

Abstract
BACKGROUND AND PURPOSE
Hydrogen sulphide (H2S), a potentially toxic gas, is also involved in the neuroprotection, neuromodulation, cardioprotection, vasodilatation and the regulation of inflammatory response and insulin secretion. We have recently reported that H2S suppresses pancreatic β-cell apoptosis induced by long-term exposure to high glucose. Here we examined the protective effects of sodium hydrosulphide (NaHS), an H2S donor, on various types of β-cell damage.
EXPERIMENTAL APPROACH
Isolated islets from mice or the mouse insulinoma MIN6 cells were cultured with palmitate, cytokines (a mixture of tumour necrosis factor-α, interferon-γ and interleukin-1β), hydrogen peroxide, thapsigargin or tunicamycin with or without NaHS. We examined DNA fragmentation, caspase-3 and -7 activities and reactive oxygen species (ROS) production in the treated cells thereafter. Apoptotic cell death in isolated islets was also assessed by the terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labelling (TUNEL) method.
KEY RESULTS
NaHS suppressed DNA fragmentation and the activities of caspase-3 and -7 induced by palmitate, the cytokines or hydrogen peroxide. In contrast, NaHS failed to protect islets and MIN6 cells from apoptosis induced by thapsigargin and tunicamycin, both of which cause endoplasmic reticulum stress. NaHS suppressed ROS production induced by cytokines or hydrogen peroxide but it had no effect on ROS production in thapsigargin-treated cells. NaHS increased Akt phosphorylation in MIN6 cells treated with cytokines but not in cells treated with thapsigargin. Treatment with NaHS decreased TUNEL-positive cells in cytokine-exposed islets.
CONCLUSIONS AND IMPLICATIONS
H2S may prevent pancreatic β-cells from cell apoptosis via an anti-oxidative mechanism and the activation of Akt signalling.
Keywords: hydrogen sulphide, pancreatic β-cell, cytoprotection, oxidative stress, apoptosis
Introduction
Hydrogen sulphide (H2S) is a pleiotropic gasotransmitter, which can be produced from L-cysteine (Maclean and Kraus, 2004; Łowicka and Bełtowski, 2007; Li and Moore, 2008; Gadalla and Snyder, 2010). Like the other two gasotransmitters, nitric oxide (NO) and carbon monoxide (CO), H2S penetrates plasma membranes and transduces intracellular and intercellular signals (Nagai et al., 2004; Łowicka and Bełtowski, 2007; Li and Moore, 2008; Qu et al., 2008; Gadalla and Snyder, 2010). H2S plays versatile roles in cell death/survival. In aortic smooth muscle cells, lung fibroblasts and pancreatic acinar cells, exposure to H2S results in apoptosis (Cao et al., 2006; Yang et al., 2006; Baskar et al., 2007). H2S, however, has a protective role in neural cells, cardiomyocytes and hepatocytes against cellular damage including inflammation and ischaemia reperfusion (Jha et al., 2008; Sivarajah et al., 2009; Kimura et al., 2010). Cytoprotection by H2S is associated with the inhibition of reactive oxygen species (ROS) production via inhibition of NADH oxidase (Samhan-Arias et al., 2009), the induction of anti-oxidative molecules such as thioredoxin (Calvert et al., 2009), and an increase in glutathione (GSH) production (Kimura and Kimura, 2004; Chen et al., 2009; Kimura et al., 2010).
Short-term stimulation of pancreatic β-cells with glucose induces insulin secretion and generates several signals involved in cell proliferation and survival in in vitro experiments (Sjöholm, 1992; Assmann et al., 2009). Chronic hyperglycaemia in diabetic conditions, however, destroys pancreatic β-cell function and decreases its mass (Chang-Chen et al., 2008; Poitout and Robertson, 2008; Robertson, 2009). The failure of β-cells is involved in the pathogenesis and progression of diabetes. Therefore, β-cell protection is the focus of new strategies for the treatment of diabetes (Del Prato et al., 2007; Wajchenberg, 2007).
We have recently reported that H2S inhibits insulin release (Kaneko et al., 2006) and prevents high glucose-induced apoptosis in mouse pancreatic islets (Kaneko et al., 2009). We also found that treatment of islets with L-cysteine improved their secretory responsiveness following stimulation with glucose (Kaneko et al., 2009). Here, we have characterized the cytoprotective effects of H2S on pancreatic β-cells.
Methods
Animals
ICR mice were purchased from Japan SLC (Shizuoka, Japan). Mice were housed in separate cages in a room with controlled temperature, humidity and lighting (12 h light and 12 h dark cycle) and allowed free access to water and standard chow (CE-2; Clea Japan, Tokyo, Japan). All animal procedures were in accordance with the Principles of Laboratory Animal Care and were approved by the University Committee on Animal Experiments in accordance with the Guideline for Animal Experimentation in Oita University.
Cell preparation
Pancreatic islets were isolated from male ICR mice by the collagenase digestion method. The solution used for the isolation was HEPES-buffered Krebs-Ringer bicarbonate buffer (HK buffer) containing 119 mmol·L−1 NaCl, 4.75 mmol·L−1 KCl, 5 mmol·L−1 NaHCO3, 2.54 mmol·L−1 CaCl2, 1.2 mmol·L−1 MgSO4, 1.2 mmol·L−1 KH2PO4 and 20 mmol·L−1 HEPES (pH 7.4 adjusted with sodium hydroxide) supplemented with 3 mmol·L−1 glucose. Mouse insulinoma MIN6 cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 15% (vol·vol−1) fetal bovine serum (FBS) at 37°C in a humidified atmosphere of 95% air/5% CO2.
