

Abstract
In the title compound, C13H16ClFN2O, the piperazine ring is flanked by 1-(2-fluorobenzyl)piperazine and adopts a chair conformation. The dihedral angle between the fluorophenyl ring and the four planar C atoms (r.m.s. = 0.0055 Å) of the piperazine chair is 78.27 (7)°, whereas the dihedral angle between the four planar C atoms of the piperazine chair and the ethanone plane is 55.21 (9) Å; the Cl atom displaced by1.589 (2) Å out of the plane.
Related literature
For the synthesis of related compounds, see: Contreras et al. (2001 ▶); Capuano et al. (2002 ▶). For their use as intermediates in the synthesis of anti-inflammatory agents or CCR1 antagonists, see: Rolland & Duhault (1989 ▶); Kaufmann (2005 ▶); Tanikawa et al. (1995 ▶); Xie et al. (2007 ▶).
Experimental
Crystal data
C13H16ClFN2O
M r = 270.73
Orthorhombic,
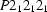
a = 7.9350 (5) Å
b = 8.4610 (4) Å
c = 19.0040 (11) Å
V = 1275.89 (12) Å3
Z = 4
Mo Kα radiation
μ = 0.30 mm−1
T = 291 K
0.30 × 0.30 × 0.20 mm
Data collection
Bruker APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2001 ▶) T min = 0.566, T max = 0.716
12886 measured reflections
3001 independent reflections
2550 reflections with I > 2σ(I)
R int = 0.033
Refinement
R[F 2 > 2σ(F 2)] = 0.035
wR(F 2) = 0.085
S = 1.01
3001 reflections
165 parameters
H-atom parameters constrained
Δρmax = 0.33 e Å−3
Δρmin = −0.18 e Å−3
Absolute structure: Flack (1983 ▶), 1255 Friedel pairs
Flack parameter: 0.03 (7)
Data collection: APEX2 (Bruker, 2003 ▶); cell refinement: SAINT (Bruker, 2001 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: DIAMOND (Brandenburg, 1999) ▶; software used to prepare material for publication: SHELXL97.
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536811006180/si2316sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536811006180/si2316Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Acknowledgments
The authors would like to thank the Ministry of Science and Technology of China (2007AA02Z160, 2009ZX09501–004) and the Chinese National Natural Science Foundation (20872077, 90813013) for financial support.
supplementary crystallographic information
Comment
Piperazine derivatives similar to the title compound are well known as being useful for a variety of pharmaceutical indication, particularly as cardiotonic, neurotropic or anti-inflammatory agents (Kaufmann, 2005). The synthesis of related pyridazine compounds and their medicinal and pharmaceutical activity were reported (Contreras et al., 2001; Capuano et al., 2002). The use of related compounds as intermediates in the synthesis of antiinflammatory agents or CCRI antagonists can be studied in various patents (Rolland & Duhault, 1989; Kaufmann, 2005; Tanikawa et al., 1995) and medicinal journal (Xie et al., 2007). Moreover, we recently identified a series of compounds bearing various substituted benzylpiperazine moiety with potent antitumor activity by virtual screening approach (paper was being revised).
Herein, we report the synthesis of the title compound as one important representative of piperazine derivatives and its X-ray crystal structure. The molecule of (I) is shown in Fig. 1. The bond lengths and angles are within normal ranges. The piperazine ring in the molecule adopts a chair conformation. The dihedral angle between the fluorophenyl ring and the four planar C atoms (r.m.s. = 0.0055 Å) of the piperazine chair is 78.27 (7)°. Whereas the dihedral angle between the four planar C atoms of the piperazine chair and the ethanone plane is 55.21 (9)Å with the Cl atom about 1.589 (2) Å out of plane. In the crystal, there are no strong intermolecular hydrogen bonds to link the molecules.
