

Abstract
Marfan syndrome (MFS) is a dominant disorder with a recognizable phenotype. In most patients with the classical phenotype mutations are found in the fibrillin-1 gene (FBN1) on chromosome 15q21. It is thought that most mutations act in a dominant negative way or through haploinsufficiency. In 9 index cases referred for MFS we detected heterozygous missense mutations in FBN1 predicted to substitute the first aspartic acid of different calcium-binding Epidermal Growth Factor-like (cbEGF) fibrillin-1 domains. A similar mutation was found in homozygous state in 3 cases in a large consanguineous family. Heterozygous carriers of this mutation had no major skeletal, cardiovascular or ophthalmological features of MFS. In the literature 14 other heterozygous missense mutations are described leading to the substitution of the first aspartic acid of a cbEGF domain and resulting in a Marfan phenotype. Our data show that the phenotypic effect of aspartic acid substitutions in the first position of a cbEGF domain can range from asymptomatic to a severe neonatal phenotype. The recessive nature with reduced expression of FBN1 in one of the families suggests a threshold model combined with a mild functional defect of this specific mutation. © 2010 Wiley-Liss, Inc.
Keywords: Marfan syndrome, fibrillin-1, FBN1 gene, autosomal recessive inheritance, pathogenesis
INTRODUCTION
Human fibrillin-1 is a large protein of approximately 350 kD and member of a family of extracellular cysteine-rich glycoproteins. Since 1991 mutations in the fibrillin-1 (FBN1) gene have been found to be responsible for Marfan syndrome (MFS; MIM# 134797) (Kainulainen et al., 1990; Dietz et al., 1991). Fibrillin-1 is characterized by a highly conserved modular domain organization. The most prominent domain is the Epidermal Growth Factor-like (EGF) domain present 46 times and containing six highly conserved cysteine residues stabilizing the structure by three disulfide bonds. Of these EGF domains, 43 have a consensus sequence for calcium binding (cb) in the N-terminal pocket of the domain which may mediate protein-protein interactions. The EGF domains are interrupted by seven transforming growth factor (TGF)-binding protein domains characterized by 8 cysteine residues involved in intra-domain disulfide bonds (Pereira et al., 1993; Robinson et al., 2006). In the last update of the Universal Marfan Database - FBN1 (UMD-FBN; http://www.umd.be) (Faivre et al., 2007) 803 different mutations are reported. Most of the mutations are missense mutations (56%) mainly substituting or creating a cysteine in a cbEGF domain. Other mutations are frameshift mutations, splice mutations and nonsense mutations. About 14% of mutations are recurring.
All cbEGF domains start with a highly conserved aspartic acid, which is crucial for binding of a positively charged Ca2+ ion (Whiteman et al., 2007). We have identified 10 index cases with a substitution of the first aspartic acid substitution of a cbEGF domain and reviewed a further 14 published cases. Most of them exhibit a complete MFS phenotype. Surprisingly, in one family the substitution only led to MFS in homozygous state in three family members, whereas 13 family members carrying the heterozygous mutation do not have Marfan syndrome after thorough clinical examination.
There is still a lot of debate how mutations in FBN1 result in the MFS phenotype, but increasing evidence for different models is emerging. Possible explanations for the observed extreme variation in expression of the substitution of the first aspartic acid of a cbEGF domain are discussed. The observation of recessive inheritance of an expected dominant mutation also underscores the fact that mutations which are predicted to have a pathogenic effect, may not always lead to clinical symptoms.
PATIENTS AND METHODS
Patients
The patients were referred for DNA analysis of the fibrillin-1 gene to confirm the clinical diagnosis of MFS. Case 1, 2, 5, 7, 9 and 10 fulfilled the clinical Ghent criteria (De Paepe A. et al., 1996) for the diagnosis MFS. All index patients fulfilled the Ghent criteria when the finding of a pathogenic mutation in FBN1 was included.
Case 9 belongs to a large Turkish pedigree (Figure 1, III-1). She was examined at the age of 22 years. At the age of 6 weeks she was operated on a right sided hernia inguinalis. She was diagnosed with bilateral subluxation of the lenses when she was 3 years old. From that time on she has been operated several times for retinal detachments and lens luxation. At the age of 14 years an aortic root replacement was performed for progressive aortic root dilatation and aortic valve regurgitation. A spontaneous pneumothorax occurred at the age of 16 years.
Figure 1.
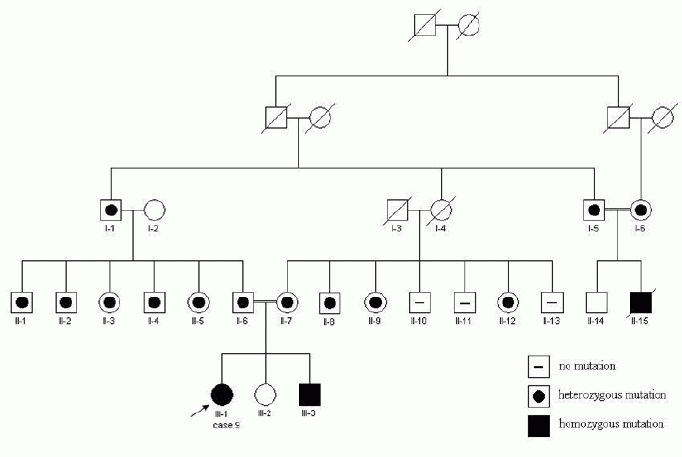
Pedigree of the family of case 9. Squares, male subjects; circles, female subjects. Affected subjects with a homozygous mutation (c.7454A>T) are represented by solid symbols. Presence or absence of the heterozygous mutation is represented by an open symbol with a black dot or a minus symbol respectively.
Clinical examination at the age of 18 years showed a marfanoid habitus, slight downslanting of palpebral fissures and a high and gothic palate. Despite long fingers, wrist and thumb signs were negative. She exhibited limited extension of her elbows, mild asymmetry of the chest, and bilateral flat feet. Her skin showed several striae on the chest, shoulders, hips and lower back. Her length was 179.5 cm (+3,7SD for Turkish descent) and an arm span of 175 cm (within normal limits). The brother of case 9 (III-3) was a 13 year old boy with Marfan syndrome. He had mild skeletal manifestations of Marfan (pes planus), a mildly dilated aortic root, ectopia lentis and dural ectasia with an anterior sacral meningocele. Furthermore he suffered from recurrent episodes of intracranial hypertension treated by drainage of cerebrospinal fluid. He has been described in a case report (Hilhorst-Hofstee et al., 2008).
The third patient (II-15) died at the age of 22 years. His case history was obtained from the medical records. At the age of 2 years bilateral subluxation of lenses was diagnosed. He developed severe aortic and mitral valve regurgitation with an aneurysm of the aortic root. He had skeletal involvement and an anterior sacral meningocele. When he was 17 years of age an aortic root replacement was performed with reconstruction of the aortic and mitral valve. Due to progressive aortic regurgitation, a re-operation was performed a year later. He died at the age of 22 after a second episode of ventricular fibrillation.
