

Abstract
In the crystal structure of the title compound, C7H6BFO2, a broad-spectrum antifungal drug (AN2690), the planar [maximum deviation 0.035 (1) Å] molecules form centrosymmetric R
2
2(8) dimers via strong O—H⋯O hydrogen bonds. The dimers are arranged into layers by weak intermolecular C—H⋯O and C—H⋯F hydrogen bonds. The symmetry of this two-dimensional supramolecular assembly can be described by the layer group p
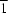 and topologically classified as a simple uninodal four-connected two-dimensional network of a (4.4.4.4.6.6) topology. Further weak C—H⋯O interactions build up the three-dimensional structure.
and topologically classified as a simple uninodal four-connected two-dimensional network of a (4.4.4.4.6.6) topology. Further weak C—H⋯O interactions build up the three-dimensional structure.
Related literature
For the review of the synthesis, properties and applications of benzoxaboroles, see: Adamczyk-Woźniak et al. (2009 ▶). For the biological activity of the title compound, see: Baker et al. (2005 ▶, 2006 ▶); Hui et al. (2007 ▶); Rock et al. (2007 ▶). For the synthesis see: Baker et al. (2006 ▶), Gunasekera et al. (2007 ▶). For related structures, see: Adamczyk-Woźniak et al. (2010 ▶); Tan et al. (2001 ▶); Yamamoto et al. (2005 ▶); Zhdankin et al. (1999 ▶). For hydrogen-bond graph-set descriptors and layer symmetry groups, see: Etter (1990 ▶) and International Tables for Crystallography (2006 ▶), respectively.
Experimental
Crystal data
C7H6BFO2
M r = 151.93
Triclinic,
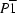
a = 3.8799 (3) Å
b = 6.3077 (5) Å
c = 14.0735 (12) Å
α = 98.068 (7)°
β = 91.564 (7)°
γ = 100.473 (7)°
V = 334.84 (5) Å3
Z = 2
Cu Kα radiation
μ = 1.06 mm−1
T = 100 K
0.60 × 0.35 × 0.20 mm
Data collection
Oxford Diffraction Gemini A Ultra diffractometer
Absorption correction: multi-scan (CrysAlis PRO; Oxford Diffraction, 2006 ▶) T min = 0.731, T max = 1.000
3451 measured reflections
1193 independent reflections
1147 reflections with I > 2σ(I)
R int = 0.016
Refinement
R[F 2 > 2σ(F 2)] = 0.032
wR(F 2) = 0.088
S = 1.07
1193 reflections
105 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.33 e Å−3
Δρmin = −0.18 e Å−3
Data collection: CrysAlis PRO (Oxford Diffraction, 2006 ▶); cell refinement: CrysAlis PRO; data reduction: CrysAlis PRO; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEPIII (Burnett & Johnson, 1996 ▶); software used to prepare material for publication: OLEX2 (Dolomanov et al., 2009 ▶), PLATON (Spek, 2009 ▶) and publCIF (Westrip, 2010 ▶).
Supplementary Material
Crystal structure: contains datablocks . DOI: 10.1107/S1600536811001632/fj2381sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536811001632/fj2381Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O2—H2⋯O1i | 0.83 (2) | 1.93 (2) | 2.7614 (13) | 175 (2) |
| C7—H7B⋯O2ii | 0.99 | 2.55 | 3.5325 (15) | 172 |
| C5—H5⋯F1iii | 0.95 | 2.58 | 3.4779 (14) | 157 |
| C7—H7A⋯O2iv | 0.99 | 2.66 | 3.2172 (14) | 116 |
| C3—H3⋯O2iv | 0.95 | 2.70 | 3.4276 (14) | 134 |
Symmetry codes: (i) 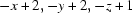 ; (ii)
; (ii) 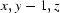 ; (iii)
; (iii) 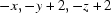 ; (iv)
; (iv) 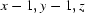 .
.
Acknowledgments
The authors acknowledge financial support by the Ministry of Science and Higher Education (grant No. N204 127938).
supplementary crystallographic information
Experimental
5-Fluoro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole (I) was synthesized according to Fig. 3.
