

Abstract
The methylene C atom in the title compound, C13H16N2S, is connected to a five-membered thiazole ring and a mesityl substituent. The rings are aligned at 75.4 (1)°. The amino substitutent interacts with the ring N atom of an adjacent molecule by an intermolecular N—H⋯N hydrogen bond, generating a helical chain running along the b axis.
Related literature
For background to the synthetic procedure,: see: Yadigarov et al. (2010 ▶).
Experimental
Crystal data
C13H16N2S
M r = 232.34
Monoclinic,
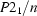
a = 5.5028 (5) Å
b = 30.832 (3) Å
c = 7.8355 (7) Å
β = 110.016 (1)°
V = 1249.08 (19) Å3
Z = 4
Mo Kα radiation
μ = 0.23 mm−1
T = 100 K
0.30 × 0.20 × 0.20 mm
Data collection
Bruker APEXII diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 1996 ▶) T min = 0.933, T max = 0.955
7129 measured reflections
2749 independent reflections
2486 reflections with I > 2σ(I)
R int = 0.024
Refinement
R[F 2 > 2σ(F 2)] = 0.045
wR(F 2) = 0.120
S = 1.06
2749 reflections
156 parameters
2 restraints
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.38 e Å−3
Δρmin = −0.24 e Å−3
Data collection: APEX2 (Bruker, 2005 ▶); cell refinement: SAINT (Bruker, 2005 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: X-SEED (Barbour, 2001 ▶); software used to prepare material for publication: publCIF (Westrip, 2010 ▶).
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536811006386/im2269sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536811006386/im2269Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H11⋯N2i | 0.88 (1) | 2.06 (1) | 2.907 (2) | 163 (2) |
Symmetry code: (i) 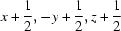 .
.
Acknowledgments
We thank Baku State University and the University of Malaya for supporting this study.
supplementary crystallographic information
Comment
A recent study reported the reaction of 1-chloro-3-(2,4,6-trimethylphenyl)-propan-2-one with primary amines. The chlorine atom in the α-chloro ketone is not replaced directly by an amino RNH– group. The intermediate product undergoes a Favorskii rearrangement that furnishes a compound having two methylene groups between the aromatic system and the amido unit (Yadigarov et al., 2010). The present study employs thiourea as the amine. One of its amino –NH2 groups is involved in the formation of the thiazolyl ring in the resulting product (Scheme I, Fig. 1). The methylene carbon is connected to the five-membered thiazolyl ring and the six-membered mesityl group. The rings are aligned at 75.4 (1) °. The amino –NH2 substitutent interacts with the ring N atom of an adjacent molecule by an N–H···N hydrogen bond generating a helical chain that runs along the b-axis of the monoclinic unit cell.
Experimental
1-Chloro-3-(2,4,6-trimethylphenyl)-propan-2-one (10 mmol) and thiourea (10 mmol) were stirred in water (100 ml) for an hour. A precipitate formed and this was collected and redissolved in hot ethanol. Slow evaporation of the solvent gave colorless crystals in 50% yield; m.p. 380–381 K.
Refinement
Carbon-bound H-atoms were placed in calculated positions [C–H 0.93 to 0.97 Å] and were included in the refinement in the riding model approximation, with Uiso(H) set to 1.2-1.5 Ueq(C).
The amino H-atoms were located in a difference Fourier map and were refined with a distance restraint of N–H 0.88±0.01 Å; their temperature factors were refined isotropically.
Figures
Fig. 1.

Thermal ellipsoid plot (Barbour, 2001) of C13H16N2S at the 70% probability level; hydrogen atoms are drawn as spheres of arbitrary radius.