All heterozygous family members had a thorough skeletal, cardiologic and ophthalmologic examination including anthropometric measurements, echocardiography and slit lamp evaluation (Table 1). Only the mother and father of case 9 had an MRI evaluation for dural ectasia. The obtained clinical data of all family members are summarized in Table 1. The father of case 9 (II-6) had no clinical signs of Marfan syndrome. The mother (II-7) was tall, with a height on +2.5 SD but with normal body proportions. She had no other skeletal, ocular or cardiovascular involvement, but had several striae on the lumbar region and around the knees. Furthermore she suffered from spontaneous pneumothorax at the age of 21 years. An MRI-scan showed a dural ectasia at S2 with otherwise a normal dural sac. The father and mother of II-15 did not exhibit any signs of Marfan syndrome. None of the nine other heterozygous family members had a major criterion in one of the organ systems. Some were found to have a non-specific or minor sign. Individual II-1 has an arm span to height ratio of 1.06 and a mild dilatation of the abdominal aorta. II-4 had recurrent inguinal hernias but this was during a period of performing heavy physical labor. II-8 had an arm span to height ratio of 1.07 and bilateral flat feet. II-9 had reduced extension of the elbows.
Table 1.
Summary of the clinical features in the family of case 9
| III-1 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case 9 | III-3 | II-6 | II-7 | II-15 | I-5 | I-6 | I-1 | II-1 | II-2 | II-3 | II-4 | II-5 | II-8 | II-9 | II-12 | |
| Age at examination (years) | 10 | 22 | 42 | 43 | 9 | 57 | 56 | 61 | 44 | 41 | 37 | 35 | 33 | 55 | 48 | 37 |
| Sex | M | F | M | F | M | M | F | M | M | M | F | M | F | M | F | F |
| height (cm) | 150,5 | 180,9 | 167,1 | 174,4 | 135,0 | 164,0 | 157,5 | 166,0 | 167,5 | 174,5 | 167,0 | 188,5 | 161,0 | 181,0 | 164,7 | 161,0 |
| height SDS (for Turkish descent) | 1,6 | 3,7 | -1,2 | 2,5 | -0,2 | -1,7 | -0,6 | -1,4 | -1,1 | 0,1 | 1,1 | 2,5 | 0,8 | 1,2 | 0,7 | 0,2 |
| arm span : height ratio | 1,02 | 0,97 | 1,04 | 1,00 | 1,00 | 1,01 | 1,03 | 1,01 | 1,06 | 1,04 | 0,98 | 0,99 | 1,02 | 1,07 | 1,02 | 1,03 |
| sitting height: height ratio | 0,488 | 0,515 | 0,536 | 0,513 | np | 0,535 | 0,528 | 0,519 | 0,527 | 0,517 | 0,531 | 0,516 | 0,523 | 0,482 | 0,520 | 0,526 |
| sitting height: height ratio SDS | -2,0 | -0,8 | 1,6 | -0,8 | np | 1,5 | 0,2 | 0,3 | 1,0 | 0,3 | 0,3 | 0,2 | 0,0 | -1,8 | -0,4 | 0,0 |
| Skeletal system | involv | involv | none | none | involv | none | none | none | none | none | none | none | none | involv | none | none |
| major | ||||||||||||||||
| pectus carinatum | no | no | no | no | yes | no | no | no | no | no | no | no | no | no | no | no |
| pectus excavatum requiring | no | no | no | no | no | no | no | no | no | no | no | no | no | no | no | no |
| surgery | ||||||||||||||||
| sitting height: height ratio <2 | ||||||||||||||||
| SD or armspan : height ratio | yes | no | no | no | np | no | no | no | yes | no | no | no | no | yes | no | no |
| >1.05 | ||||||||||||||||
| wrist and thumbsigns | no | no | no | no | no | no | no | no | no | no | no | no | no | no | no | no |
| scoliosis of >20° or | ||||||||||||||||
| spondylolisthesis | no | no | no | no | np | no | no | no | no | no | no | no | no | no | no | no |
| reduced extension at the elbow | ||||||||||||||||
| (<170°) | no | yes | no | yes | np | no | no | no | no | no | no | no | no | no | yes | no |
| pes planus | yes | yes | no | no | yes | no | no | no | no | no | no | yes | no | yes | no | no |
| protrusio acetabulae | np | np | np | np | np | np | np | np | np | np | np | np | np | np | np | np |
| minor | ||||||||||||||||
| pectus excavatum of moderate severity | no | no | no | no | yes | no | no | no | no | no | no | no | no | no | no | no |
| joint hypermobility | no | no | no | no | np | no | no | no | no | no | no | no | no | no | no | no |
| highly arched palate with crowding | no | yes | no | no | yes | no | no | no | no | no | no | no | no | no | no | no |
| facial appearance | no | yes | no | no | yes | no | no | no | no | no | no | no | no | no | no | no |
| Ocular system | major | major | none | none | major | none | none | none | none | none | none | none | none | none | none | none |
| major | ||||||||||||||||
| ectopia lentis | yes | yes | no | no | yes | no | no | no | no | no | no | no | no | no | no | no |
| minor | ||||||||||||||||
| abnormally flat cornea | np | np | np | np | np | no | no | no | no | no | no | no | no | no | no | no |
| increased axial length of globe | np | np | np | np | np | np | np | np | np | np | np | np | no | no | no | no |
| hypoplastic iris or ciliary muscle | np | np | no | no | yes | no | no | no | no | no | no | no | no | no | no | no |
| Cardiovascular system | major | major | none | none | major | none | none | none | involv | none | none | none | none | none | none | none |
| major | ||||||||||||||||
| Z-score aortic root diameter | 10,51 | 4 | 0.3 | -1.4 | >22 | -0.9 | -0.8 | -1.8 | 1.4 | -1.1 | -0.8 | -2.9 | -2.5 | 0.4 | 0.5 | 0.8 |
| dilatation ascending aorta | yes (arr) | yes | no | no | yes (arr) | no | no | no | no | no | no | no | no | no | no | no |
| dissection of ascending aorta | no | no | no | no | no | no | no | no | no | no | no | no | no | no | no | no |
| minor | ||||||||||||||||
| mitral valve prolaps | yes | yes | no | no | yes | no | no | no | no | no | no | no | no | no | no | no |
| dilatation of main pulmonary artery | no | no | no | no | np | no | no | no | no | no | no | no | no | no | no | no |
| calcification of the mitral annulus < 40 years | no | no | no | no | np | no | no | np | no | no | no | no | no | no | no | no |
| dilatation or dissection of descending aorta < 50 years | no | no | np | no | np | no | no | no | yes | no | no | no | no | no | no | no |
| Pulmonary system | involv | none | none | involv | none | none | none | none | none | none | none | none | none | none | none | none |
| minor | ||||||||||||||||
| spontanous pneumothorax or apical blebs | yes | no | no | yes | no | no | no | no | no | no | no | no | no | no | no | no |
| Skin and integument | involv | none | none | involv | np | none | none | none | none | none | involv | involv | none | none | none | none |
| minor | ||||||||||||||||
| striae atrophicae recurrent or incisional herniae | yes no | no no | no no | yes no | np np | no no | no no | no no | no no | no no | yes no | no yes3 | no no | no no | no no | no no |
| Dura | np | major | none | major | np | np | np | np | np | np | np | np | np | np | np | np |
| major | ||||||||||||||||
| lumbosacral dural ectasia | np | yes | no | yes | np | np | np | np | np | np | np | np | np | np | np | np |
| Family or genetic history | major4 | major4 | major4 | major4 | major4 | major4 | major4 | major4 | major4 | major4 | major4 | major4 | major4 | major4 | major4 | major |
| major | ||||||||||||||||
| 1st degree relative with Marfan syndrome pathogenic mutation in FBN1 | no hom | yes hom | yes het | yes het | no hom | yes het | yes het | no het | no het | no het | no het | no het | no het | no het | no het | no het |
F female; M male; involve involvement; np not performed; arr aortic root replacement; homh mutation; Z-score related to body surface area and age according to Roman et al. (Roman et al., 1989); 1) aortic root measurement at the age of 14 years just before aortic root replacement; replacement; 2) no exact measurement available; 3) recurrent inguinal hernias during a period member or the presence of a pathogenic FBN1 mutation.