2-Bromo-5-fluorobenzaldehyde was purchased from Sigma-Aldrich and used as received. 2-Bromo-5-fluorobenzaldehyde (5.00 g, 0.025 mol) and 2.69 g (0.025 mol) of trimethoxymethane was dissolved in 100 ml of methanol in a two-necked vessel. 0.4 ml of concentrated H2SO4 was added. The solution was refluxed for one hour and left to cool down. Then the solution was brought to pH≈11 with a concentrated solution of NaOMe in methanol. The reaction mixture was distilled under vacuum to give 5.90 g of 1-Bromo-2-(dimethoxymethyl)-4-fluorobenzene as a colorless liquid (yield 96%; 1H NMR (CDCl3, 400 MHz): 7.49 (dd, 1H), 7.34 (dd, 1H), 6.91 (td, 1H), 5.49 (s, 1H), 3.37 (s, 6H) p.p.m.). The product was dissolved in 100 ml of dry Et2O in a three-necked vessel under argon flow. The solution was cooled down to -78°C using dry ice/acetone bath. n-Butyllithium in hexane (2.5 M, 11 ml) was added dropwise to keep the temperature under -70°C. The solution was stirred for one hour, then 3.80 g (0.026 mol, 4.4 ml) of triethyl borate was added slowly, keeping the temperature under -70°C. The dry ice/acetone bath was removed and the solution was stirred for one hour. The solution was brought to pH≈3 with 3 M aq. HCl. The aqueous layer was separated and extracted with Et2O (2 × 100 ml). The organic layers were combined and the solvent was partially removed under vacuum. The remaining thick solution was dissolved in hot water. Yellowish crystals of 4-fluoro-2-formylphenylboronic acid were filtered after a few hours. Recrystallization from water gave 1.79 g of the product (yield 49%; 1H NMR (CDCl3, 400 MHz): 9.89 (s, 1H), 8.31 (dd, 1H), 7.62 (dd, 1H), 7.40 (td, 1H) p.p.m.). The product (1.79 g, 0.011 mol) was dissolved in 100 ml of methanol in a one-necked vessel. 0.44 g (0.012 mol) of NaBH4 was added in small portions. The solution was mixed for 12 h. Another portion of 0.22 g of NaBH4 was added and the solution was mixed for 3 days. The solvent was removed under vacuum. The crude product was dissolved in water. Crystallization gave 0.82 g of 5-Fluoro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole (I) as yellowish crystals (yield 51%; 1H NMR (CDCl3, 400 MHz): 7.72 (dd, 1H), 7.06 (m, 2H), 5.08 (s, 2H) p.p.m.; 19F NMR (CDCl3, 376.3 MHz): -113.51 (q) p.p.m.; 11B NMR ((CdD3)2CO, 64.1 MHz): 32.0 p.p.m.; m.p. 135–136°C).
Refinement
H2 atom bonded to O2 atom was located in a difference map and freely refined. Other H atoms were positioned geometrically and refined using a riding model with C—H = 0.95–0.99 Å and with Uiso(H) = 1.2 times Ueq(C).
Figures
Fig. 1.

ORTEP plot of the hydrogen bonded dimer of (I), showing the atom-labelling scheme. Displacement ellipsoids are drawn at the 50% probability level.
Fig. 2.

Projection on (1 0 2) plane showing layers of molecules linked by O—H···O (dashed lines), C—H···O and C—H···F (dotted lines) H-bonds.
Fig. 3.

Synthesis of 5-fluoro-1,3-dihydro-1-hydroxy-2,1-benzoxaborole (I).