Measurement of fragmented DNA
Isolated islets (25–30 islets per dish) were transferred to RPMI 1640 medium (Sigma-Aldrich, St. Louis, MO, USA) containing 10% FBS and cultured for 18 h with 5 mmol·L−1 glucose alone or 5 mmol·L−1 glucose plus cytokines (1000 U·mL−1 tumour necrosis factor-α (TNF-α), 1000 U·mL−1 interferon-γ (IFN-γ) and 50 U·mL−1 interleukin-1β (IL-1β)), 10 µmol·L−1 hydrogen peroxide (H2O2), 1 µmol·L−1 thapsigargin or 5 µg·mL−1 tunicamycin in the presence or absence of 0.1 mmol·L−1 sodium hydrosulphide (NaHS). For treatment with free fatty acids, islets were cultured for 72 h in RPMI 1640 medium containing 1% (wt·vol−1) BSA and 5 mmol·L−1 glucose alone or 5 mmol·L−1 glucose plus 0.5 mmol·L−1 palmitate. Palmitate was dissolved at 500 mmol·L−1 in ethanol, heated to 60°C, and then added to RPMI 1640 medium containing 10% (wt·vol−1) fatty acid-free BSA to generate a 5 mmol·L−1 palmitate stock solution. The stock solution was used in a 1:10 dilution in RPMI 1640 medium. MIN6 cells (1 × 106 to 2 × 106 cells per dish) were cultured for 18 h in DMEM (supplemented with 15% FBS) containing 20 mmol·L−1 glucose alone or 20 mmol·L−1 glucose plus cytokines, H2O2, thapsigargin or tunicamycin. For treatment with free fatty acids, MIN6 cells were cultured for 72 h in DMEM containing 1% BSA and 20 mmol·L−1 glucose alone or 20 mmol·L−1 glucose plus 0.5 mmol·L−1 palmitate. Cultured islets and MIN6 cells were washed 3 times with HK buffer containing 3 mmol·L−1 glucose. Cytoplasmic histone-associated DNA fragments were quantified by the cell death-detection elisa kit (Roche Diagnostics, Mannheim, Germany), which detects DNA-laddering derived from apoptotic cell death with antihistone and antiDNA monoclonal antibodies.
Measurement of caspase activity
Caspase-3 and -7 activities were analysed with the Caspase-Glo 3/7 assay kit (Promega, Madison, WI, USA). Cleavage of the luminogenic substrate (aminoluciferin containing DEVD sequence) by caspase-3 or -7 releases a substrate for luciferase generating a luminescent signal. The islets were cultured as described above, and the caspase activities therein were detected according to the manufacturer's instructions.
Terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labelling (TUNEL) and insulin staining
Isolated islets (100 islets/dish) were transferred to RPMI 1640 medium containing 10% FBS and cultured for 18 h with 5 mmol·L−1 glucose alone or 5 mmol·L−1 glucose plus the cytokine mixture in the presence or absence of 0.1 mmol·L−1 NaHS. Cultured islets were fixed with phosphate-buffered 3% (wt·vol−1) paraformaldehyde and cryosectioned at every 5 µm. The TUNEL assay was performed using the In Situ Cell Detection Kit (Roche Diagnostics, Mannheim, Germany). Cryosections of islets were incubated with the TUNEL reaction mixture for 1 h at 37°C and then incubated with anti-insulin guinea pig antibody (diluted 1:1000), followed by incubation with Alexa-Fluor-568 anti-guinea-pig IgG (diluted 1:100).
Measurement of ROS
The ROS production in pancreatic islets and MIN6 cells was assessed using dichlorodihydrofluorescein diacetate (H2DCFDA). H2DCFDA is a cell-permeable indicator of ROS that is hydrolysed by intracellular esterases to the nonfluorescent dichlorodihydrofluorescein (H2DCF). In the presence of ROS, H2DCF is oxidized to the fluorescent dichlorofluorescein (DCF) in the cells. Isolated islets were incubated with HK buffer containing 10 µmol·L−1 H2DCFDA for 30 min at 37°C in the dark. Islets were then washed twice with HK buffer containing 0.1% BSA and further incubated for 12 h in the BSA-containing HK buffer supplemented with 5 mmol·L−1 glucose alone or 5 mmol·L−1 glucose plus one of the following – the cytokine mixture or 1 µmol·L−1 thapsigargin – in the presence or absence of 0.1 mmol·L−1 NaHS. After 12 h, islets were immediately frozen in liquid nitrogen and thawed. The extracts were briefly centrifuged to remove cell debris. MIN6 cells (4 × 104 cells per well) were seeded in 96-well plates and cultured for 3 days. The cells were incubated with HK buffer containing 10 µmol·L−1 H2DCFDA for 30 min at 37°C in the dark. The cells were washed twice with HK buffer containing 0.1% BSA and further incubated for 12 h in the BSA-containing HK buffer supplemented with 20 mmol·L−1 glucose alone or 20 mmol·L−1 glucose plus one of the following – 30 µmol·L−1 H2O2, the cytokine mixture or 1 µmol·L−1 thapsigargin – in the presence or absence of 0.1 mmol·L−1 NaHS. The fluorescence intensity in the islet extracts or MIN6 cells was measured at an excitation/emission wavelength of 485/530 nm using a Spectra Fluor Plus microplate reader (TECAN, Männedorf, Switzerland).