The clinical data of cases 1-10 and published cases are summarized in Table 2 together with the molecular data.
Table 2.
Published and observed missense mutations leading to the substitution of the first aspartic acid of a cbEGF domain
| Nucleotide change | Amino acid change | Exon | cbEGF domain | Diagnosis | Phenotype | Reference | Aberant mRNA splicing | Predicted aberant mRNA splicinga | Inheritance |
|---|---|---|---|---|---|---|---|---|---|
| C.1468OT | p.Asp490Tyr | 11 | #3 | Classical MFS | ard, el, sk | (Hayward and Brock, 1997) | np | yes | Unknown |
| c.2168A>C | p.Asp723Ala | 18 | #7 | Severe classical MFS | ard, mvp, el, myop, sk | (Dietz etal., 1993) | np | no | De novo |
| c.2168A>T | p.Asp723Val | 18 | #7 | Classical MFS | ard, mvp, el, sk, myop | (Katzke etal., 2002) | np | no | De novo |
| c.2728G>A | p.Asp910Asn | 22 | #10 | Classical MFS | unknown | UMD | np | yes | Unknown |
| c.3209A>G | p.Asp1070Gly | 26 | #12 | Neonatal MFS | unknown | UMD | np | no | Unknown |
| c.3338A>G | p.Asp1113Gly | 27 | #13 | Phenotype unknown | unknown | (Liu etal., 1997) | np | no | Unknown |
| c.3463G>A | p.Asp1155Asn | 27 | #14 | Thoracic aortic aneurysm | ard, mvp, diss | (Milewicz et al., 1996) | no | yes | De novo |
| c.3464A>G | p.Asp1155Gly | 28 | #14 | Classical MFS | ard, el | Case 1 | no | no | Parents not available, 5 sibs neg for mutation |
| c.3712G>A | p.Asp1238Asn | 29 | #16 | Phenotype unknown | unknown | (Yuan etal., 1999) | np | no | Unknown |
| c.3713A>G | p.Asp1238Gly | 30 | #16 | Classical MFS | ard, mvp, sk | (Tiecke etal., 2001) | np | no | Familial |
| c.3964G>A | p.Asp1322Asn | 31 | #18 | Neonatal MFS | ard, mi, ti, myop, sk | Case 2 | np | yes | De novo |
| c.4210G>T | p.Asp1 404Tyr | 33 | #20 | Classical MFS | ard, el, sk | (Hayward et al., 1997) | yes | yes | Familial |
| c.5422G>C | p.Asp1808His | 43 | #26 | Lens luxation and striae | el, str | Case 3 | np | no | Parents not available |
| c.5671G>A | p.Asp1891His | 45 | #28 | Classical MFS | ard, sk | Case 4 | np | no | De novo |
| c.5788G>C | p.Asp1930His | 45 | #29 | Classical MFS | ard, el, sk | Case 5 | np | yes | De novo |
| c.5788G>A | p.Asp1930Asn | 46 | #29 | Phenotype unknown | unknown | (Liu etal., 1997) | np | yes | Unknown |
| c.5788G>A | p.Asp1930Asn | 46 | #29 | Classical MFS | ard, el, sk, de, str | Case 6 | np | yes | Parents not available |
| c.6037G>T | p.Asp2013 Tyr | 48 | #31 | Classical MFS | ard, el, sk, de, str | Case 7 | np | yes | Familial |
| c.6379G>T | p.Asp2127Tyr | 51 | #32 | Classical MFS | ard, el | (Matsukawa et al., 2001) | np | yes | Familial |
| c.6381T>A | p.Asp2127Glu | 52 | #32 | Classical MFS | ard, sk | (Kainulainen etal., 1994) | np | no | Familial |
| c.7331A>G | p.Asp2444Gly | 59 | #38 | Classical MFS | ard, sk | Case 8 | np | no | De novo |
| c.7454A>T | p.Asp2485Val | 60 | #39 | Classical MFS in homozygous state | ard, el, sk, str, her, pn | Case 9 | no | Familial | |
| c.7819G>A | p.Asp2607Asn | 62 | #42 | Classical MFS | ard, mvp, sk | Case 10 | no | no | Mother suspect for MFS |
| c.7820A>G | p.Asp2607Gly | 63 | #42 | Phenotype unknown | unkown | (Liu etal., 1997) | np | no | Unknown |
UMD Universal Marfan Database - FBN1 (UMD-FBN; http://www.umd.be); cbEGF calcium binding Epidermal Growth Factor domain; bp basepair; np not performed; neg negative; pos positive; ard aortic root dilatation; diss aortic dissection; mvp mitral valve prolapse, mi mitral valve insufficiency; el ectopia lentis; pal high arched palate; ti tricuspid valve insufficiency; myop high myopia; ar arachnodactyly; hm hypermobility; contr contractures; str striae; her hernia; sk skeletal involvement; de dural ectasia; pn pneumothorax; unknown. The gray row represents the recessive mutation described in this article.
Mutation numbering refers to the FBN1 cDNA GenBank reference sequence: NM_000138.3, with the A of the ATG translation initiation codon as nucleotide +1 (http://www.hgvs.org/mutnomen).a) Alamut mutation interpretation software version 1.5; Interactive Biosoftware, Rouen France.
Molecular studies
DNA was extracted from peripheral blood or paraffin embedded tissue, using standard techniques, analyzed by DHPLC and direct DNA sequencing as described previously (Matyas et al., 2002).