Crystal data
| C7H6BFO2 | Z = 2 |
| Mr = 151.93 | F(000) = 156 |
| Triclinic, P1 | Dx = 1.507 Mg m−3 |
| Hall symbol: -P 1 | Melting point: 408 K |
| a = 3.8799 (3) Å | Cu Kα radiation, λ = 1.5418 Å |
| b = 6.3077 (5) Å | Cell parameters from 3116 reflections |
| c = 14.0735 (12) Å | θ = 3.2–67.1° |
| α = 98.068 (7)° | µ = 1.06 mm−1 |
| β = 91.564 (7)° | T = 100 K |
| γ = 100.473 (7)° | Prism, light yellow |
| V = 334.84 (5) Å3 | 0.60 × 0.35 × 0.20 mm |
Data collection
| Oxford Diffraction Gemini A Ultra diffractometer | 1193 independent reflections |
| Radiation source: Enhance Ultra (Cu) X-ray Source | 1147 reflections with I > 2σ(I) |
| mirror | Rint = 0.016 |
| Detector resolution: 10.3347 pixels mm-1 | θmax = 67.1°, θmin = 3.2° |
| ω scans | h = −4→4 |
| Absorption correction: multi-scan (CrysAlis PRO; Oxford Diffraction, 2006) | k = −7→7 |
| Tmin = 0.731, Tmax = 1.000 | l = −16→14 |
| 3451 measured reflections |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.032 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.088 | w = 1/[σ2(Fo2) + (0.0511P)2 + 0.1152P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.07 | (Δ/σ)max < 0.001 |
| 1193 reflections | Δρmax = 0.33 e Å−3 |
| 105 parameters | Δρmin = −0.18 e Å−3 |
| 0 restraints | Extinction correction: SHELXL97 (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.046 (5) |
Special details
| Experimental. Empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm. (Oxford Diffraction, 2006) |
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O2 | 0.9636 (2) | 1.22613 (14) | 0.60638 (7) | 0.0189 (3) | |
| F1 | 0.0385 (2) | 0.70856 (13) | 0.93059 (5) | 0.0282 (3) | |
| O1 | 0.7364 (2) | 0.84381 (13) | 0.56821 (6) | 0.0171 (3) | |
| C3 | 0.2566 (3) | 0.6725 (2) | 0.77652 (9) | 0.0185 (3) | |
| H3 | 0.1624 | 0.5207 | 0.7660 | 0.022* | |
| C4 | 0.2168 (3) | 0.8036 (2) | 0.86129 (9) | 0.0200 (3) | |
| C2 | 0.4422 (3) | 0.7757 (2) | 0.70750 (9) | 0.0162 (3) | |
| C1 | 0.5825 (3) | 0.9983 (2) | 0.72234 (9) | 0.0167 (3) | |
| C5 | 0.3485 (3) | 1.0251 (2) | 0.87950 (9) | 0.0207 (3) | |
| H5 | 0.3121 | 1.1083 | 0.9388 | 0.025* | |
| C6 | 0.5346 (3) | 1.1237 (2) | 0.80968 (9) | 0.0186 (3) | |
| H6 | 0.6291 | 1.2754 | 0.8210 | 0.022* | |
| B1 | 0.7783 (3) | 1.0404 (2) | 0.63011 (10) | 0.0164 (3) | |
| C7 | 0.5248 (3) | 0.6711 (2) | 0.61026 (9) | 0.0170 (3) | |
| H7B | 0.6562 | 0.5526 | 0.6164 | 0.020* | |
| H7A | 0.3064 | 0.6096 | 0.5701 | 0.020* | |
| H2 | 1.061 (5) | 1.212 (3) | 0.5545 (15) | 0.040 (5)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O2 | 0.0245 (5) | 0.0147 (5) | 0.0169 (5) | 0.0025 (4) | 0.0053 (4) | 0.0011 (3) |
| F1 | 0.0329 (5) | 0.0323 (5) | 0.0196 (4) | 0.0035 (4) | 0.0106 (3) | 0.0065 (3) |
| O1 | 0.0208 (5) | 0.0145 (5) | 0.0154 (5) | 0.0019 (3) | 0.0046 (3) | 0.0017 (3) |
| C3 | 0.0178 (6) | 0.0188 (6) | 0.0193 (7) | 0.0037 (5) | 0.0009 (5) | 0.0033 (5) |