Immunoblot analysis
The MIN6 cells were cultured for 6 h with 20 mmol·L−1 glucose alone or 20 mmol·L−1 glucose plus the cytokine mixture or 1 µmol·L−1 thapsigargin in the presence or absence of 0.1 mmol·L−1 NaHS. The cells were sonicated in an ice-cold homogenization buffer [20 mmol·L−1 Tris-HCl (pH 7.4), 2 mmol·L−1 EDTA, 2 mmol·L−1 2-mercaptoethanol, 50 µg·mL−1 phenylmethylsulphonylfluoride, 10 µg·mL−1 aprotinin, 10 µg·mL−1 leupeptin, 250 mmol·L−1 sucrose, 10 mmol·L−1 sodium fluoride and 2 mmol·L−1β-glycerophosphate]. The protein samples were boiled in a buffer containing 62.5 mmol·L−1 Tris-HCl (pH 6.8), 2% (wt·vol−1) sodium dodecyl sulphate (SDS), 10% (wt·vol−1) glycerol, 5% (vol·vol−1) 2-mercaptoethanol and 0.002% (wt·vol−1) bromophenol blue. The proteins in the samples were then separated on a 10% (wt·vol−1) polyacrylamide gel and transferred to polyvinylidene difluoride membrane. The membrane was blocked with 1% (wt·vol−1) skim milk in a buffer [Tris-buffered saline containing Tween 20 (T-TBS)] containing 10 mmol·L−1 Tris-HCl (pH 7.4), 137 mmol·L−1 NaCl and 0.1% (vol·vol−1) Tween 20, and incubated overnight at 4°C with rabbit antiphospho (Ser473)-Akt antibody (diluted 1:200). The membrane was washed several times in T-TBS and further incubated with a horseradish peroxidase-conjugated donkey anti-rabbit IgG antibody (diluted 1:10 000). Immunopositive bands were visualized using a commercial kit (ECL plus Western Blotting Reagent Pack, GE Healthcare, Piscataway, NJ, USA). For standardization, the blots were stripped and reprobed with anti-Akt antibodies (diluted 1:2000). The intensity of each band was measured with the NIH image software.
Statistical analysis
Results are expressed as the mean ± SE. Statistical analyses of DNA fragmentation, caspase activity and ROS production were performed by one-way anova followed by the Tukey's method. Statistical significance of immunoblot analysis was calculated using Student's unpaired t-test. Significance was set at P < 0.05.
Materials
TNF-α, IFN-γ and IL-1β were purchased from Peprotech (Rocky Hill, NJ, USA). The ROS indicator H2DCFDA was obtained from Molecular Probes (Eugene, OR, USA). The primary antibodies against Akt and phospho (Ser473)-Akt were purchased from Cell Signaling (Danvers, MA, USA), and the secondary antibody against rabbit IgG was from GE Healthcare (Piscataway, NJ, USA). The anti-insulin guinea-pig antibody was purchased from Seikagaku Corporation (Tokyo, Japan), and Alexa-Fluor-568 anti-guinea-pig IgG was from Molecular Probes. All other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) or Wako (Osaka, Japan).
Results
Effects of NaHS on DNA fragmentation in mouse pancreatic islets treated with palmitate or cytokines
We measured DNA fragmentation to determine whether NaHS protects against the islet cell apoptosis induced by palmitate or inflammatory cytokines. Palmitate at 0.5 mmol·L−1 caused a 2.9-fold increase over control treatments (5 mmol·L−1 glucose) in DNA fragmentation within islets (Figure 1A). Treatment with the cytokine mixture (1000 U·mL−1 TNF-α, 1000 U·mL−1 IFN-γ and 50 U·mL−1 IL-1β) also increased DNA fragmentation in islets by 60% (Figure 1B). The increase in DNA fragmentation induced by palmitate or cytokine treatment was inhibited by co-treatment with 0.1 mmol·L−1 NaHS (Figure 1A and B).
Figure 1.
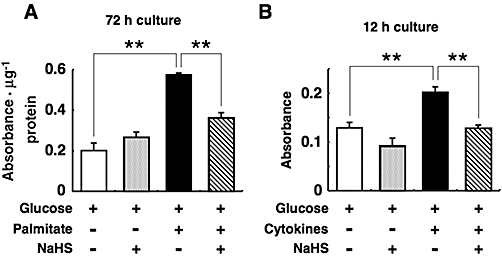
Cytoprotective effects of sodium hydrosulphide (NaHS) against pancreatic islet cell death induced by palmitate (A) and cytokines (B). Mouse pancreatic islets were cultured for the indicated durations with 5 mmol·L−1 glucose alone or 5 mmol·L−1 glucose plus 0.5 mmol·L−1 palmitate or the cytokine mixture (1000 U·mL−1 tumour necrosis factor-α, 1000 U·mL−1 interferon-γ and 50 U·mL−1 interleukin-1β) in the presence or absence of 0.1 mmol·L−1 NaHS. After this, histone-associated DNA fragments were quantified by elisa to evaluate apoptotic cell death. Each column represents the mean ± SE from three to five separate experiments. **P < 0.01.