Experimental
All chemicals and solvents were obtained from commercial supplies and used without purification. To a solution of chloroacetic chloride (0.58 ml, 7.15 mmol) in CH2Cl2 (10 ml) was added, at 0 °C, 1-(2-fluorobenzyl)piperazine(II) (1.15 g, 5.90 mmol) dissolved in CH2Cl2 (20 ml) which was prepared from the reaction of anhydrous piperazine(III) and 1-(chloromethyl)-2-fluorobenzene(IV). The reaction mixture was stirred at room temperature for about 30 min until no 1-(2-fluorobenzyl)piperazine remained, as monitored by TLC. The mixture was poured into cold H2O (50 ml) and rendered alkaline with a 10% NaHCO3 aqueous solution and separated. The organic layer, dried over Na2SO4, was evaporated under reduced pressure to give 1.44 g of pure title compound as a yellow oil. Yield 90%; 1H NMR (400 MHz, CDCl3) δ 7.35 (dd, J = 7.4, 1.4 Hz, 1H), δ 7.23–7.29 (m, 1H), δ 7.12 (t, J = 7.2 Hz, 1H), δ 7.04 (t, J = 9.2 Hz, 1H), δ 4.05 (s, 2H), δ 3.64 (d, J = 5.2 Hz, 2H), δ 3.62 (d, J = 4.4 Hz, 2H), 3.52 (t, J = 5.0 Hz, 2H), δ 2.51 (dt, J = 15.6, 4.8 Hz, 4H); 13C NMR (100.6 MHz, CDCl3) δ 161.16, 131.24, 128.88, 123.88, 123.75, 115.27, 115.05, 54.87, 52.41, 52.00, 46.06, 41.93, 40.67.
Refinement
All H atoms were positioned geometrically and refined using a riding model approximation with distances C—H = 0.93 Å for the benzene ring and 0.97 Å for Csp3 carbon atoms and Uiso(H) = 1.2 times Ueq(C). The absolute structure was determined by using the Flack parameter refinement with the TWIN/BASF instruction, and the coordinates of all atoms were inverted by instruction MOVE 1 1 1 - 1 in the final refinement with SHELXL97.
Figures
Fig. 1.

Molecular structure of the title compound showing displacement ellipsoids at the 30% probability level.
Fig. 2.

Synthesis of the title compound
Crystal data
| C13H16ClFN2O | F(000) = 568 |
| Mr = 270.73 | Dx = 1.409 Mg m−3 |
| Orthorhombic, P212121 | Mo Kα radiation, λ = 0.71069 Å |
| Hall symbol: P 2ac 2ab | Cell parameters from 25 reflections |
| a = 7.9350 (5) Å | θ = 7.5–15° |
| b = 8.4610 (4) Å | µ = 0.30 mm−1 |
| c = 19.0040 (11) Å | T = 291 K |
| V = 1275.89 (12) Å3 | Block, colorless |
| Z = 4 | 0.30 × 0.30 × 0.20 mm |
Data collection
| Bruker APEXII CCD diffractometer | 3001 independent reflections |
| Radiation source: fine-focus sealed tube | 2550 reflections with I > 2σ(I) |
| graphite | Rint = 0.033 |
| ω scans | θmax = 28.4°, θmin = 3.2° |
| Absorption correction: multi-scan (SADABS; Bruker, 2001) | h = −10→10 |
| Tmin = 0.566, Tmax = 0.716 | k = −10→10 |
| 12886 measured reflections | l = −25→25 |
Refinement
| Refinement on F2 | Hydrogen site location: inferred from neighbouring sites |
| Least-squares matrix: full | H-atom parameters constrained |
| R[F2 > 2σ(F2)] = 0.035 | w = 1/[σ2(Fo2) + (0.028P)2 + 0.4P] where P = (Fo2 + 2Fc2)/3 |