The reference sequence used to describe the mutations is the FBN1 cDNA GenBank reference sequence: NM_000138.3. Nucleotide numbering reflects cDNA numbering with +1 corresponding to the A of the ATG translation initiation codon in the reference sequence, according to journal guidelines (http://www.hgvs.org/mutnomen).
Skin biopsies of case 1, 9 and her mother and case 10 were available. Fibroblasts were cultured and mRNA was isolated from confluent monolayers. For each individual two RNA isolations were performed: one cell culture of each was incubated with cycloheximide (0.25 mg/ml) for 4.5 hours prior to RNA isolation, to prevent possible nonsense mediated decay (NMD) of aberrantly spliced mRNA.
RNA was isolated using the RNA isolation minikit (Qiagen) according to the manufacturers instructions. Full length single stranded cDNA was prepared with oligo-dT-primer and SuperscriptTMII RT reverse trancriptase (Invitrogen). To detect possible splice errors, the complete coding sequence of FBN1 was analyzed by direct sequencing in 24 overlapping PCR fragments. The primers for overlapping fragments were positioned in different exons, to avoid allele dropout in case of exon skipping. For analysis of the mutation in exon 60, primers in exon 55 (forward) and 63 (reverse) were used.
Primer sequences are given in Supp. Table S1.
RESULTS
Heterozygous mutations leading to a substitution of the first aspartic acid of a cbEGF domain were found in nine index cases and a homozygous mutation was found in 1 index case. These mutations and 14 comparable mutations described in the literature or in the UMD-FBN1 (UMD-FBN; http://www.umd.be) are listed in Table 2. The phenotypes were classical MFS in 15 cases, neonatal MFS in two cases, thoracic aortic aneurysm in one case and lens luxation with striae in one case. In four cases described in the literature the phenotype is not clear.
The mutation was de novo in cases 2, 4, 5, and 8. In case 1, 3 and 6 parental DNA was not available. In case 1 five sibs were tested negative for the mutation. In case 6 the parents and seven sibs had no clinical symptoms of MFS. Case 7 has an affected sister with the same mutation. Their father died as a consequence of his third aortic dissection when he was 52 years of age. He was thought to have MFS. The mother of case 10 is probably affected but was not molecularly tested. Of the published mutations three were de novo, three were familial and in eight inheritance was unknown (Table 2).
Using DHPLC for mutation scanning of all 65 exons of FBN1, initially no mutation was found in case 9. As the parents are consanguineous, a recessive mutation was suspected. Testing one of the parents, a heterozygous mutation was detected in exon 60: c.7454A>T, leading to the amino acid substitution p.Asp2485Val. This mutation was found in a homozygous state in the patients III-1, III-3 and II-15. The parents (II-6 and II-7) of patients III-1 and III-3 are first cousins. The parents of patient II-15 are distantly related and both are related to II-6 and II-7 (Figure 1). In the parents of patients III-1, III-3 and II-15 and 9 family members the mutation was heterozygous. The mutation was not detected in 1000 Caucasian and 60 Turkish control chromosomes.
Sequence analysis of cDNA, made from mRNA from cultured fibroblasts of case 1, case 9 and her mother, and case 10 showed no evidence of erroneous splicing of exon 60. Treatment with cycloheximide, to prevent possible nonsense mediated decay of erroneously spliced mRNA, gave the same results. Fibrobasts of the other patients were not available.
DISCUSSION
We identified a heterozygous substitution of the first aspartic acid of a cbEGF domain in FBN1 in nine index patients and a homozygous substitution in one index patient with MFS. Reviewing the literature we found 12 reports of substitution of the first aspartic acid, and a further two unpublished cases are quoted in the UMD database (UMD-FBN; http://www.umd.be) (Collod-Beroud et al., 2003) as summarized in Table 2. All 10 index patients found in our center fulfilled the Ghent criteria when the finding of a pathogenic mutation was included. Of the 14 published cases, eight were reported to have a classical Marfan phenotype, one was a neonatal Marfan and one had a thoracic aortic aneurysm. Of four cases the phenotype was not reported. In all cases the acidic amino acid aspartic acid is replaced by a nonpolar or noncharged polar amino acid, apart from one mutation where aspartic acid is replaced by another acidic amino acid (p.Asp2127Glu) (Kainulainen et al., 1994). The codons of the first aspartic acids in the cbEGF domains always contain the last base of one exon and the first two bases of the next. Consequently, mutations of these codons may affect splicing. Aberrant splicing was excluded in cases 1, 9, and 10 and in one of the published cases (Milewicz et al., 1996). The mutation c.4210G>T was shown to destroy a donor splice site with abnormal splicing as a consequence (Hayward et al., 1997). Of the 19 cases in which no cDNA analysis was performed, prediction software predicted aberrant splicing in eight cases (Table 2). Exon skipping, as a result of these mutations, may have more severe effects than missense mutations, because the exons are all in frame and consequently, skipping will lead to a shorter protein that may exert a dominant negative effect (Robinson etal., 2002).
There are several reasons to argue that a substitution of the first aspartic acid of a cbEGF domain will lead to a MFS phenotype in the heterozygous state. Calcium binding of cbEGF domains is necessary for stabilization of the secondary structure, prevention of proteolytic degradation and for protein-protein interaction (Dietz et al., 1993; Handford et al., 1991; Cooke et al., 1987). The first aspartic acid of a cbEGF domain is highly conserved in evolution and in the human fibrillin-1 gene all cbEGF domains start with an aspartic acid, which is crucial for binding of a positively charged Ca2+ ion (Figure 2) (Whiteman et al., 2007). Furthermore mutations of the first amino acid of a cbEGF domain of coagulation factor IX in haemophilia B have been proven to reduce calcium binding even if the aspartic acid is replaced by the acidic amino acid glutamic acid (Handford et al., 1991; Winship and Dragon, 1991). The finding of 23 MFS or MFS-like cases with a heterozygous substitution of an aspartic acid in this position of the cbEGF domain underscores the crucial role of this amino acid. In this view the recessive nature of the mutation p.Asp2485Val in the family of case 9 came as a surprise. The family of case 9 (Figure 1) has been thoroughly investigated. Patients III-1 (case 9), III-3 and II-15 have the classical type of Marfan syndrome according to the Ghent criteria (De Paepe A. et al., 1996). Based on the pedigree with three affected patients and healthy consanguineous parents recessive inheritance could be expected. This was confirmed by finding a homozygous missense mutation in all three affected patients. The four unaffected parents and nine other unaffected relatives were found to be carriers of the mutation. Unexpectedly in none of the investigated heterozygous carriers obvious signs of Marfan syndrome could be found. Only after thorough clinical examination one of them (II-7) was found to have a dural ectasia at S2, which as yet is considered a major symptom in the Ghent criteria. Together with the family history and some minor signs (pneumothorax, striae and reduced extension of the elbows) this classifies II-7 as having Marfan syndrome. However, compared to the homozygous cases, the cardinal Marfan features in the skeletal, cardiac and ophthalmological systems are absent.
Figure 2.