Effects of NaHS on DNA fragmentation by H2O2, thapsigargin and tunicamycin
H2O2 at 10 µmol·L−1 increased DNA fragmentation in mouse islets as a result of oxidative stress. Treatment of islets with 0.1 mmol·L−1 NaHS suppressed H2O2-induced DNA fragmentation (Figure 2). We also examined the effects of NaHS on DNA fragmentation induced by thapsigargin (1 µmol·L−1) and tunicamycin (5 µg·mL−1) both of which cause endoplasmic reticulum (ER) stress. Treatment with thapsigargin or tunicamycin markedly increased DNA fragmentation; however, co-treatment with NaHS did not inhibit DNA fragmentation induced by thapsigargin or tunicamycin treatment (Figure 3).
Figure 2.
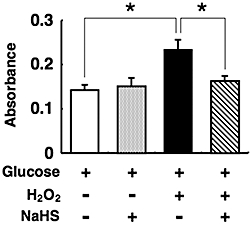
Protection by sodium hydrosulphide (NaHS) of pancreatic islets from oxidative stress induced by hydrogen peroxide (H2O2). Mouse pancreatic islets were cultured for the indicated durations with 5 mmol·L−1 glucose alone or 5 mmol·L−1 glucose plus 10 µmol·L−1 H2O2 in the presence or absence of 0.1 mmol·L−1 NaHS. After this, histone-associated DNA fragments were quantified by elisa to evaluate apoptotic cell death. Each column represents the mean ± SE from three to five separate experiments. *P < 0.05.
Figure 3.
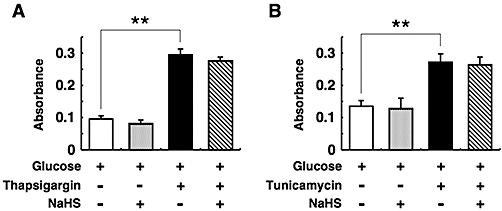
Lack of effect of sodium hydrosulphide (NaHS) on cell death induced by thapsigargin (A) or tunicamycin (B). Pancreatic islets were cultured for 18 h with or without 1 µmol·L−1 thapsigargin or 5 µg·mL−1 tunicamycin in the presence or absence of 0.1 mmol·L−1 NaHS. These experiments were carried out consistently in the presence of 5 mmol·L−1 glucose. After the culture, fragmented DNA was measured by elisa. Each column represents the mean ± SE from three to five separate experiments. *P < 0.05 and **P < 0.01.
Effects of NaHS on the activities of caspase-3 and -7
Islet cell death was also assayed by measuring the caspase-3 and -7 activities. Caspase-3 and -7 activities in the islets increased by 19–31% on exposure to 10 µmol·L−1 H2O2, the cytokine mixture or 1 µmol·L−1 thapsigargin as compared to control (Figure 4). NaHS at 0.1 mmol·L−1 suppressed the caspase-3 and -7 activities induced by H2O2 or cytokines (Figure 4A and B), but NaHS failed to suppress caspase-3 and -7 activities induced by thapsigargin (Figure 4C). These results were in good agreement with the results of the DNA fragmentation assays.
Figure 4.
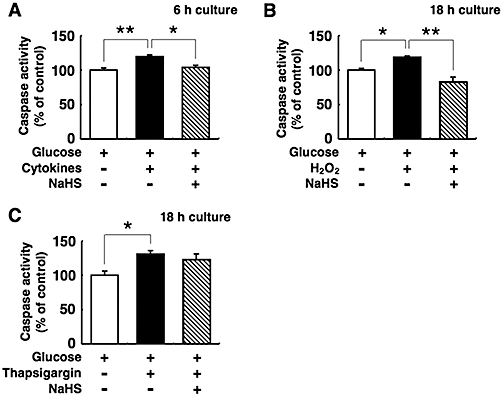
Effects of sodium hydrosulphide (NaHS) on caspase-3 and -7 activities in pancreatic islets. Mouse pancreatic islets were cultured with 5 mmol·L−1 glucose alone or 5 mmol·L−1 glucose plus the cytokine mixture (A), 10 µmol·L−1 hydrogen peroxide (H2O2) (B) or 1 µmol·L−1 thapsigargin (C) with or without 0.1 mmol·L−1 NaHS. After the culturing, the activities of caspase-3 and -7 were analysed. Each column represents the mean ± SE from three to five separate experiments. *P < 0.05 and **P < 0.01.
Effects of NaHS on the proportion of TUNEL-positive pancreatic β-cells
To determine whether NaHS protects pancreatic β-cells from apoptosis, mouse isolated islets were cultured with the cytokine mixture and then were double stained using TUNEL reagent and anti-insulin antibody. Treatment with the cytokine mixture increased the proportion of insulin- and TUNEL-positive cells. NaHS at 0.1 mmol·L−1, decreased TUNEL-positive β-cells induced by the cytokine mixture (Figure 5).
Figure 5.
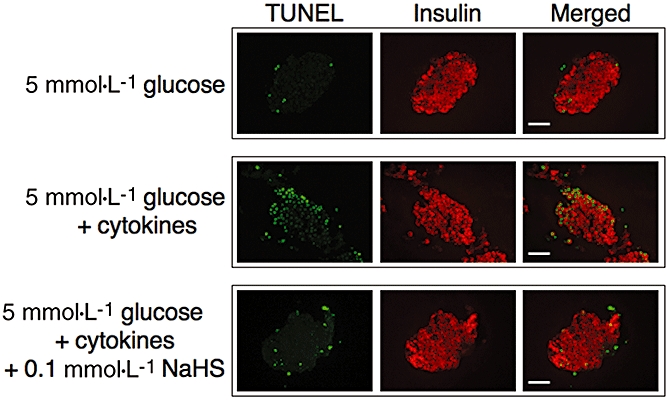
Terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labelling (TUNEL) staining of mouse islets. Isolated pancreatic islets were cultured for 18 h with 5 mmol·L−1 glucose alone (upper lane) or 5 mmol·L−1 glucose plus the cytokine mixture with (lower lane) or without (middle lane) 0.1 mmol·L−1 NaHS. Apoptotic cell death is demonstrated by the TUNEL staining in green. Insulin-positive cells are shown in red. Scale bar = 50 µm.