| wR(F2) = 0.085 | (Δ/σ)max < 0.001 |
| S = 1.01 | Δρmax = 0.33 e Å−3 |
| 3001 reflections | Δρmin = −0.18 e Å−3 |
| 165 parameters | Extinction correction: SHELXL97 (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| 0 restraints | Extinction coefficient: 0.077 (3) |
| Primary atom site location: structure-invariant direct methods | Absolute structure: Flack (1983), 1255 Friedel pairs |
| Secondary atom site location: difference Fourier map | Flack parameter: 0.03 (7) |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 1.51486 (16) | 0.40256 (17) | 0.67832 (8) | 0.0512 (3) | |
| Cl1 | 1.23456 (9) | 0.09826 (6) | 0.64207 (3) | 0.06647 (19) | |
| F1 | 0.6313 (2) | 0.88072 (19) | 0.64912 (8) | 0.0801 (4) | |
| N1 | 0.96049 (18) | 0.62572 (16) | 0.64509 (8) | 0.0378 (3) | |
| N2 | 1.2433 (2) | 0.46187 (17) | 0.70180 (8) | 0.0437 (4) | |
| C1 | 0.8356 (2) | 0.8253 (2) | 0.56706 (9) | 0.0415 (4) | |
| C2 | 0.7317 (3) | 0.9332 (2) | 0.59859 (10) | 0.0492 (5) | |
| C3 | 0.7224 (3) | 1.0901 (3) | 0.58058 (12) | 0.0622 (6) | |
| H3 | 0.6497 | 1.1582 | 0.6041 | 0.075* | |
| C4 | 0.8215 (3) | 1.1433 (3) | 0.52783 (12) | 0.0623 (6) | |
| H4 | 0.8177 | 1.2491 | 0.5146 | 0.075* | |
| C5 | 0.9276 (3) | 1.0405 (3) | 0.49380 (12) | 0.0588 (6) | |
| H5 | 0.9960 | 1.0766 | 0.4574 | 0.071* | |
| C6 | 0.9328 (3) | 0.8832 (3) | 0.51363 (10) | 0.0524 (5) | |
| H6 | 1.0047 | 0.8148 | 0.4898 | 0.063* | |
| C7 | 0.8427 (2) | 0.6546 (2) | 0.58860 (10) | 0.0459 (4) | |
| H7A | 0.7312 | 0.6211 | 0.6034 | 0.055* | |
| H7B | 0.8746 | 0.5912 | 0.5482 | 0.055* | |
| C8 | 1.1333 (2) | 0.6613 (2) | 0.62433 (10) | 0.0424 (4) | |
| H8A | 1.1409 | 0.7723 | 0.6118 | 0.051* | |
| H8B | 1.1613 | 0.6000 | 0.5828 | 0.051* | |
| C9 | 1.2590 (2) | 0.6266 (2) | 0.68052 (10) | 0.0459 (4) | |
| H9A | 1.3719 | 0.6464 | 0.6631 | 0.055* | |
| H9B | 1.2395 | 0.6951 | 0.7207 | 0.055* | |
| C10 | 1.0718 (2) | 0.4225 (3) | 0.72213 (11) | 0.0500 (5) | |
| H10A | 1.0413 | 0.4821 | 0.7638 | 0.060* | |
| H10B | 1.0655 | 0.3109 | 0.7336 | 0.060* | |
| C11 | 0.9506 (2) | 0.4583 (2) | 0.66487 (11) | 0.0460 (5) | |
| H11A | 0.9758 | 0.3931 | 0.6242 | 0.055* | |
| H11B | 0.8372 | 0.4337 | 0.6804 | 0.055* | |
| C12 | 1.3729 (2) | 0.3613 (2) | 0.69620 (9) | 0.0388 (4) | |
| C13 | 1.3417 (3) | 0.1882 (2) | 0.71208 (10) | 0.0476 (5) | |
| H13A | 1.2753 | 0.1787 | 0.7547 | 0.057* | |
| H13B | 1.4484 | 0.1351 | 0.7197 | 0.057* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0326 (7) | 0.0537 (8) | 0.0671 (9) | 0.0000 (6) | −0.0001 (6) | 0.0034 (7) |
| Cl1 | 0.0760 (4) | 0.0506 (3) | 0.0729 (3) | −0.0109 (3) | −0.0059 (3) | −0.0099 (3) |
| F1 | 0.0805 (10) | 0.0910 (10) | 0.0688 (8) | 0.0264 (8) | 0.0272 (8) | 0.0187 (8) |
| N1 | 0.0326 (7) | 0.0339 (7) | 0.0471 (8) | 0.0024 (6) | −0.0027 (6) | 0.0003 (6) |
