

Abstract
Background
Ischemia-reperfusion injury impairs lung transplant outcomes. The transcription factors, activator protein-1, and nuclear factor kappa B, are activated early in reperfusion and drive the development of injury. Thrombin inhibition with hirudin, and calcineurin inhibition with tacrolimus have independently been shown to ameliorate lung ischemia-reperfusion injury by reducing activator protein-1 and nuclear factor kappa B activation, respectively. However, high doses were required to achieve protection using individual agents, raising concerns about potential toxicities. We sought to determine if low-dose combination therapy reduced injury through synergistic inhibition of pretranscriptional signaling events.
Methods
Rats were pretreated with either intravenous hirudin or tacrolimus at low doses or high doses, or both at low doses, prior to undergoing left lung ischemia and reperfusion. Lungs were assessed for markers of lung injury, including bronchoalveolar lavage cytokine-chemokine content and transcription factor transactivation of activator protein-1 and nuclear factor kappa B.
Results
High-dose monotherapy with hirudin or tacrolimus reduced lung injury and transactivation of activator protein-1 and nuclear factor kappa B activation, respectively, whereas low-dose monotherapy with either agent did not alter transcription factor activation or lung injury compared with positive controls. Low-dose combination therapy was more protective than high-dose monotherapy with either drug, and correlated with a reduction in activation of both transcription factors and their associated cytokines.
Conclusions
The significant decrease in lung injury severity and transcription factor activation with combined pathway inhibition suggests pretranscriptional signaling redundancy between the calcineurin and thrombin dependent pathways in lung reperfusion injury.
Introduction
Lung transplantation is a life-saving treatment option for select patients with end-stage lung disease. Unfortunately, the inevitable period of ischemia and subsequent reperfusion can lead to significant lung injury in up to 20% of patients [1]. The development of lung ischemia-reperfusion injury (LIRI) is dependent upon transactivation of the proinflammatory transcription factors nuclear factor kappa B (NFκB) and activator protein1(AP-1) and the subsequent cytokine-chemokine secretion by resident pulmonary cell populations [2 – 4]. Ultimately, significant pulmonary edema, inflammatory cell infiltration, and impaired compliance and gas exchange develop as full injury manifests.
Thrombin, a serine protease involved in the coagulation cascade, is also known to activate multiple inflammatory cell populations. Through activation of proteinase-activated receptors on such cell types as endothelial cells, macrophages, and platelets, thrombin promotes loss of endothelial integrity, leukocyte migration, and proinflammatory cytokine release [5]. Utilizing an in vivo model of lung reperfusion injury, we have previously demonstrated thrombin localization to alveolar macrophages as well as pulmonary endothelial and epithelial cells within 15 minutes of reperfusion [6]. Hirudin, a direct thrombin inhibitor, was shown to ameliorate lung injury in our model by reducing AP-1 dependent proinflammatory transcriptional activation, including cytokineinduced neutrophil chemoattractant (CINC), but did not alter tumor necrosis factor-alpha (TNF-α) secretion [6].
Calcineurin inhibition with tacrolimus is a cornerstone of current immunosuppressive regimens in solid organ transplantation. By limiting calcineurin-dependent activation of the transcription factor, nuclear factor of activated T cell, tacrolimus effectively modulates the cellular immune response responsible for acute rejection. Calcineurin is also known to promote acute nonalloimmune specific proinflammatory signaling through NFκB dependent transcriptional activation. We have shown that calcineurin inhibition with either cyclosporine or tacrolimus diminished the transactivation of NFκB in LIRI, and subsequently reduced pulmonary capillary leak, leukocyte infiltration, as well as CINC and TNFα secretion without altering interleukin-1β (IL-1β) secretion [7,8]
Given individually at the doses we employed, both hirudin and tacrolimus provided significant protection in our in vivo model of LIRI, through modulation of AP-1 and NFκB dependent mechanisms respectively. Secondary effects of each agent, when given to a donor in the setting of multiorgan procurement, must be taken into account. High-dose thrombin inhibition will clinically alter the coagulation profile, potentially causing significant intraoperative bleeding. Intratracheal administration of tacrolimus decreases, but does not eliminate, system absorption at the doses previously used [8]. To circumvent these potential side effects, we proposed using low-dose combination therapy with intratracheal tacrolimus and intravenous hirudin. We hypothesized that the low-dose combination therapy of hirudin and tacrolimus would provide improved protection from lung injury when compared with either agent alone through a synergistic effect on the inhibition of AP-1 and NFκB and the subsequent production of proinflammatory mediators.