Effects of NaHS against DNA fragmentation of MIN6 cells
To further examine the cytoprotective effects of NaHS, DNA fragmentation in MIN6 cells was analysed in MIN6 cells treated with palmitate, the cytokine mixture, H2O2 or thapsigargin. Treatment of MIN6 cells with 0.5 mmol·L−1 palmitate, the cytokine mixture, 40 µmol·L−1 H2O2 or 1 µmol·L−1 thapsigargin caused a 1.3- to 5-fold increase over control in DNA fragmentation (Figure 6). Co-treatment with 0.1 mmol·L−1 NaHS prevented the accumulation of fragmented DNA induced by palmitate, cytokines and H2O2 (Figure 6A–C), but it did not prevent DNA fragmentation induced by thapsigargin (Figure 6D).
Figure 6.
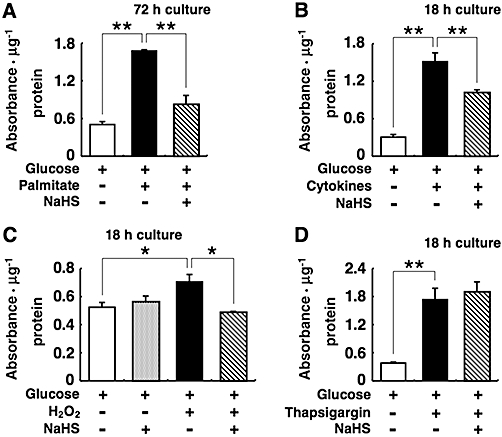
Effects of sodium hydrosulphide (NaHS) on MIN6 cell death induced by palmitate, cytokines, hydrogen peroxide (H2O2) or thapsigargin. MIN6 were cultured with 20 mmol·L−1 glucose alone or 20 mmol·L−1 glucose plus 0.5 mmol·L−1 palmitate (A), the cytokine mixture (B), 40 µmol·L−1 H2O2 (C) or 1 µmol·L−1 thapsigargin (D) in the presence or absence of 0.1 mmol·L−1 NaHS. Fragmented DNA in MIN6 cells was quantified by elisa to evaluate apoptotic cell death. Each column represents the mean ± SE from three to five separate experiments. *P < 0.05 and **P < 0.01.
Effects of NaHS on ROS production
Oxidative stress increases ROS production in cells. We investigated whether NaHS suppresses ROS production. Co-incubation of islets with 0.1 mmol·L−1 NaHS suppressed cytokine-induced ROS production, although treatment with the cytokine mixture did not significantly increase the production (Figure 7A). Treatment with H2O2 or cytokines significantly increased ROS production in MIN6 cells, and this increase was inhibited by co-treatment with NaHS (Figure 7B). Thapsigargin did not increase ROS production, and addition of NaHS did not change the ROS production in thapsigargin-treated islets and MIN6 cells (Figure 7A and B). One of the reasons why the cytokine mixture failed to induce a statistically significant increase in islet ROS production may be because pancreatic islets are composed of different types cells other than β-cells.
Figure 7.
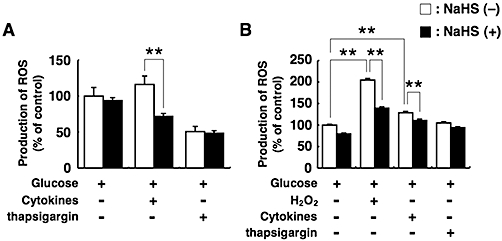
Production of reactive oxygen species (ROS) in pancreatic islets and MIN6 cells. (A) Pancreatic islets were preincubated for 30 min with 10 µmol·L−1 dichlorodihydrofluorescein diacetate (H2DCFDA) fluorescein, and then incubated for 12 h with 5 mmol·L−1 glucose alone or 5 mmol·L−1 glucose plus the cytokine mixture or 1 µmol·L−1 thapsigargin in the presence or absence of 0.1 mmol·L−1 sodium hydrosulphide (NaHS). (B) MIN6 cells were preincubated for 30 min with 10 µmol·L−1 H2DCFDA fluorescein, and then incubated for 12 h with 20 mmol·L−1 glucose alone or 20 mmol·L−1 glucose plus 30 µmol·L−1 hydrogen peroxide (H2O2), the cytokine mixture or 1 µmol·L−1 thapsigargin in the presence or absence of 0.1 mmol·L−1 NaHS. The ROS levels were measured using a dichlorofluorescein fluorescence assay. Each column represents the mean ± SE from three to five separate experiments. **P < 0.01.
Effects of NaHS on Akt phosphorylation
Akt signalling is activated by phosphorylation, and this activation promotes cell proliferation and survival. We examined whether Akt activation contributes to the cytoprotection mediated by NaHS (Figure 8A and B). NaHS increased Akt phosphorylation in MIN6 cells cultured with cytokines, but not in cells cultured with thapsigargin.
Figure 8.