| N2 | 0.0343 (8) | 0.0416 (7) | 0.0552 (9) | 0.0042 (7) | 0.0010 (8) | 0.0090 (6) |
| C1 | 0.0373 (9) | 0.0480 (10) | 0.0393 (9) | 0.0030 (8) | −0.0091 (7) | −0.0007 (8) |
| C2 | 0.0465 (11) | 0.0587 (12) | 0.0423 (9) | 0.0063 (10) | −0.0002 (9) | 0.0045 (8) |
| C3 | 0.0751 (15) | 0.0544 (11) | 0.0573 (12) | 0.0190 (12) | 0.0006 (12) | −0.0014 (10) |
| C4 | 0.0801 (17) | 0.0479 (12) | 0.0590 (13) | −0.0008 (11) | −0.0167 (11) | 0.0099 (10) |
| C5 | 0.0596 (14) | 0.0677 (14) | 0.0492 (11) | −0.0068 (11) | −0.0017 (10) | 0.0128 (10) |
| C6 | 0.0502 (11) | 0.0624 (13) | 0.0447 (10) | 0.0049 (10) | −0.0006 (9) | −0.0006 (10) |
| C7 | 0.0400 (10) | 0.0464 (10) | 0.0512 (10) | −0.0017 (8) | −0.0089 (9) | −0.0047 (9) |
| C8 | 0.0363 (9) | 0.0337 (8) | 0.0570 (11) | −0.0023 (7) | −0.0006 (8) | 0.0053 (8) |
| C9 | 0.0370 (9) | 0.0362 (9) | 0.0645 (11) | −0.0006 (8) | −0.0076 (9) | 0.0016 (8) |
| C10 | 0.0393 (11) | 0.0497 (11) | 0.0610 (12) | 0.0068 (8) | 0.0094 (9) | 0.0158 (10) |
| C11 | 0.0328 (9) | 0.0397 (9) | 0.0656 (13) | −0.0015 (8) | 0.0056 (9) | 0.0061 (9) |
| C12 | 0.0364 (9) | 0.0433 (9) | 0.0366 (8) | 0.0023 (8) | −0.0050 (7) | −0.0010 (7) |
| C13 | 0.0479 (11) | 0.0442 (11) | 0.0506 (11) | 0.0067 (9) | −0.0049 (9) | 0.0071 (9) |
Geometric parameters (Å, °)
| O1—C12 | 1.228 (2) | C5—H5 | 0.9300 |
| Cl1—C13 | 1.753 (2) | C6—H6 | 0.9300 |
| F1—C2 | 1.324 (2) | C7—H7A | 0.9700 |
| N1—C7 | 1.444 (2) | C7—H7B | 0.9700 |
| N1—C8 | 1.459 (2) | C8—C9 | 1.490 (3) |
| N1—C11 | 1.467 (2) | C8—H8A | 0.9700 |
| N2—C12 | 1.339 (2) | C8—H8B | 0.9700 |
| N2—C10 | 1.454 (2) | C9—H9A | 0.9700 |
| N2—C9 | 1.457 (2) | C9—H9B | 0.9700 |
| C1—C6 | 1.366 (3) | C10—C11 | 1.483 (3) |
| C1—C2 | 1.369 (3) | C10—H10A | 0.9700 |
| C1—C7 | 1.502 (3) | C10—H10B | 0.9700 |
| C2—C3 | 1.373 (3) | C11—H11A | 0.9700 |
| C3—C4 | 1.351 (3) | C11—H11B | 0.9700 |
| C3—H3 | 0.9300 | C12—C13 | 1.515 (3) |
| C4—C5 | 1.373 (3) | C13—H13A | 0.9700 |
| C4—H4 | 0.9300 | C13—H13B | 0.9700 |
| C5—C6 | 1.384 (3) | ||
| C7—N1—C8 | 111.87 (15) | C9—C8—H8A | 108.9 |
| C7—N1—C11 | 108.62 (14) | N1—C8—H8B | 108.9 |
| C8—N1—C11 | 108.59 (13) | C9—C8—H8B | 108.9 |
| C12—N2—C10 | 126.45 (15) | H8A—C8—H8B | 107.7 |
| C12—N2—C9 | 121.38 (16) | N2—C9—C8 | 109.27 (15) |
| C10—N2—C9 | 111.92 (15) | N2—C9—H9A | 109.8 |
| C6—C1—C2 | 115.21 (18) | C8—C9—H9A | 109.8 |
| C6—C1—C7 | 121.77 (18) | N2—C9—H9B | 109.8 |
| C2—C1—C7 | 123.02 (18) | C8—C9—H9B | 109.8 |
| F1—C2—C1 | 117.11 (18) | H9A—C9—H9B | 108.3 |
| F1—C2—C3 | 118.22 (19) | N2—C10—C11 | 111.40 (16) |
| C1—C2—C3 | 124.7 (2) | N2—C10—H10A | 109.3 |
| C4—C3—C2 | 118.4 (2) | C11—C10—H10A | 109.3 |
| C4—C3—H3 | 120.8 | N2—C10—H10B | 109.3 |
| C2—C3—H3 | 120.8 | C11—C10—H10B | 109.3 |
| C3—C4—C5 | 119.7 (2) | H10A—C10—H10B | 108.0 |
| C3—C4—H4 | 120.2 | N1—C11—C10 | 110.55 (16) |