Material and Methods
Reagents
All reagents were purchased from Sigma-Aldrich Chemical (St. Louis, MO) unless otherwise specified. Recombinant hirudin (Bayer Healthcare Pharmaceuticals, Wayne, NJ) and the calcineurin inhibitor tacrolimus (Astellas Pharma Inc, Deerfield, IL) were obtained from the pharmacy at the University of Washington Medical Center.
Animal Model
Pathogen-free Long-Evans rats (Harlan Sprague Dawley, Indianapolis, IN), weighing between 275 grams and 300 grams, were used for all experiments. Approval for all experimental protocols was granted by the University of Washington Animal Care Committee. All animals received humane care in compliance with the “Principles of Laboratory Animal Care” established by the National Society for Medical Research and the “Guide for the Care and Use of Laboratory Animals” developed by the Institute of Laboratory Animal Resources and published by the National Institute of Health (NIH Publication No. 86-23, revised 1996).
A well-established, warm, in situ, ischemia-reperfusion model was used as previously described [4, 9]. In brief, animals were anesthetized with 30 mg of intraperitoneal pentobarbital after which a 14G angiocatheter was placed under direct vision into the trachea through a midline neck incision. The catheter was secured with a suture and the animal placed on a Harvard Rodent Ventilator (Harvard Apparatus, Boston, MA). Ventilator settings were standardized with an inspired oxygen content of 60%, 2 cm H2O of positive end-expiratory pressure, and a respiratory rate of 80. Peak pressures were monitored and did not exceed 10 cm H2O. A left thoracotomy was performed and the pleural space entered sharply through the fifth interspace. The left lung was mobilized atraumatically and the inferior pulmonary ligament sharply divided. After mobilization, 50 units of heparin in a volume of 500πL was then administered through the penile vein and allowed to circulate for five minutes. A noncrushing clamp was subsequently placed across the left lung hilum, occluding the pulmonary artery, vein, and main-stem bronchus, for a period of 90 minutes. This period of ischemia and hypoxia was held constant for all experimental groups, after which the clamp was removed and the left lung allowed to reventilate and reperfuse for up to four hours. During reperfusion 500 πL of warm saline was injected subcutaneously every hour to maintain hydration. At the end of the reperfusion period, a midline laparotomy and sternotomy were performed. Blood samples were taken from the inferior vena cava and the animals sacrificed by aortic transection. The heart-lung block was excised and the pulmonary circulation flushed with 20 mL of saline. The left lung was separated from other mediastinal structures. Rats were treated with 0.1 mg/kg (high dose) or 0.0125 mg/kg (low dose) tacrolimus through their endotracheal tube 1 hour prior to the onset of ischemia as previously described [8]. Recombinant hirudin was given at a bolus dose of 1 mg/kg (high dose) or 0.125 mg/kg (low dose) intravenously 30 minutes before ischemia. The higher doses of hirudin and tacrolimus had previously been shown to reduce LIRI severity [6–8] while preliminary studies showed that treatment with low-dose (one-eighth of the high dose) therapy with either hirudin or tacrolimus was not protective. Tacrolimus and hirudin were administered 1 hour and 30 minutes prior to ischemia, respectively, based on prior studies showing that administration prior to ischemia, and specifically at these time points, was most beneficial in comparison to at initiation of reperfusion or after 2 hours of reperfusion [6, 7]. In addition, pretreatment allows for translation to clinical lung transplantation given that the timing of the onset of ischemia is predictable.