| C4 | 0.0181 (6) | 0.0273 (7) | 0.0160 (6) | 0.0052 (5) | 0.0038 (5) | 0.0061 (5) |
| C2 | 0.0149 (6) | 0.0174 (6) | 0.0168 (6) | 0.0055 (5) | −0.0004 (4) | 0.0014 (5) |
| C1 | 0.0154 (6) | 0.0174 (6) | 0.0178 (7) | 0.0051 (5) | −0.0011 (5) | 0.0021 (5) |
| C5 | 0.0212 (6) | 0.0256 (7) | 0.0155 (6) | 0.0083 (5) | 0.0008 (5) | −0.0016 (5) |
| C6 | 0.0187 (6) | 0.0179 (6) | 0.0188 (6) | 0.0049 (5) | −0.0001 (5) | −0.0007 (5) |
| B1 | 0.0162 (6) | 0.0162 (7) | 0.0172 (7) | 0.0055 (5) | −0.0011 (5) | 0.0009 (5) |
| C7 | 0.0197 (6) | 0.0135 (6) | 0.0174 (6) | 0.0017 (5) | 0.0036 (5) | 0.0021 (5) |
Geometric parameters (Å, °)
| O2—B1 | 1.3483 (18) | C2—C1 | 1.3948 (18) |
| O2—H2 | 0.83 (2) | C2—C7 | 1.5025 (17) |
| F1—C4 | 1.3562 (15) | C1—C6 | 1.4013 (17) |
| O1—B1 | 1.3922 (17) | C1—B1 | 1.5522 (18) |
| O1—C7 | 1.4471 (15) | C5—H5 | 0.9500 |
| C3—H3 | 0.9500 | C5—C6 | 1.3856 (18) |
| C3—C4 | 1.3822 (19) | C6—H6 | 0.9500 |
| C3—C2 | 1.3897 (18) | C7—H7B | 0.9900 |
| C4—C5 | 1.3829 (19) | C7—H7A | 0.9900 |
| O2—B1—O1 | 121.51 (12) | C2—C3—H3 | 121.9 |
| O2—B1—C1 | 130.25 (12) | C2—C1—C6 | 119.16 (12) |
| F1—C4—C3 | 117.85 (12) | C2—C1—B1 | 104.93 (11) |
| F1—C4—C5 | 118.27 (12) | C2—C7—H7B | 110.7 |
| O1—B1—C1 | 108.24 (11) | C2—C7—H7A | 110.7 |
| O1—C7—C2 | 105.45 (9) | C1—C2—C7 | 110.88 (11) |
| O1—C7—H7B | 110.7 | C1—C6—H6 | 120.2 |
| O1—C7—H7A | 110.7 | C5—C6—C1 | 119.66 (12) |
| C3—C4—C5 | 123.88 (12) | C5—C6—H6 | 120.2 |
| C3—C2—C1 | 122.36 (12) | C6—C1—B1 | 135.86 (12) |
| C3—C2—C7 | 126.75 (11) | C6—C5—H5 | 120.6 |
| C4—C3—H3 | 121.9 | B1—O2—H2 | 115.3 (13) |
| C4—C3—C2 | 116.12 (12) | B1—O1—C7 | 110.46 (10) |
| C4—C5—H5 | 120.6 | H7B—C7—H7A | 108.8 |
| C4—C5—C6 | 118.82 (12) | ||
| F1—C4—C5—C6 | 179.28 (10) | C2—C1—B1—O2 | −179.25 (12) |
| C3—C4—C5—C6 | −0.58 (19) | C2—C1—B1—O1 | 0.71 (13) |
| C3—C2—C1—C6 | −0.23 (17) | C1—C2—C7—O1 | 2.08 (13) |
| C3—C2—C1—B1 | 177.60 (11) | C6—C1—B1—O2 | −2.0 (2) |
| C3—C2—C7—O1 | −177.19 (11) | C6—C1—B1—O1 | 178.00 (12) |
| C4—C3—C2—C1 | 0.24 (18) | B1—O1—C7—C2 | −1.57 (13) |
| C4—C3—C2—C7 | 179.42 (11) | B1—C1—C6—C5 | −177.18 (12) |
| C4—C5—C6—C1 | 0.57 (18) | C7—O1—B1—O2 | −179.45 (10) |
| C2—C3—C4—F1 | −179.69 (9) | C7—O1—B1—C1 | 0.59 (13) |
| C2—C3—C4—C5 | 0.17 (19) | C7—C2—C1—C6 | −179.53 (10) |
| C2—C1—C6—C5 | −0.19 (17) | C7—C2—C1—B1 | −1.70 (13) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O2—H2···O1i | 0.83 (2) | 1.93 (2) | 2.7614 (13) | 175 (2) |
| C7—H7B···O2ii | 0.99 | 2.55 | 3.5325 (15) | 172 |
| C5—H5···F1iii | 0.95 | 2.58 | 3.4779 (14) | 157 |
| C7—H7A···O2iv | 0.99 | 2.66 | 3.2172 (14) | 116 |
| C3—H3···O2iv | 0.95 | 2.70 | 3.4276 (14) | 134 |
Symmetry codes: (i) −x+2, −y+2, −z+1; (ii) x, y−1, z; (iii) −x, −y+2, −z+2; (iv) x−1, y−1, z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: FJ2381).
