

Abstract
The title compound, C6H4Br2S, represents a versatile building block for the preparation of π-conjugated redox-active thienyl oligomers and metal-mediated cross-coupling reactions. This is due to the presence of an electrochemically active thienyl heterocycle and a reactive dibromovinyl substituent, which easily undergoes dehydrobromination in the presence of n-butyllithium to afford 2-ethynylthiophene. In the molecule, the alkenyl unit and the thiophene ring are almost coplanar with an angle of 3.5 (2)° between the normals of the best planes of the thiophene ring and the vinyl moiety.
Related literature
The title compound was first prepared in 1980, see: Bestmann et al. (1980 ▶). For an alternative synthesis using a Corey–Fuchs reaction, see: Beny et al. (1982 ▶). For a structural comparison with 2,2-dibromovinyl[2,2]paracyclophane [PCP—C(H)=CBr2], (2,2-dibromovinyl)ferrocene [Fc—C(H)=CBr2], and 2-thienylmethylenemalononitrile [C4H3S—C(H)=C(CN)2], see: Clément et al. (2007a
▶,b
▶) and Mukherjee et al. (1984 ▶), respectively. For recent applications, see: Herz et al. (1999 ▶); Rao et al. (2010 ▶); Zhang et al. (2010 ▶).
Experimental
Crystal data
C6H4Br2S
M r = 267.97
Monoclinic,
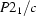
a = 9.6843 (19) Å
b = 7.2379 (14) Å
c = 11.484 (2) Å
β = 109.16 (3)°
V = 760.4 (3) Å3
Z = 4
Mo Kα radiation
μ = 10.84 mm−1
T = 173 K
0.4 × 0.4 × 0.2 mm
Data collection
Stoe IPDS diffractometer
Absorption correction: numerical (FACEIT in IPDS; Stoe & Cie, 1999 ▶) T min = 0.188, T max = 0.658
6492 measured reflections
1667 independent reflections
1444 reflections with I > 2σ(I)
R int = 0.064
Refinement
R[F 2 > 2σ(F 2)] = 0.058
wR(F 2) = 0.139
S = 1.04
1667 reflections
82 parameters
H-atom parameters not refined
Δρmax = 1.36 e Å−3
Δρmin = −1.73 e Å−3
Data collection: EXPOSE in IPDS (Stoe & Cie, 1999 ▶); cell refinement: CELL in IPDS; data reduction: INTEGRATE in IPDS; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 (Farrugia, 1997 ▶); software used to prepare material for publication: WinGX (Farrugia, 1999 ▶).
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536811002522/im2256sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536811002522/im2256Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Acknowledgments
We thank the Fonds der Chemischen Industrie (FCI) and the Deutsche Forschungsgemeinschaft (DFG) for financial support.
supplementary crystallographic information
Comment
The title compound (Scheme 1, Fig. 1), which is easily accessible from thiophene-2-carbaldehyde via the Corey-Fuchs reaction, has over the last 30 years become a versatile starting material for a variety of organic transformations and a precursor in material science. This interest is due to the conjugation between the electrochemically active thienyl heterocycle with the reactive halogenated olefin moiety. Originally it was used for the preparation of terthiophenes (Beny et al., 1982). Recent applications include Pd-catalyzed cross-coupling reactions (Herz et al., 1999; Rao et al., 2010) as well as the synthesis of imidazo[1,5-α]pyridines (Zhang et al., 2010).
In the course of our interest in developing new π-conjugated dihalovinyl compounds R—C(H)═CX2 with functional groups (R = imine, ferrocenyl, [2,2]paracyclophane) as substrates for oxidative addition reactions across low-valent noble metals, we have recently reported the synthesis and crystal structures of 4-2',2'-dibromovinyl[2,2]paracyclophane (Clément et al. 2007a) and (2,2-dibromovinyl)ferrocene (Clément et al. 2007b). With this aim in mind, we also prepared the title compound 2-(2,2-dibromoethenyl)thiophene. A survey of the CSD data base revealed that neither 2-vinylthiophene nor a halogenated derivative of the types [C4H3S—C(H)═C(H)X] or [C4H3S—C(H)═CX2] (X = halogen) had been structurally characterized yet. The most related molecule found is 2-thienylmethylenemalononitrile [C4H3S—C(H)═C(CN)2] (Mukherjee et al., 1984). In the latter compound, the angle between the normals of the two planar parts of the molecule, the thiophene cycle and the dicyanovinyl moiety, amounts to 3.6 (5)°. In the title compound, the corresponding angle lies in the same range [3.5 (2)°]. A somewhat larger angle of 10.4° has been determined for (2,2-dibromovinyl)ferrocene [Fc—C(H)═CBr2] (Clément et al., 2007b), whereas in 4-(2',2'-dibromovinyl)[2,2]paracyclophane [PCP—C(H)═CBr2] an angle of 51.1° has been observed significantly deviating from coplanarity (Clément et al., 2007a). The length of the vinylic C1—C2 double bond [1.335 (7) Å] matches well with those of [PCP—C(H)═CBr2] [1.320 (3)°] and [Fc—C(H)═ CBr2] [1.318 (4) Å] (Clément et al., 2007a; Clément et al., 2007b). A similar bond length of 1.353 (5) Å has also been reported for [C4H3S—C(H)═C(CN)2] (Mukherjee et al., 1984).