| C5—C4—H4 | 120.2 | N1—C11—H11A | 109.5 |
| C4—C5—C6 | 120.0 (2) | C10—C11—H11A | 109.5 |
| C4—C5—H5 | 120.0 | N1—C11—H11B | 109.5 |
| C6—C5—H5 | 120.0 | C10—C11—H11B | 109.5 |
| C1—C6—C5 | 122.1 (2) | H11A—C11—H11B | 108.1 |
| C1—C6—H6 | 119.0 | O1—C12—N2 | 123.07 (18) |
| C5—C6—H6 | 119.0 | O1—C12—C13 | 118.68 (17) |
| N1—C7—C1 | 112.92 (15) | N2—C12—C13 | 118.24 (17) |
| N1—C7—H7A | 109.0 | C12—C13—Cl1 | 110.34 (13) |
| C1—C7—H7A | 109.0 | C12—C13—H13A | 109.6 |
| N1—C7—H7B | 109.0 | Cl1—C13—H13A | 109.6 |
| C1—C7—H7B | 109.0 | C12—C13—H13B | 109.6 |
| H7A—C7—H7B | 107.8 | Cl1—C13—H13B | 109.6 |
| N1—C8—C9 | 113.25 (16) | H13A—C13—H13B | 108.1 |
| N1—C8—H8A | 108.9 | ||
| C6—C1—C2—F1 | 177.86 (18) | C11—N1—C8—C9 | −57.8 (2) |
| C7—C1—C2—F1 | −1.6 (3) | C12—N2—C9—C8 | 120.59 (19) |
| C6—C1—C2—C3 | −0.9 (3) | C10—N2—C9—C8 | −54.0 (2) |
| C7—C1—C2—C3 | 179.7 (2) | N1—C8—C9—N2 | 56.1 (2) |
| F1—C2—C3—C4 | −178.3 (2) | C12—N2—C10—C11 | −118.1 (2) |
| C1—C2—C3—C4 | 0.4 (3) | C9—N2—C10—C11 | 56.2 (2) |
| C2—C3—C4—C5 | 0.1 (3) | C7—N1—C11—C10 | 179.10 (16) |
| C3—C4—C5—C6 | 0.0 (3) | C8—N1—C11—C10 | 57.2 (2) |
| C2—C1—C6—C5 | 0.9 (3) | N2—C10—C11—N1 | −57.6 (2) |
| C7—C1—C6—C5 | −179.60 (19) | C10—N2—C12—O1 | 179.59 (18) |
| C4—C5—C6—C1 | −0.5 (3) | C9—N2—C12—O1 | 5.8 (3) |
| C8—N1—C7—C1 | −63.4 (2) | C10—N2—C12—C13 | 0.2 (3) |
| C11—N1—C7—C1 | 176.72 (16) | C9—N2—C12—C13 | −173.64 (16) |
| C6—C1—C7—N1 | 93.1 (2) | O1—C12—C13—Cl1 | −103.48 (18) |
| C2—C1—C7—N1 | −87.4 (2) | N2—C12—C13—Cl1 | 75.97 (19) |
| C7—N1—C8—C9 | −177.68 (15) |
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: SI2316).
References
- Brandenburg, K. (1999). DIAMOND Crystal Impact GbR, Bonn, Germany.
- Bruker (2001). SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Bruker (2003). APEX2 Bruker AXS Inc., Madison, Wisconsin, USA.
- Capuano, B., Crosby, I. T., Lloyd, E. J. & Taylor, D. A. (2002). Aust. J. Chem. 55, 565–576.
- Contreras, J. M., Parrot, I., Sippl, W., Rival, Y. M. & Wermuth, C. G. (2001). J. Med. Chem. 44, 2707–2718. [DOI] [PubMed]
- Flack, H. D. (1983). Acta Cryst. A39, 876–881.
- Kaufmann, U. (2005). WO Patent No. 2005079769, 291 pp.
- Rolland, Y. & Duhault, J. (1989). EP Patent No. 319412, 44 pp.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Tanikawa, K., Saito, A., Hirotsuka, M. & Shikada, K. (1995). WO Patent No. 9501343, 127 pp.
- Xie, Y. F., Lake, K., Ligsay, K., Komandla, M., Sircar, I., Nagarajan, G., Li, J., Xu, K., Parise, J., Schneider, L., Huang, D., Liu, J. P., Dines, K., Sakurai, N., Barbosa, M. & Jack, R. (2007). Bioorg. Med. Chem. Lett. 17, 3367–3372. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S1600536811006180/si2316sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536811006180/si2316Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report