Crystal data
| C13H16N2S | F(000) = 496 |
| Mr = 232.34 | Dx = 1.235 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2yn | Cell parameters from 3598 reflections |
| a = 5.5028 (5) Å | θ = 2.6–29.1° |
| b = 30.832 (3) Å | µ = 0.23 mm−1 |
| c = 7.8355 (7) Å | T = 100 K |
| β = 110.016 (1)° | Prism, colorless |
| V = 1249.08 (19) Å3 | 0.30 × 0.20 × 0.20 mm |
| Z = 4 |
Data collection
| Bruker APEXII diffractometer | 2749 independent reflections |
| Radiation source: fine-focus sealed tube | 2486 reflections with I > 2σ(I) |
| graphite | Rint = 0.024 |
| φ and ω scans | θmax = 27.5°, θmin = 2.6° |
| Absorption correction: multi-scan (SADABS; Sheldrick, 1996) | h = −7→7 |
| Tmin = 0.933, Tmax = 0.955 | k = −40→24 |
| 7129 measured reflections | l = −9→10 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.045 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.120 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.06 | w = 1/[σ2(Fo2) + (0.060P)2 + 0.6878P] where P = (Fo2 + 2Fc2)/3 |
| 2749 reflections | (Δ/σ)max = 0.001 |
| 156 parameters | Δρmax = 0.38 e Å−3 |
| 2 restraints | Δρmin = −0.24 e Å−3 |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| S1 | 0.65720 (8) | 0.220399 (14) | 0.75764 (6) | 0.02680 (15) | |
| N1 | 0.2450 (3) | 0.26387 (5) | 0.7802 (2) | 0.0283 (3) | |
| H11 | 0.351 (3) | 0.2757 (6) | 0.8795 (19) | 0.030 (5)* | |
| H12 | 0.080 (2) | 0.2637 (8) | 0.764 (3) | 0.038 (6)* | |
| N2 | 0.1780 (3) | 0.20379 (5) | 0.58369 (19) | 0.0241 (3) | |
| C1 | 0.3278 (3) | 0.23024 (5) | 0.7040 (2) | 0.0215 (3) | |
| C2 | 0.5821 (3) | 0.17807 (6) | 0.6029 (2) | 0.0267 (4) | |
| H2 | 0.7056 | 0.1604 | 0.5759 | 0.032* | |
| C3 | 0.3239 (3) | 0.17382 (5) | 0.5263 (2) | 0.0244 (3) | |
| C4 | 0.1797 (4) | 0.13975 (6) | 0.3931 (3) | 0.0338 (4) | |
| H4A | 0.0544 | 0.1542 | 0.2862 | 0.041* | |
| H4B | 0.0800 | 0.1217 | 0.4502 | 0.041* | |
| C5 | 0.3520 (3) | 0.11046 (6) | 0.3288 (2) | 0.0282 (4) | |
| C6 | 0.3814 (4) | 0.11748 (6) | 0.1605 (2) | 0.0301 (4) | |
| C7 | 0.5436 (4) | 0.09018 (7) | 0.1069 (3) | 0.0344 (4) | |
| H7 | 0.5633 | 0.0950 | −0.0075 | 0.041* | |
| C8 | 0.6772 (4) | 0.05629 (6) | 0.2137 (3) | 0.0344 (4) | |
| C9 | 0.6454 (4) | 0.04995 (6) | 0.3799 (3) | 0.0365 (4) | |
| H9 | 0.7352 | 0.0269 | 0.4557 | 0.044* | |
| C10 | 0.4858 (4) | 0.07644 (6) | 0.4388 (3) | 0.0336 (4) | |
| C11 | 0.2363 (5) | 0.15279 (7) | 0.0330 (3) | 0.0473 (6) | |
| H11A | 0.2694 | 0.1807 | 0.0966 | 0.071* | |