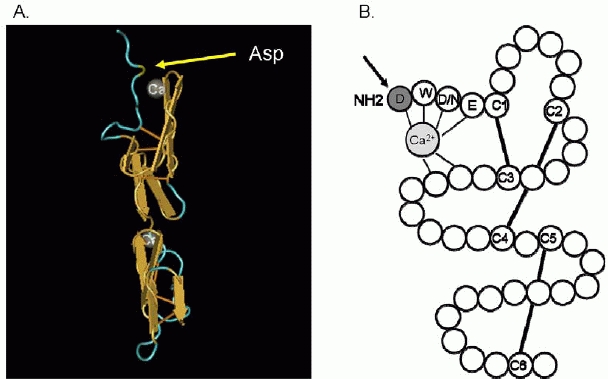
Class I cbEGF domain showing the position of the first Asp in relation to the calcium molecule. (A) 3D picture of a cbEGF domain of fibrillin-1. The yellow arrow points to the first Asp. Picture derived from the NCBI database (http://www.ncbi.nlm.nih.gov/) (Downing et al., 1996). (B) cbEGD like domain (Handford et al., 1991). The arrow points to the first aspartic acid residue. Solid lines are the disulphide bridges between cysteine residues.
To our knowledge very little is known about recessive FBN1 mutations. Only one family has been reported in which Marfan syndrome was found in two affected cousins homozygous for a FBN1 mutation while the four normal parents were heterozygous carriers, indicating recessive inheritance of the syndrome (De Vries et al., 2007). The mutation is located in exon 11 and leads to an amino-acid substitution creating a cysteine. Like us the authors expected this mutation to have a dominant effect in the heterozygous state. Two of the parents were sibs and exhibited minor signs of Marfan syndrome (increased arm span to height ratio and a highly arched palate). No other family members have been investigated. Only one other probably recessively inherited form of Marfan syndrome has been described but could not be proven by molecular analysis as the gene was not yet known (Fried and Krakowsky, 1977).
Three families have been described in the literature in which both parents are affected with more severely affected children. The first is an Italian couple of first cousins, both affected with Marfan syndrome, who had 4 affected children. Two of the four affected children showed more severe manifestations than other affected family members, presumably due to homozygosity (Capotorti L. et al., 1959). In 1984, Chemke described a family with Marfan syndrome. Two sibs suffered from a severe phenotype reminiscent of neonatal Marfan syndrome. Their parents were cousins and had a much milder phenotype. Remarkable is that the probably homozygous sibs were the only patients in the family with ectopia lentis (Chemke et al., 1984). In 1988 a severely affected boy has been described (Schollin et al., 1988). Both parents were affected and were found to carry amissense mutation in FBN1. Compound heterozygosity was identified in the severely affected child (Karttunen et al., 1994).
In the recessive family described here the heterozygous mutation does not exert an important effect on the phenotype. Only in the homozygous state the abnormal fibrillin causes the classical clinical phenotype of Marfan syndrome. This observation together with the few other described families with bi-allelic inheritance, may support both alternative pathogenetic models of Marfan syndrome. A dominant negative effect of FBN1 mutations has been the leading hypothesis for the pathogenesis of Marfan syndrome for a long time. However, several manifestations of Marfan syndrome like bone overgrowth, craniofacial features, lung disease, and muscle and fat hypoplasia could not be explained by a structurally abnormal protein. The observation that fibrillin interacts with a variety of proteins, including the latent TGFβ binding proteins (LTBP's) has lead to several investigations indicating that fibrillin-1 can interact with TGFβ signaling (Annes et al., 2003; Azhar et al., 2003; Isogai et al., 2003; Hubmacher et al., 2006; Kaartinen and Warburton, 2003; Loeys et al., 2006; Neptune et al., 2003; Ng et al., 2004; Ten Dijke and Arthur, 2007).
According to splice site prediction software (Alamut mutation interpretation software version 1.5; Interactive Biosoftware, Rouen, France) the c.7454A>T mutation, found in the family described here, is not predicted to cause erroneous splicing. This was confirmed by cDNA studies, showing no evidence of splice error. The position of the mutation (exon 60) may explain the lack of expression as mutations in the more C-terminal end of the gene are expected to give a milder phenotype (Faivre et al., 2007; Robinson et al., 2002). However the mutation in case 10 is even more terminal and still leads to a classical MFS phenotype.
Hutchinson et al. (Hutchinson et al., 2003) hypothesized that variable expression of the normal FBN1 allele could moderate the phenotypic effect of the mutant allele. A compensatory higher level of FBN1 expression from the normal allele would explain a milder phenotype. As the normal alleles are inherited from 5 different parents in our recessive family, this mechanism is highly unlikely.
We hypothesize that the p.Asp2485Val mutation acts as a hypomorphic allele with a minimal dominant negative effect. Reduction of gene expression of both alleles could be the main determinant of the phenotype in homozygotes. The observation of only one major clinical sign in one of the heterozygotes (dural ectasia in II-7, Figure 1) and no major clinical signs in 12 other heterozygotes could be explained by sufficient gene expression with only a mild functional defect of the mutant allele product. This was also shown in a mouse model, however in this model the mutation had a severe dominant negative effect (Pereira et al., 1997). In this model with a deletion of 272 amino acids in the central part of fibrillin-1, a tenfold reduction in expression of the mutant allele was shown in heterozygous mice, resulting in a normal phenotype. Homozygous mutant mice however died shortly after birth due to severe vascular complications. The other mouse model of Pereira was a targeted FBN1 mutation leading to 15% expression of a normal product with no abnormal phenotype in heterozygous mice, while mice homozygous for this mutation have severe abnormalities comparable with the neonatal MFS phenotype. These mouse models suggests that there is a threshold of expression of the normal allele below which the abnormal phenotype will develop (Pereira et al., 1999; Dietz and Mecham, 2000).
To understand the exact pathogenetic mechanism expression studies and studies on protein synthesis, secretion and matrix incorporation of the Asp2486Val mutation are necessary For comprehensive studies of this type, a mouse model should be created.
The finding of a homozygous substitution of A>T, as described here, has implications for mutation screening in MFS. Homozygous substitutions will not have an effect on denaturation of double stranded DNA, because the basepair remains the same. Consequently, heteroduplex based testing, such as DHPLC, working with the principle of differential denaturation of double stranded heteroduplex DNA, cannot detect this mutation in homozygous state. Formerly, based on the presumed dominant inheritance mode, only heterozygous mutations were expected. Now it is clear that recessive inheritance is also possible and mutations may have been missed in similar cases, because heteroduplex based testing has been used until recently in many large diagnostic centers. Most laboratories nowadays use direct sequencing, which avoids this problem.
In conclusion we have shown that the first aspartic acid of a cbEGF domain in FBN1 is important for the function of fibrillin-1, but may not always lead to a clinical effect in the heterozygous state. This underscores that missense mutations must be interpreted with care.
