 1996;56:264–267. [PubMed] [Google Scholar]
- Yang S, Lin L, Chen JX, Lee CR, Seubert JM, Wang Y, et al. Cytochrome P450 epoxygenases protect endothelial cells from apoptosis induced by tumor necrosis factor-α via MAPK and PI3K/Akt signaling pathways. Am J Physiol Heart Circ Physiol. 2007;293:H142–H151. doi: 10.1152/ajpheart.00783.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, et al. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res. 2000;87:992–998. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005;15:11–18. doi: 10.1038/sj.cr.7290257. [DOI] [PubMed] [Google Scholar]
- Zhang C, Harder DR. Cerebral capillary endothelial cell mitogenesis and morphogenesis induced by astrocytic epoxyeicosatrienoic acid. Stroke. 2002;33:2957–2964. doi: 10.1161/01.str.0000037787.07479.9a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.