| H11B | 0.0505 | 0.1465 | −0.0085 | 0.071* | |
| H11C | 0.2946 | 0.1541 | −0.0719 | 0.071* | |
| C12 | 0.8478 (4) | 0.02682 (8) | 0.1499 (3) | 0.0487 (6) | |
| H12A | 0.9348 | 0.0438 | 0.0820 | 0.073* | |
| H12B | 0.7419 | 0.0042 | 0.0711 | 0.073* | |
| H12C | 0.9776 | 0.0133 | 0.2552 | 0.073* | |
| C13 | 0.4571 (7) | 0.06770 (8) | 0.6208 (3) | 0.0609 (8) | |
| H13A | 0.5687 | 0.0434 | 0.6802 | 0.091* | |
| H13B | 0.2767 | 0.0605 | 0.6029 | 0.091* | |
| H13C | 0.5074 | 0.0936 | 0.6974 | 0.091* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S1 | 0.0157 (2) | 0.0299 (2) | 0.0334 (3) | −0.00014 (16) | 0.00646 (16) | −0.00426 (18) |
| N1 | 0.0173 (7) | 0.0329 (8) | 0.0320 (8) | 0.0012 (6) | 0.0049 (6) | −0.0077 (7) |
| N2 | 0.0193 (7) | 0.0246 (7) | 0.0269 (7) | −0.0006 (6) | 0.0061 (5) | −0.0005 (6) |
| C1 | 0.0171 (7) | 0.0248 (8) | 0.0214 (7) | 0.0014 (6) | 0.0051 (6) | 0.0038 (6) |
| C2 | 0.0229 (8) | 0.0246 (8) | 0.0342 (9) | 0.0014 (7) | 0.0118 (7) | −0.0012 (7) |
| C3 | 0.0241 (8) | 0.0230 (8) | 0.0267 (8) | 0.0000 (7) | 0.0096 (6) | 0.0010 (7) |
| C4 | 0.0296 (9) | 0.0303 (9) | 0.0407 (10) | −0.0038 (8) | 0.0110 (8) | −0.0094 (8) |
| C5 | 0.0298 (9) | 0.0229 (8) | 0.0295 (9) | −0.0032 (7) | 0.0072 (7) | −0.0051 (7) |
| C6 | 0.0343 (9) | 0.0249 (9) | 0.0255 (9) | −0.0034 (7) | 0.0031 (7) | −0.0002 (7) |
| C7 | 0.0414 (10) | 0.0374 (11) | 0.0234 (9) | −0.0047 (9) | 0.0098 (8) | −0.0049 (8) |
| C8 | 0.0328 (10) | 0.0319 (10) | 0.0339 (10) | −0.0014 (8) | 0.0055 (8) | −0.0129 (8) |
| C9 | 0.0458 (11) | 0.0245 (9) | 0.0292 (9) | 0.0056 (8) | 0.0000 (8) | −0.0014 (8) |
| C10 | 0.0489 (11) | 0.0239 (9) | 0.0260 (9) | −0.0024 (8) | 0.0102 (8) | −0.0025 (7) |
| C11 | 0.0621 (14) | 0.0328 (11) | 0.0353 (11) | 0.0052 (10) | 0.0016 (10) | 0.0071 (9) |
| C12 | 0.0402 (12) | 0.0481 (13) | 0.0552 (14) | 0.0031 (10) | 0.0127 (10) | −0.0213 (11) |
| C13 | 0.118 (2) | 0.0327 (12) | 0.0400 (13) | 0.0024 (13) | 0.0379 (15) | 0.0050 (10) |
Geometric parameters (Å, °)
| S1—C2 | 1.7323 (18) | C7—C8 | 1.383 (3) |
| S1—C1 | 1.7415 (16) | C7—H7 | 0.9500 |
| N1—C1 | 1.351 (2) | C8—C9 | 1.386 (3) |
| N1—H11 | 0.88 (1) | C8—C12 | 1.509 (3) |
| N1—H12 | 0.87 (1) | C9—C10 | 1.389 (3) |
| N2—C1 | 1.304 (2) | C9—H9 | 0.9500 |
| N2—C3 | 1.396 (2) | C10—C13 | 1.512 (3) |
| C2—C3 | 1.346 (2) | C11—H11A | 0.9800 |
| C2—H2 | 0.9500 | C11—H11B | 0.9800 |
| C3—C4 | 1.502 (2) | C11—H11C | 0.9800 |
| C4—C5 | 1.515 (2) | C12—H12A | 0.9800 |