Acknowledgments
We thank the patients and their families for their very kind cooperation, Prof. Dr. E. Bakker for his helpful suggestions and Jacqueline Egthuijsen for technical assistance.
Supplementary material
Supp. Table S1.
Primers for cDNA sequencing of FBN1: F=forward; R=reverse; numbers refer to position in cDNA sequence
| fragment | primername | Sequence 5’ > 3’ | Length bp |
|---|---|---|---|
| 1 | FBN1F1 | ATGCGTCGAGGGCGTCTGCT | 384 |
| FBN1R384 | GCTACCTCCATTCATACAGCGA | ||
| 2 | FBN1F318 | GATAGCTCCTTCCTGTGGCTCC | 406 |
| FBN1R728 | CCGTGCGGATATTTGGAATG | ||
| 3 | FBN1F657 | CCCCTGTGAGATGTGTCCTG | 407 |
| FBN1R1064 | TTGGTTATGGACTGTGGCAGC | ||
| 4 | FBN1F1012 | ACAGCTCTGACAAACGGGCG | 384 |
| FBN1R1396 | TGCAGCGTCCATTTTGACAG | ||
| 5 | FBN1F1358 | GCCAGTTGGTCCGCTATCTC | 330 |
| FBN1R1688 | ACATGAAAGCCCGCATTACAC | ||
| 6 | FBN1F1609 | AATGGCCGGATCTGCAATAA | 398 |
| FBN1R2007 | CTGGCCTCTCTTGTATCCACCA | ||
| 7 | FBN1F1927 | CTGGCTGTGGGTCTGGATGG | 362 |
| FBN1R2289 | GCAGTTTTTCCCAGTTGAATCC | ||
| 8 | FBN1F2212 | ATCTGTGAAAACCTTCGTGGGA | 399 |
| FBN1R2611 | AGGTGGCTCCATTGATGTTGA | ||
| 9 | FBN1F2458 | GTCTGCAAGAACAGCCCAGG | 357 |
| FBN1R2815 | TGGGACACTGACACTTGAATGA | ||
| 10 | FBN1F2699 | TACTCAAGAATTAAAGGAACA | 525 |
| FBN1R3224 | CGGCATTCGTCAATGTCTGTGC | ||
| 11 | FBN1F3141 | CATTGGCAGCTTTAAGTGCAGG | 460 |
| FBN1R3621 | ACCACCATTCATTATGCTGCA | ||
| 12 | FBN1F3558 | CCATTCAACTCCCGATAGGCT | 335 |
| FBN1R3893 | TTTTCACAGGTCCCACTTAGGC | ||
| 13 | FBN1F3783 | CAGGTGCTTGTGTTATGATG | 392 |
| FBN1R4173 | GCACAGACAGCGGTAAGA | ||
| 14 | FBN1F4062 | GATTGGAGATGGCATTAAGTGC | 451 |
| FBN1R4513 | TGTTGACACAGTTCCCACTGA | ||
| 15 | FBN1F4425 | CTACGAACTGGACAGAAGCGG | 593 |
| FBN1R5018 | ATACAGGTGTAGTTGCCAACGG | ||
| 16 | FBN1F4910 | ACTACCTGAATGAAGATACACG | 626 |
| FBN1R5536 | GACCTGTGGAGGTGAAGCGGTAG | ||
| 17 | FBN1F5348 | TCAACATGGTTGGCAGCTTCC | 476 |
| FBN1R5824 | AAAGATTCCCATTTCCACTTGC | ||
| 18 | FBN1F5722 | ACAATTGGTTCCTTCAACTG | 356 |
| FBN1R6074 | GCACAAATTTCTGGCTCTT | ||
| 19 | FBN1F5973 | CTTGGATGGGTCCTACAGATGC | 579 |
| FBN1R6552 | CACATTCTTGCAGGTTCCATT | ||
| 20 | FBN1F6466 | GGTTATACTCTAGCGGGAATG | 450 |
| FBN1R6937 | TCCCACGGGTGTTGAGGCAGCG | ||
| 21 | FBN1F6842 | AGCGGAGACCTGATGGAGAGG | 645 |
| FBN1R7487 | CAGTTGTGTTGCTTGGTTGCA | ||
| 22 | FBN1F7429 | CAAGAGGATGGAAGGAGCTGC | 476 |
| FBN1R7905 | GAACTGTTCATACTGGAAGCCG | ||
| 23 | FBN1F7785 | CTACCTCCAGCACTACCAGTGG | 384 |
| FBN1R8169 | GTAGCCATTGATCTTACACTCG | ||
| 24 | FBN1F8024 | CACCTGGTTACTTCCGCATAGG | 672 |
| FBN1R8696 | ATGATTCTGATTGGGGGAAAA |
REFERENCES
- Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- Azhar M, Schultz JJ, Grupp I, Dorn GW, Meneton P, Molin DG, Gittenberger-de Groot AC, Doetschman T. Transforming growth factor beta in cardiovascular development and function. Cytokine Growth Factor Rev. 2003;14:391–407. doi: 10.1016/s1359-6101(03)00044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capotorti L, de Benedetti Gaddini R, Rizzo P. Contribution to the study of the heredity of Marfan's syndrome: description of a family tree of 4 generations with marriage between consanguineous parents. Acta Genet Med Gemellol. 1959;8:455–482. [Google Scholar]
- Chemke J, Nisani R, Feigl A, Garty R, Cooper M, Barash Y, Duksin D. Homozygosity for autosomal dominant Marfan syndrome. J Med Genet. 1984;21:173–177. doi: 10.1136/jmg.21.3.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collod-Beroud G, Le Bourdelles S, Ades L, Ala-Kokko L, Booms P, Boxer M, Child A, Comeglio P, De Paepe A, Hyland JC, Holman K, Kaitila I, Loeys B, Matyas G, Nuytinck L, Peltonen L, Rantamaki T, Robinson P, Steinmann B, Junien C, Beroud C, Boileau C. Update of the UMD-FBN1 mutation database and creation of an FBN1 polymorphism database. Hum Mutat. 2003;22:199–208. doi: 10.1002/humu.10249. [DOI] [PubMed] [Google Scholar]
- Cooke RM, Wilkinson AJ, Baron M, Pastore A, Tappin MJ, Campbell ID, Gregory H, Sheard B. The solution structure of human epidermal growth factor. Nature. 1987;327:339–341. doi: 10.1038/327339a0. [DOI] [PubMed] [Google Scholar]
- De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet. 1996;62:417–426. doi: 10.1002/(SICI)1096-8628(19960424)62:4<417::AID-AJMG15>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- De Vries BB, Pals G, Odink R, Hamel BC. Homozygosity for a FBN1 missense mutation: clinical and molecular evidence for recessive Marfan syndrome. Eur J Hum Genet. 2007;15:930–935. doi: 10.1038/sj.ejhg.5201865. [DOI] [PubMed] [Google Scholar]
- Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- Dietz HC, McIntosh I, Sakai LY, Corson GM, Chalberg SC, Pyeritz RE, Francomano CA. Four novel FBN1 mutations: significance for mutant transcript level and EGF-like domain calcium binding in the pathogenesis of Marfan syndrome. Genomics. 1993;17:468–475. doi: 10.1006/geno.1993.1349. [DOI] [PubMed] [Google Scholar]