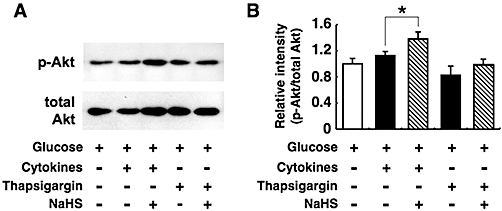
Effects of sodium hydrosulphide (NaHS) on Akt phosphorylation. MIN6 cells were cultured for 6 h with the cytokine mixture or 1 µmol·L−1 thapsigargin in the presence or absence of 0.1 mmol·L−1 NaHS. The concomitant concentration of glucose was 20 mmol·L−1. Protein extracts from these cells were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis and immunoblotted for the detection of phosphorylated (upper lane) and total (lower lane) Akt (A). The phosphorylated Akt (p-Akt) and total Akt levels were then analysed by densitometrical measurement of the intensity of each band with the NIH image software (B). The ratio of p-Akt to total Akt was calculated. Each column represents the mean ± SE from four to five separate experiments. *P < 0.05.
Discussion and conclusions
We have demonstrated that H2S has a protective effect against fatty acid-, cytokine- and H2O2-induced β-cell apoptosis. We previously reported that H2S provides cytoprotection to β-cells from high glucose-induced damage (Kaneko et al., 2009). High glucose and free fatty acids cause β-cell apoptosis through various mechanisms including oxidative stress, ER stress, free radical-induced damage, persistent elevation of the intracellular Ca2+ levels and increases in the production of cytokines (Efanova et al., 1998; Chang-Chen et al., 2008; Poitout and Robertson, 2008; Robertson, 2009). In particular, β-cells are sensitive to oxidative stress because they express low levels of anti-oxidative enzymes such as superoxide dismutase and catalase (Lenzen et al., 1996). Chronic exposure to high concentrations of glucose, free fatty acids or cytokines cause mitochondrial dysfunction and the production of ROS, which induces oxidative stress (Lowell and Shulman, 2005; Lenzen, 2008). Overproduction of ROS and a redox imbalance result in DNA damage, protein oxidation, lipid peroxidation and inhibition of gene expression. The effects of NaHS on ROS production suggest that H2S protects β-cells via its anti-oxidant activity.
H2S has many physiological effects including the regulation of apoptosis, inflammatory response, synaptic transmission and smooth muscle relaxation. Anti-apoptotic effects of H2S may result from a direct chemical reaction between H2S and oxidative substances such as superoxide and hydrogen peroxide. However, other metabolic and signalling pathways may also participate in H2S-mediated anti-apoptotic effects. For example, NaHS increased the production of GSH in MIN6 cells (Kaneko et al., 2009), and GSH is a potent intracellular anti-oxidant that attenuates oxidative stress. In neuronal cells, the protective effects of H2S are also mediated by increased GSH levels (Kimura and Kimura, 2004; Kimura et al., 2010). Therefore, the increase of GSH by H2S may contribute to the protection of β-cells from oxidative stress and to the maintenance of cellular redox homeostasis. H2S is reported to have cardioprotective effects in the events of ischaemia-reperfusion injury (Elrod et al., 2007; Calvert et al., 2009; Sivarajah et al., 2009). The cardioprotection induced by H2S is mediated by activation of the phosphatidylinositol 3-kinase/Akt pathway (Hu et al., 2008; Yong et al., 2008). In pancreatic β-cells, Akt plays important roles in the regulation of cell mass and function (Dickson and Rhodes, 2004; Elghazi and Bernal-Mizrachi, 2009). In this study, Akt phosphorylation was increased after NaHS treatment. We suggest that the activation of Akt signalling may be involved in the H2S-mediated suppression of β-cell apoptosis.
Unexpectedly, H2S failed to influence ER stress-induced apoptosis. This observation contradicted a previous report demonstrating that H2S stimulated ER stress-induced apoptosis of INS-1E, a rat insulinoma cell line (Yang et al., 2007). This discrepancy may result from species-specific differences in the sensitivity of the β-cells to H2S or in the experimental conditions used for the induction of apoptosis.
H2S is endogenously produced by cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) (Maclean and Kraus, 2004). Recent studies suggest that 3-mercaptopyruvate sulphurtransferase may be involved in the enzymatical production of endogenous H2S (Shibuya et al., 2009), although the distribution of this enzyme has not been examined thoroughly. We have recently reported that CBS is constitutively expressed in mouse pancreatic islets but that the expression of CSE is induced by glucose stimulation (Kaneko et al., 2009). This reminds us of distinct roles of constitutive and inducible NO synthases (Nathan and Xie, 1994; Umar and van der Laarse, 2010). Constitutive NO synthase produces NO in a Ca2+/calmodulin-dependent manner and generates NO to transduce the Ca2+ signal. On the other hand, inducible NO synthase functions in apoptosis. Therefore, H2S-producing enzymes have specific and distinct functions; the constitutive enzyme, CBS, acutely inhibits insulin exocytosis, whereas CSE modulates cell survival/death (Yang et al., 2007; Kaneko et al., 2009). This hypothesis requires additional experimental confirmation. Together with our recent findings that H2S suppresses insulin secretion over the short-term (Kaneko et al., 2006) and prevents glucotoxicity during long-term exposure to high glucose (Kaneko et al., 2009), the present findings also support our hypothesis that CSE may be an ‘intrinsic brake’ which functions as a safety device in pancreatic β-cells.