| C4—H4A | 0.9900 | C12—H12B | 0.9800 |
| C4—H4B | 0.9900 | C12—H12C | 0.9800 |
| C5—C10 | 1.397 (3) | C13—H13A | 0.9800 |
| C5—C6 | 1.399 (3) | C13—H13B | 0.9800 |
| C6—C7 | 1.393 (3) | C13—H13C | 0.9800 |
| C6—C11 | 1.508 (3) | ||
| C2—S1—C1 | 89.03 (8) | C6—C7—H7 | 118.8 |
| C1—N1—H11 | 119.3 (14) | C7—C8—C9 | 117.63 (18) |
| C1—N1—H12 | 115.1 (16) | C7—C8—C12 | 121.1 (2) |
| H11—N1—H12 | 118 (2) | C9—C8—C12 | 121.3 (2) |
| C1—N2—C3 | 110.84 (14) | C8—C9—C10 | 121.79 (18) |
| N2—C1—N1 | 125.03 (15) | C8—C9—H9 | 119.1 |
| N2—C1—S1 | 114.43 (12) | C10—C9—H9 | 119.1 |
| N1—C1—S1 | 120.50 (13) | C9—C10—C5 | 119.78 (17) |
| C3—C2—S1 | 110.36 (13) | C9—C10—C13 | 119.37 (19) |
| C3—C2—H2 | 124.8 | C5—C10—C13 | 120.84 (19) |
| S1—C2—H2 | 124.8 | C6—C11—H11A | 109.5 |
| C2—C3—N2 | 115.33 (15) | C6—C11—H11B | 109.5 |
| C2—C3—C4 | 127.16 (16) | H11A—C11—H11B | 109.5 |
| N2—C3—C4 | 117.47 (15) | C6—C11—H11C | 109.5 |
| C3—C4—C5 | 113.94 (15) | H11A—C11—H11C | 109.5 |
| C3—C4—H4A | 108.8 | H11B—C11—H11C | 109.5 |
| C5—C4—H4A | 108.8 | C8—C12—H12A | 109.5 |
| C3—C4—H4B | 108.8 | C8—C12—H12B | 109.5 |
| C5—C4—H4B | 108.8 | H12A—C12—H12B | 109.5 |
| H4A—C4—H4B | 107.7 | C8—C12—H12C | 109.5 |
| C10—C5—C6 | 119.39 (17) | H12A—C12—H12C | 109.5 |
| C10—C5—C4 | 120.04 (17) | H12B—C12—H12C | 109.5 |
| C6—C5—C4 | 120.56 (17) | C10—C13—H13A | 109.5 |
| C5—C6—C7 | 118.95 (17) | C10—C13—H13B | 109.5 |
| C5—C6—C11 | 122.00 (18) | H13A—C13—H13B | 109.5 |
| C7—C6—C11 | 119.01 (18) | C10—C13—H13C | 109.5 |
| C8—C7—C6 | 122.45 (18) | H13A—C13—H13C | 109.5 |
| C8—C7—H7 | 118.8 | H13B—C13—H13C | 109.5 |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H11···N2i | 0.88 (1) | 2.06 (1) | 2.907 (2) | 163 (2) |
Symmetry codes: (i) x+1/2, −y+1/2, z+1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: IM2269).
References
- Barbour, L. J. (2001). J. Supramol. Chem. 1, 189–191.
- Bruker (2005). APEX2 and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Sheldrick, G. M. (1996). SADABS University of Göttingen, Germany.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
- Yadigarov, R. R., Khalilov, A. N., Mamedov, I. G., Nagiev, F. N., Magerramov, A. M. & Allakhverdiev, M. A. (2010). Russ. J. Org. Chem. 45, 1856–1858.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536811006386/im2269sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536811006386/im2269Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report