- Dietz HC, Mecham RP. Mouse models of genetic diseases resulting from mutations in elastic fiber proteins. Matrix Biol. 2000;19:481–488. doi: 10.1016/s0945-053x(00)00101-3. [DOI] [PubMed] [Google Scholar]
- Downing AK, Knott V, Werner JM, Cardy CM, Campbell ID, Handford PA. Solution structure of a pair of calcium-binding epidermal growth factor-like domains: implications for the Marfan syndrome and other genetic disorders. Cell. 1996;85:597–605. doi: 10.1016/s0092-8674(00)81259-3. [DOI] [PubMed] [Google Scholar]
- Faivre L, Collod-Beroud G, Loeys BL, Child A, Binquet C, Gautier E, Callewaert B, Arbustini E, Mayer K, Arslan-Kirchner M, Kiotsekoglou A, Comeglio P, Marziliano N, Dietz HC, Halliday D, Beroud C, Bonithon-Kopp C, Claustres M, Muti C, Plauchu H, Robinson PN, Ades LC, Biggin A, Benetts B, Brett M, Holman KJ, De Backer J, Coucke P, Francke U, De Paepe A, Jondeau G, Boileau C. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007;81:454–466. doi: 10.1086/520125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried K, Krakowsky D. Probable autosomal recessive Marfan syndrome. J Med Genet. 1977;14:359–361. doi: 10.1136/jmg.14.5.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handford PA, May hew M, Baron M, Winship PR, Campbell ID, Brownlee GG. Key residues involved in calcium-binding motifs in EGF-like domains. Nature. 1991;351:164–167. doi: 10.1038/351164a0. [DOI] [PubMed] [Google Scholar]
- Hayward C, Porteous ME, Brock DJ. Mutation screening of all 65 exons of the fibrillin-1 gene in 60 patients with Marfan syndrome: report of 12 novel mutations. Hum Mutat. 1997;10:280–289. doi: 10.1002/(SICI)1098-1004(1997)10:4<280::AID-HUMU3>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Hilhorst-Hofstee Y, Kroft LJ, Pals G, Van Vugt JP, Overweg-Plandsoen WC. Intracranial hypertension in 2 children with marfan syndrome. J Child Neurol. 2008;23:954–955. doi: 10.1177/0883073808315341. [DOI] [PubMed] [Google Scholar]
- Hubmacher D, Tiedemann K, Reinhardt DP. Fibrillins: from biogenesis of microfibrils to signaling functions. Curr Top Dev Biol. 2006;75:93–123. doi: 10.1016/S0070-2153(06)75004-9. [DOI] [PubMed] [Google Scholar]
- Hutchinson S, Furger A, Halliday D, Judge DP, Jefferson A, Dietz HC, Firth H, Handford PA. Allelic variation in normal human FBN1 expression in a family with Marfan syndrome: a potential modifier of phenotype? Hum Mol Genet. 2003;12:2269–2276. doi: 10.1093/hmg/ddg241. [DOI] [PubMed] [Google Scholar]
- Isogai Z, Ono RN, Ushiro S, Keene DR, Chen Y, Mazzieri R, Charbonneau NL, Reinhardt DP, Rifkin DB, Sakai LY. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J Biol Chem. 2003;278:2750–2757. doi: 10.1074/jbc.M209256200. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Warburton D. Fibrillin controls TGF-beta activation. Nat Genet. 2003;33:331–332. doi: 10.1038/ng0303-331. [DOI] [PubMed] [Google Scholar]
- Kainulainen K, Karttunen L, Puhakka L, Sakai L, Peltonen L. Mutations in the fibrillin gene responsible for dominant ectopia lentis and neonatal Marfan syndrome. Nat Genet. 1994;6:64–69. doi: 10.1038/ng0194-64. [DOI] [PubMed] [Google Scholar]
- Kainulainen K, Pulkkinen L, Savolainen A, Kaitila I, Peltonen L. Location on chromosome 15 of the gene defect causing Marfan syndrome. N Engl J Med. 1990;323:935–939. doi: 10.1056/NEJM199010043231402. [DOI] [PubMed] [Google Scholar]
- Karttunen L, Raghunath M, Lonnqvist L, Peltonen L. A compound-heterozygous Marfan patient: two defective fibrillin alleles result in a lethal phenotype. Am J Hum Genet. 1994;55:1083–1091. [PMC free article] [PubMed] [Google Scholar]
- Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, De Backer JF, Oswald GL, Symoens S, Manouvrier S, Roberts AE, Faravelli F, Greco MA, Pyeritz RE, Milewicz DM, Coucke PJ, Cameron DE, Braverman AC, Byers PH, De Paepe AM, Dietz HC. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355:788–798. doi: 10.1056/NEJMoa055695. [DOI] [PubMed] [Google Scholar]
- Matyas G, De Paepe A, Halliday D, Boileau C, Pals G, Steinmann B. Evaluation and application of denaturing HPLC for mutation detection in Marfan syndrome: Identification of 20 novel mutations and two novel polymorphisms in the FBN1 gene. Hum Mutat. 2002;19:443–456. doi: 10.1002/humu.10054. [DOI] [PubMed] [Google Scholar]
- Milewicz DM, Michael K, Fisher N, Coselli JS, Markello T, Biddinger A. Fibrillin-1 (FBN1) mutations in patients with thoracic aortic aneurysms. Circulation. 1996;94:2708–2711. doi: 10.1161/01.cir.94.11.2708. [DOI] [PubMed] [Google Scholar]
- Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- Ng CM, Cheng A, Myers LA, Martinez-Murillo F, Jie C, Bedja D, Gabrielson KL, Hausladen JM, Mecham RP, Judge DP, Dietz HC. TGF-beta-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. J Clin Invest. 2004;114:1586–1592. doi: 10.1172/JCI22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira L, Andrikopoulos K, Tian J, Lee SY, Keene DR, Ono R, Reinhardt DP, Sakai LY, Biery NJ, Bunton T, Dietz HC, Ramirez F. Targetting of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nat Genet. 1997;17:218–222. doi: 10.1038/ng1097-218. [DOI] [PubMed] [Google Scholar]