In conclusion, H2S protects pancreatic β-cells against oxidative stress via an anti-oxidative mechanism and by the activation of Akt signalling. We suggest that H2S production via glucose-induced CSE expression may be a self-protecting mechanism that functions under diabetic conditions.
Acknowledgments
The authors thank Professor J-I. Miyazaki (Osaka University) for his gift of MIN6 cells and Dr Y Kaneko for her helpful advice. This work was partly supported by KAKENHI (21591146 & 22790255) from Japan Society Promotion of Science and Research Fund at the Discretion of President, Oita University, and grants from Oita Broadcasting System Cultural Foundation and from Oita University Venture Business Laboratory.
Glossary
Abbreviations
- CBS
cystathionine β-synthase
- CO
carbon monoxide
- CSE
cystathionine γ-lyase
- DCF
dichlorofluorescein
- DMEM
Dulbecco's modified Eagle's medium
- ER
endoplasmic reticulum
- FBS
fetal bovine serum
- GSH
glutathione
- H2DCF
dichlorodihydrofluorescein
- H2DCFDA
dichlorodihydrofluorescein diacetate
- H2O2
hydrogen peroxide
- H2S
hydrogen sulphide
- HK
buffer, HEPES-buffered Krebs-Ringer bicarbonate buffer
- IFN-γ
interferon-γ
- IL-1β
interleukin-1β
- NaHS
sodium hydrosulphide
- NO
nitric oxide
- p-Akt
phosphorylated Akt
- ROS
reactive oxygen species
- SDS
sodium dodecyl sulphate
- T-TBS
Tris-buffered saline containing Tween 20
- TNF-α
tumour necrosis factor-α
- TUNEL
terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labelling
Conflicts of interest
The authors state no conflict of interest.
Supporting Information
Supporting Information: Teaching Materials; Figs 1–8 as PowerPoint slide.
References
- Assmann A, Ueki K, Winnay JN, Kadowaki T, Kulkarni RN. Glucose effects on beta-cell growth and survival require activation of insulin receptors and insulin receptor substrate 2. Mol Cell Biol. 2009;29:3219–3228. doi: 10.1128/MCB.01489-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskar R, Li L, Moore PK. Hydrogen sulfide-induces DNA damage and changes in apoptotic gene expression in human lung fibroblast cells. FASEB J. 2007;21:247–255. doi: 10.1096/fj.06-6255com. [DOI] [PubMed] [Google Scholar]
- Calvert JW, Jha S, Gundewar S, Elrod JW, Ramachandran A, Pattillo CB, et al. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ Res. 2009;105:365–374. doi: 10.1161/CIRCRESAHA.109.199919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Adhikari S, Ang AD, Bhatia M. Mechanism of induction of pancreatic acinar cell apoptosis by hydrogen sulfide. Am J Physiol Cell Physiol. 2006;291:C503–C510. doi: 10.1152/ajpcell.00547.2005. [DOI] [PubMed] [Google Scholar]
- Chang-Chen KJ, Mullur R, Bernal-Mizrachi E. β-cell failure as a complication of diabetes. Rev Endocr Metab Disord. 2008;9:329–343. doi: 10.1007/s11154-008-9101-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SL, Yang CT, Yang ZL, Guo RX, Meng JL, Cui Y, et al. Hydrogen sulfide protects H9c2 cells against chemical hypoxia-induced cell injuries. Clin Exp Pharmacol Physiol. 2009;37:316–321. doi: 10.1111/j.1440-1681.2009.05289.x. [DOI] [PubMed] [Google Scholar]
- Del Prato S, Bianchi C, Marchetti P. β-cell function and anti-diabetic pharmacotherapy. Diabetes Metab Res Rev. 2007;23:518–527. doi: 10.1002/dmrr.770. [DOI] [PubMed] [Google Scholar]
- Dickson LM, Rhodes CJ. Pancreatic β-cell growth and survival in the onset of type 2 diabetes: a role for protein kinase B in the Akt? Am J Physiol Endocrinol Metab. 2004;287:E192–E198. doi: 10.1152/ajpendo.00031.2004. [DOI] [PubMed] [Google Scholar]
- Efanova IB, Zaitsev SV, Zhivotovsky B, Kohler M, Efendic S, Orrenius S, et al. Glucose and tolbutamide induce apoptosis in pancreatic β-cells: a process dependent on intracellular Ca2+ concentration. J Biol Chem. 1998;273:33501–33507. doi: 10.1074/jbc.273.50.33501. [DOI] [PubMed] [Google Scholar]
- Elghazi L, Bernal-Mizrachi E. Akt and PTEN: β-cell mass and pancreas plasticity. Trends Endocrinol Metab. 2009;20:243–251. doi: 10.1016/j.tem.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci U S A. 2007;104:15560–15565. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadalla MM, Snyder SH. Hydrogen sulfide as a gasotransmitter. J Neurochem. 2010;113:14–26. doi: 10.1111/j.1471-4159.2010.06580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Chen X, Pan TT, Neo KL, Lee SW, Khin ES, et al. Cardioprotection induced by hydrogen sulfide preconditioning involves activation of ERK and PI3K/Akt pathways. Pflügers Arch. 2008;455:607–616. doi: 10.1007/s00424-007-0321-4. [DOI] [PubMed] [Google Scholar]