References
- Adamczyk-Woźniak, A., Cyrański, M. K., Jakubczyk, M., Klimentowska, P., Koll, A., Kołodziejczak, J., Pojmaj, G., Żubrowska, A., Żukowska, Z. & Sporzyński, A. (2010). J. Phys. Chem. A, 114, 2324–2330. [DOI] [PubMed]
- Adamczyk-Woźniak, A., Cyrański, M. K., Żubrowska, A. & Sporzyński, A. (2009). J. Organomet. Chem. 694, 3533–3541.
- Baker, S. J., Hui, X. & Maibach, H. I. (2005). Annu. Rep. Med. Chem. 40, 323–335.
- Baker, S. J., Zhang, Y.-K., Akama, T., Lau, A., Zhou, H., Hernandez, V., Mao, W., Alley, M. R. K., Sanders, V. & Plattner, J. (2006). J. Med. Chem. 49, 4447–4450. [DOI] [PubMed]
- Burnett, M. N. & Johnson, C. K. (1996). ORTEPIII Report ORNL-6895. Oak Ridge National Laboratory, Tennessee, USA.
- Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. (2009). J. Appl. Cryst. 42, 339–341.
- Etter, M. C. (1990). Acc. Chem. Res. 23, 120–126.
- Gunasekera, D. S., Gerold, D. J., Aalderks, N. S., Chandra, J. S., Maanu, C. A., Kiprof, P., Zhdankin, V. V. & Reddy, M. V. R. (2007). Tetrahedron, 63, 9401–9405.
- Hui, X., Baker, S. J., Wester, R. C., Barbadillo, S., Cashmore, A. K., Sanders, V., Hold, K. M., Akama, T., Zhang, Y.-K., Plattner, J. J. & Maibach, H. I. (2007). J. Pharm. Sci. 96, 2622–2631. [DOI] [PubMed]
- International Tables for Crystallography (2006). Vol. E, Subperiodic Groups, edited by V. Kopsky & D. B. Litvin. Dordrecht: Kluwer Academic Publishers.
- Oxford Diffraction (2006). CrysAlis PRO Oxford Diffraction Ltd, Abingdon, Oxfordshire, England.
- Rock, F. L., Mao, W., Yaremchuk, A., Tukalo, M., Crepin, T., Zhou, H., Zhang, Y.-K., Hernandez, V., Akama, T., Baker, S. J., Plattner, J. J., Shapiro, L., Martinis, S. A., Benkovic, S. J., Cusack, S. & Alley, M. R. K. (2007). Science, 316, 1759–1761. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Tan, Y.-L., White, A. J. P., Widdowson, D. A., Wilhelm, R. & Williams, D. J. (2001). J. Chem. Soc. Perkin Trans. 1, pp. 3269–3280.
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
- Yamamoto, Y., Ishii, J., Nishiyama, H. & Itoh, K. (2005). J. Am. Chem. Soc. 127, 9625–9631. [DOI] [PubMed]
- Zhdankin, V. V., Persichini, P. J. III, Zhang, L., Fix, S. & Kiprof, P. (1999). Tetrahedron Lett. 40, 6705–6708.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks . DOI: 10.1107/S1600536811001632/fj2381sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536811001632/fj2381Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report