- Pereira L, D'Alessio M, Ramirez F, Lynch JR, Sykes B, Pangilinan T, Bonadio J. Genomic organization of the sequence coding for fibrillin, the defective gene product in Marfan syndrome. Hum Mol Genet. 1993;2:1762. doi: 10.1093/hmg/2.10.1762. [DOI] [PubMed] [Google Scholar]
- Pereira L, Lee SY, Gayraud B, Andrikopoulos K, Shapiro SD, Bunton T, Biery NJ, Dietz HC, Sakai LY, Ramirez F. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. ProcNatl Acad SciU S A. 1999;96:3819–3823. doi: 10.1073/pnas.96.7.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PN, Arteaga-Solis E, Baldock C, Collod-Beroud G, Booms P, De Paepe A, Dietz HC, Guo G, Handford PA, Judge DP, Kielty CM, Loeys B, Milewicz DM, Ney A, Ramirez F, Reinhardt DP, Tiedemann K, Whiteman P, Godfrey M. The molecular genetics of Marfan syndrome and related disorders. J Med Genet. 2006;43:769–787. doi: 10.1136/jmg.2005.039669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PN, Booms P, Katzke S, Ladewig M, Neumann L, Palz M, Pregla R, Tiecke F, Rosenberg T. Mutations of FBN1 and genotype-phenotype correlations in Marfan syndrome and related fibrillinopathies. Hum Mutat. 2002;20:153–161. doi: 10.1002/humu.10113. [DOI] [PubMed] [Google Scholar]
- Roman MJ, Devereux RB, Kramer-Fox R, O'Loughlin J. Two-dimensional echocardiographic aortic root dimensions in normal children and adults. Am J Cardiol. 1989;64:507–512. doi: 10.1016/0002-9149(89)90430-x. [DOI] [PubMed] [Google Scholar]
- Schollin J, Bjarke B, Gustavson KH. Probable homozygotic form of the Marfan syndrome in a newborn child. Acta Paediatr Scand. 1988;77:452–456. doi: 10.1111/j.1651-2227.1988.tb10679.x. [DOI] [PubMed] [Google Scholar]
- Ten Dijke P, Arthur HM. Extracellular control of TGFbeta signalling in vascular development and disease. Nat Rev Mol Cell Biol. 2007;8:857–869. doi: 10.1038/nrm2262. [DOI] [PubMed] [Google Scholar]
- Whiteman P, Willis AC, Warner A, Brown J, Redfield C, Handford PA. Cellular and molecular studies of Marfan syndrome mutations identify co-operative protein folding in the cbEGF12-13 region of fibrillin-1. Hum Mol Genet. 2007;16:907–918. doi: 10.1093/hmg/ddm035. [DOI] [PubMed] [Google Scholar]
- Winship PR, Dragon AC. Identification of haemophilia B patients with mutations in the two calcium binding domains of factor IX: importance of a beta-OH Asp 64——Asn change. Br J Haematol. 1991;77:102–109. doi: 10.1111/j.1365-2141.1991.tb07955.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supp. Table S1.
Primers for cDNA sequencing of FBN1: F=forward; R=reverse; numbers refer to position in cDNA sequence
| fragment | primername | Sequence 5’ > 3’ | Length bp |
|---|---|---|---|
| 1 | FBN1F1 | ATGCGTCGAGGGCGTCTGCT | 384 |
| FBN1R384 | GCTACCTCCATTCATACAGCGA | ||
| 2 | FBN1F318 | GATAGCTCCTTCCTGTGGCTCC | 406 |
| FBN1R728 | CCGTGCGGATATTTGGAATG | ||
| 3 | FBN1F657 | CCCCTGTGAGATGTGTCCTG | 407 |
| FBN1R1064 | TTGGTTATGGACTGTGGCAGC | ||
| 4 | FBN1F1012 | ACAGCTCTGACAAACGGGCG | 384 |
| FBN1R1396 | TGCAGCGTCCATTTTGACAG | ||
| 5 | FBN1F1358 | GCCAGTTGGTCCGCTATCTC | 330 |
| FBN1R1688 | ACATGAAAGCCCGCATTACAC | ||
| 6 | FBN1F1609 | AATGGCCGGATCTGCAATAA | 398 |
| FBN1R2007 | CTGGCCTCTCTTGTATCCACCA | ||
| 7 | FBN1F1927 | CTGGCTGTGGGTCTGGATGG | 362 |
| FBN1R2289 | GCAGTTTTTCCCAGTTGAATCC | ||
| 8 | FBN1F2212 | ATCTGTGAAAACCTTCGTGGGA | 399 |
| FBN1R2611 | AGGTGGCTCCATTGATGTTGA | ||
| 9 | FBN1F2458 | GTCTGCAAGAACAGCCCAGG | 357 |
| FBN1R2815 | TGGGACACTGACACTTGAATGA | ||
| 10 | FBN1F2699 | TACTCAAGAATTAAAGGAACA | 525 |
| FBN1R3224 | CGGCATTCGTCAATGTCTGTGC | ||
| 11 | FBN1F3141 | CATTGGCAGCTTTAAGTGCAGG | 460 |
| FBN1R3621 | ACCACCATTCATTATGCTGCA | ||
| 12 | FBN1F3558 | CCATTCAACTCCCGATAGGCT | 335 |
| FBN1R3893 | TTTTCACAGGTCCCACTTAGGC | ||
| 13 | FBN1F3783 | CAGGTGCTTGTGTTATGATG | 392 |
| FBN1R4173 | GCACAGACAGCGGTAAGA | ||
| 14 | FBN1F4062 | GATTGGAGATGGCATTAAGTGC | 451 |
| FBN1R4513 | TGTTGACACAGTTCCCACTGA | ||
| 15 | FBN1F4425 | CTACGAACTGGACAGAAGCGG | 593 |
| FBN1R5018 | ATACAGGTGTAGTTGCCAACGG | ||
| 16 | FBN1F4910 | ACTACCTGAATGAAGATACACG | 626 |
| FBN1R5536 | GACCTGTGGAGGTGAAGCGGTAG | ||
| 17 | FBN1F5348 | TCAACATGGTTGGCAGCTTCC | 476 |
| FBN1R5824 | AAAGATTCCCATTTCCACTTGC | ||
| 18 | FBN1F5722 | ACAATTGGTTCCTTCAACTG | 356 |
| FBN1R6074 | GCACAAATTTCTGGCTCTT | ||
| 19 | FBN1F5973 | CTTGGATGGGTCCTACAGATGC | 579 |
| FBN1R6552 | CACATTCTTGCAGGTTCCATT | ||
| 20 | FBN1F6466 | GGTTATACTCTAGCGGGAATG | 450 |
| FBN1R6937 | TCCCACGGGTGTTGAGGCAGCG | ||
| 21 | FBN1F6842 | AGCGGAGACCTGATGGAGAGG | 645 |
| FBN1R7487 | CAGTTGTGTTGCTTGGTTGCA | ||
| 22 | FBN1F7429 | CAAGAGGATGGAAGGAGCTGC | 476 |
| FBN1R7905 | GAACTGTTCATACTGGAAGCCG | ||
| 23 | FBN1F7785 | CTACCTCCAGCACTACCAGTGG | 384 |
| FBN1R8169 | GTAGCCATTGATCTTACACTCG | ||
| 24 | FBN1F8024 | CACCTGGTTACTTCCGCATAGG | 672 |
| FBN1R8696 | ATGATTCTGATTGGGGGAAAA |