- Jha S, Calvert JW, Duranski MR, Ramachandran A, Lefer DJ. Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: role of antioxidant and antiapoptotic signaling. Am J Physiol Heart Circ Physiol. 2008;295:H801–H806. doi: 10.1152/ajpheart.00377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko Y, Kimura Y, Kimura H, Niki I. L-cysteine inhibits insulin release from the pancreatic β-cell: possible involvement of metabolic production of hydrogen sulfide, a novel gasotransmitter. Diabetes. 2006;55:1391–1397. doi: 10.2337/db05-1082. [DOI] [PubMed] [Google Scholar]
- Kaneko Y, Kimura T, Taniguchi S, Souma M, Kojima Y, Kimura Y, et al. Glucose-induced production of hydrogen sulfide may protect the pancreatic beta-cells from apoptotic cell death by high glucose. FEBS Lett. 2009;583:377–382. doi: 10.1016/j.febslet.2008.12.026. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004;18:1165–1167. doi: 10.1096/fj.04-1815fje. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Goto Y, Kimura H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid Redox Signal. 2010;12:1–13. doi: 10.1089/ars.2008.2282. [DOI] [PubMed] [Google Scholar]
- Lenzen S. Oxidative stress: the vulnerable β-cell. Biochem Soc Trans. 2008;36:343–347. doi: 10.1042/BST0360343. [DOI] [PubMed] [Google Scholar]
- Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radical Biol Med. 1996;20:463–466. doi: 10.1016/0891-5849(96)02051-5. [DOI] [PubMed] [Google Scholar]
- Li L, Moore PK. Putative biological roles of hydrogen sulfide in health and disease: a breath of not so fresh air? Trends Pharmacol Sci. 2008;29:84–90. doi: 10.1016/j.tips.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- Łowicka E, Bełtowski J. Hydrogen sulfide (H2S) – the third gas of interest for pharmacologists. Pharmacol Rep. 2007;59:4–24. [PubMed] [Google Scholar]
- Maclean KN, Kraus JP. Hydrogen sulfide production and metabolism in mammalian tissues. In: Wang R, editor. In Signal Transduction and the Gasotransmitters. Totowa, NJ: Humana Press; 2004. pp. 275–292. [Google Scholar]
- Nagai Y, Tsugane M, Oka J, Kimura H. Hydrogen sulfide induces calcium waves in astrocytes. FASEB J. 2004;18:557–559. doi: 10.1096/fj.03-1052fje. [DOI] [PubMed] [Google Scholar]
- Nathan C, Xie Q-W. Regulation of biosynthesis of nitric oxide. J Biol Chem. 1994;269:13725–13728. [PubMed] [Google Scholar]
- Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and β-cell dysfunction. Endocr Rev. 2008;29:351–366. doi: 10.1210/er.2007-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu K, Lee SW, Bian JS, Low CM, Wong PT. Hydrogen sulfide: neurochemistry and neurobiology. Neurochem Int. 2008;52:155–165. doi: 10.1016/j.neuint.2007.05.016. [DOI] [PubMed] [Google Scholar]
- Robertson RP. β−cell deterioration during diabetes: what's in the gun? Trends Endocrinol Metab. 2009;20:388–393. doi: 10.1016/j.tem.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samhan-Arias AK, Garcia-Bereguiain MA, Gutierrez-Merino C. Hydrogen sulfide is a reversible inhibitor of the NADH oxidase activity of synaptic plasma membranes. Biochem Biophys Res Commun. 2009;388:718–722. doi: 10.1016/j.bbrc.2009.08.076. [DOI] [PubMed] [Google Scholar]
- Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K, et al. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid Redox Signal. 2009;11:703–714. doi: 10.1089/ars.2008.2253. [DOI] [PubMed] [Google Scholar]
- Sivarajah A, Collino M, Yasin M, Benetti E, Gallicchio M, Mazzon E, et al. Anti-apoptotic and anti-inflammatory effects of hydrogen sulfide in a rat model of regional myocardial I/R. Shock. 2009;31:267–274. doi: 10.1097/SHK.0b013e318180ff89. [DOI] [PubMed] [Google Scholar]
- Sjöholm Å. Intracellular signal transduction pathways that control pancreatic β-cell proliferation. FEBS Lett. 1992;311:85–90. doi: 10.1016/0014-5793(92)81373-t. [DOI] [PubMed] [Google Scholar]
- Umar S, van der Laarse A. Nitric oxide and nitric oxide synthase isoforms in the normal, hypertrophic, and failing heart. Mol Cell Biochem. 2010;333:191–201. doi: 10.1007/s11010-009-0219-x. [DOI] [PubMed] [Google Scholar]
- Wajchenberg BL. β−cell failure in diabetes and preservation by clinical treatment. Endocr Rev. 2007;28:187–218. doi: 10.1210/10.1210/er.2006-0038. [DOI] [PubMed] [Google Scholar]
- Yang G, Wu L, Wang R. Pro-apoptotic effect of endogenous H2S on human aorta smooth muscle cells. FASEB J. 2006;20:553–555. doi: 10.1096/fj.05-4712fje. [DOI] [PubMed] [Google Scholar]
- Yang G, Yang W, Wu L, Wang R. H2S, endoplasmic reticulum stress, and apoptosis of insulin-secreting beta cells. J Biol Chem. 2007;282:16567–16576. doi: 10.1074/jbc.M700605200. [DOI] [PubMed] [Google Scholar]
- Yong QC, Lee SW, Foo CS, Neo KL, Chen X, Bian JS. Endogenous hydrogen sulphide mediates the cardioprotection induced by ischemic postconditioning. Am J Physiol Heart Circ Physiol. 2008;295:H1330–H1340. doi: 10.1152/ajpheart.00244.2008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.