Bond lenths and angles of the thienyl moiety may be considered as normal and deserve no further comment. The unit cell consists of 4 molecules which are held together by weak interactions only (Fig. 2). These consist of the short Br1—Br2 distance [3.6501 (9) Å, Br1_5-Br2_2] as well as the short distances between Br2 and the carbon atoms of the thiophene ring [3.604 (5) Å, Br2_2-C4; 3.479 (6) Å, Br2_2-C5; 3.624 (5) Å, Br2_2-C6].
Experimental
Triphenylphosphine (4.20 g, 16.0 mmol), CBr4 (5.31 g, 16.0 mmol) and zinc dust (1.05 g, 16.0 mmol) were placed in a Schlenk tube and 40 ml of CH2Cl2 were slowly added. The mixture was stirred at room temperature for 28 h. Then, 2-thiophenecarboxaldehyde (0.89 g, 8.00 mmol) in CH2Cl2 (10 ml) was added and stirring was continued for further 2 h. The reaction mixture was extracted with three 50 ml portions of pentane. CH2Cl2 was added when the reaction mixture became too viscous for further extractions. The extracts were filtered and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel with CH2Cl2/petroleum ether (1:4). Slow evaporation afforded white crystals of 2-(2,2-dibromoethenyl)thiophene (yield: 90%). Characterization data have been previously described in the literature. (Beny et al., 1982)
Refinement
H atoms were refined using a riding model in their ideal geometric positions using the riding model approximation with Uiso(H) = 1.2Ueq(C) for all H atoms.
Figures
Fig. 1.

Molecular structure of the title compound with thermal ellipsoids drawn at the 50% probability level.
Fig. 2.

Crystal packing of 2-(2,2-dibromoethenyl)thiophene. Symmetry operations: (1) x, y, z; (2) -x, y - 1/2, -z - 1/2; (3) -x, -y - 1, -z; (4) x, -y + 1/2, z - 1/2; (5) x - 1, y, z; (6) x, y -1, z; (7) x - 1, -y + 1/2, z - 1/2; (8) -x, -y - 1, -z - 1.
Crystal data
| C6H4Br2S | F(000) = 504 |
| Mr = 267.97 | Dx = 2.341 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 980 reflections |
| a = 9.6843 (19) Å | θ = 2.2–27.0° |
| b = 7.2379 (14) Å | µ = 10.84 mm−1 |
| c = 11.484 (2) Å | T = 173 K |
| β = 109.16 (3)° | Plates, colourless |
| V = 760.4 (3) Å3 | 0.4 × 0.4 × 0.2 mm |
| Z = 4 |
Data collection
| Stoe IPDS diffractometer | 1444 reflections with I > 2σ(I) |
| graphite | Rint = 0.064 |
| φ scans | θmax = 27.0°, θmin = 2.2° |
| Absorption correction: numerical (FACEIT in IPDS; Stoe & Cie, 1999) | h = −11→12 |
| Tmin = 0.188, Tmax = 0.658 | k = −9→9 |
| 6492 measured reflections | l = −14→14 |
| 1667 independent reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.058 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.139 | H-atom parameters not refined |
| S = 1.04 | w = 1/[σ2(Fo2) + (0.1099P)2] where P = (Fo2 + 2Fc2)/3 |
| 1667 reflections | (Δ/σ)max < 0.001 |
| 82 parameters | Δρmax = 1.36 e Å−3 |
| 0 restraints | Δρmin = −1.73 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Br1 | 0.90429 (5) | 0.69525 (7) | 0.15399 (5) | 0.0306 (2) | |
| Br2 | 0.75506 (5) | 0.94572 (7) | 0.30330 (4) | 0.0290 (2) | |
| C1 | 0.7287 (5) | 0.7946 (6) | 0.1647 (4) | 0.0214 (9) | |
| C2 | 0.5999 (5) | 0.7559 (7) | 0.0803 (4) | 0.0225 (9) | |
| H2 | 0.6061 | 0.6818 | 0.0139 | 0.027* | |
| C3 | 0.4530 (5) | 0.8068 (6) | 0.0720 (4) | 0.0188 (8) | |
| C4 | 0.3277 (5) | 0.7471 (7) | −0.0213 (4) | 0.0225 (9) | |
| H4 | 0.3305 | 0.6695 | −0.0873 | 0.027* | |
| C5 | 0.1970 (6) | 0.8134 (7) | −0.0080 (5) | 0.0307 (11) | |
| H5 | 0.1026 | 0.7857 | −0.0637 | 0.037* | |
| C6 | 0.2213 (5) | 0.9216 (7) | 0.0939 (5) | 0.0289 (10) | |
| H6 | 0.1455 | 0.9771 | 0.1174 | 0.035* | |
| S | 0.40366 (13) | 0.94655 (17) | 0.17497 (12) | 0.0265 (3) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Br1 | 0.0220 (3) | 0.0379 (4) | 0.0324 (3) | 0.00528 (18) | 0.0097 (2) | −0.0004 (2) |
| Br2 | 0.0268 (3) | 0.0342 (3) | 0.0250 (3) | −0.00466 (18) | 0.0072 (2) | −0.00800 (17) |
| C1 | 0.024 (2) | 0.021 (2) | 0.0202 (19) | 0.0009 (16) | 0.0089 (17) | 0.0037 (16) |
| C2 | 0.024 (2) | 0.020 (2) | 0.024 (2) | 0.0027 (18) | 0.0104 (18) | 0.0016 (17) |
| C3 | 0.024 (2) | 0.0157 (19) | 0.0183 (19) | 0.0008 (15) | 0.0095 (17) | 0.0015 (15) |
| C4 | 0.024 (2) | 0.020 (2) | 0.024 (2) | 0.0029 (17) | 0.0083 (18) | 0.0041 (17) |
| C5 | 0.020 (2) | 0.034 (3) | 0.036 (3) | −0.0005 (18) | 0.005 (2) | 0.006 (2) |
| C6 | 0.022 (2) | 0.028 (2) | 0.037 (3) | −0.0003 (18) | 0.011 (2) | 0.003 (2) |
| S | 0.0256 (6) | 0.0287 (6) | 0.0277 (6) | 0.0006 (4) | 0.0121 (5) | −0.0054 (4) |
Geometric parameters (Å, °)
| Br1—C1 | 1.887 (5) | C4—C5 | 1.408 (7) |
| Br2—C1 | 1.878 (5) | C4—H4 | 0.95 |
| C1—C2 | 1.335 (7) | C5—C6 | 1.362 (8) |
| C2—C3 | 1.442 (6) | C5—H5 | 0.95 |
| C2—H2 | 0.95 | C6—S | 1.714 (5) |
| C3—C4 | 1.397 (7) | C6—H6 | 0.95 |
| C3—S | 1.738 (4) | ||
| C2—C1—Br2 | 124.9 (4) | C3—C4—H4 | 123.3 |
| C2—C1—Br1 | 121.3 (4) | C5—C4—H4 | 123.3 |
| Br2—C1—Br1 | 113.7 (3) | C6—C5—C4 | 112.4 (5) |
| C1—C2—C3 | 131.4 (4) | C6—C5—H5 | 123.8 |
| C1—C2—H2 | 114.3 | C4—C5—H5 | 123.8 |
| C3—C2—H2 | 114.3 | C5—C6—S | 112.6 (4) |
| C4—C3—C2 | 124.1 (4) | C5—C6—H6 | 123.7 |
| C4—C3—S | 109.8 (3) | S—C6—H6 | 123.7 |
| C2—C3—S | 126.1 (3) | C6—S—C3 | 91.9 (2) |
| C3—C4—C5 | 113.3 (4) |
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: IM2256).
References
- Beny, J.-P., Dhawan, S. N., Kagan, J. & Sundlass, S. (1982). J. Org. Chem. 47, 2201–2204.
- Bestmann, H. J. & Frey, H. (1980). Liebigs Ann. Chem. pp. 2061–2071.
- Clément, S., Guyard, L., Knorr, M., Dilsky, S., Strohmann, C. & Arroyo, M. (2007a). J. Organomet. Chem. 692, 839–850.
- Clément, S., Guyard, L., Knorr, M., Vilafane, F., Strohmann, C. & Kubicki, M. M. (2007b). Eur. J. Inorg. Chem. pp. 5052–5061.
- Farrugia, L. J. (1997). J. Appl. Cryst. 30, 565.
- Farrugia, L. J. (1999). J. Appl. Cryst. 32, 837–838.
- Herz, H.-G., Queiroz, M. J. R. P. & Maas, G. (1999). Synthesis, pp. 1013–1016
- Mukherjee, A. K., Mukherjee (née Mondal), M., De, A. & Bhattacharyya, S. P. (1984). Acta Cryst. C40, 991–992.
- Rao, M. L. N., Jadhav, D. N. & Dasgupta, P. (2010). Org. Lett. 12, 2048–2051. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Stoe & Cie (1999). IPDS Stoe & Cie, Darmstadt, Germany.
- Zhang, A., Zheng, X., Fan, J. & Shen, W. (2010). Tetrahedron Lett. 51, 828–831.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536811002522/im2256sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536811002522/im2256Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report