Seven different experimental groups were studied in vivo. Negative controls were sacrificed without undergoing surgical manipulation. The other six groups underwent 90 minutes of ischemia followed by 4 hours of reperfusion. Positive control animals received an equivalent volume of intravenous or intratracheal vehicle prior to ischemia. Treated groups were then either given high-dose tacrolimus or hirudin, low-dose tacrolimus or hirudin or combined low-dose therapy. Combination high-dose therapy was not tested due to concern regarding their combined toxicities. Assessment of lung injury severity and molecular analyses of tissue samples were performed with the following protocols.
Lung Permeability Index
Changes in lung vascular permeability were assessed as a ratio of 125I (symbol for iodine-125) radiolabeled bovine serum albumin (BSA) in the left lung to that of 1 mL. The 125I-BSA was purchased from NEN Life Sciences (Boston, MA) and was serially diluted to obtain an activity of approximately 800,000 counts per minute (cpm). A 1% BSA/phosphate-buffered saline solution was added to make a final volume of 500 πL which was injected intravenously 5 minutes prior to removal of the left hilar clamp. The lung was allowed to reperfuse for four hours and, immediately before sacrifice, 1 mL of blood was aspirated from the inferior vena cava. The left lung was subsequently harvested as described previously. The cpm for the left lung and the blood were then quantified in a gamma counter. The permeability index is then calculated as follows:
This ratio corrects for any variation in the systemic distribution of 125I-BSA.
Bronchoalveolar Lavage (BAL) and Inflammatory Cell Counts
After four hours of reperfusion, the left lung was selectively lavaged with 5 mL of sterile saline via the endotracheal tube. A clamp was placed across the right hilum to achieve selective left lung lavage. The recovered lavage fluid was centrifuged at 1,800 rpm for ten minutes at 4°C to pellet the cells. The supernatant was frozen at −80°C for later cytokine analysis and the pellet was resuspended in 10 mL of sterile phosphate buffered saline. One milliliter of resuspended cells were subsequently stained with Gill’s solution and counted using a hemacytometer (Hausser Scientific, Horsham, PA). Differential cell counts were not performed because previous studies demonstrated that nearly all of the cells were neutrophils [9].
Electrophoretic Mobility Shift Assay
Lungs were allowed to reperfuse for 15 minutes and nuclear protein was then extracted from whole lung homogenates as previously described [4, 6]. Protein concentrations were determined using the bicinchoninic assay. Five micrograms of nuclear protein was incubated with biotin labeled NFκB or AP-1 oligonucleotide probe (Panomics Inc, Redwood City, CA) for 20 minutes at room temperature. Running unlabeled probe in a cold competition reaction was performed to validate the specificity of each probe. Samples were resolved on a 6% sodium dodecyl sulfate polyacrylamide gel electrophoresis run at 80 volts for one to two hours. After transfer to a nylon membrane (Amersham, Rockford, IL), membranes were incubated for 15 minutes in a streptavidin-horseradish peroxidase conjugate solution and subsequently visualized using pierce enhancer solution (Pierce Biotechnology, Inc, Rockford, IL) and autoradiography. Densitometry was performed to assess relative differences in activation between groups using the image analysis software, Image Pro Plus (Media Cybernetics, Silver Spring, MD).
Enzyme-Linked Immunosorbent Assay
The BAL CINC, IL-1β, and TNF-α concentrations were analyzed using sandwich enzyme-linked immunosorbent assays (R&D Systems, Minneapolis, MN) as previously described [4, 6].
Activated Partial Thromboplastin Time (aPTT) Measurement
The aPTT was measured in animals given no hirudin or either high-dose or low-dose hirudin 30 minutes after administration. All measurements were performed by the University of Washington clinical laboratory.
Statistical Analysis
All data are presented as mean values ± standard error of the mean unless otherwise designated. Statistical differences between groups were assessed using the two-tailed Student’s t test (Microsoft Excel 2002; Microsoft Corp, Redmond, WA) with post-hoc Bonferroni adjustment for multiple comparisons. The p values refer to comparisons between vehicle treated positive controls and each experimental group.
Results
Markers of left lung injury, including permeability index and BAL cell counts, are summarized in Table 1. Left lung BAL cytokine-chemokine content are summarized in Table 2.
Table 1.
Markers of Left Lung Injurya
| Experimental Group | Permeability Index | BAL Cell Count (×10^5 Cells) |
|---|---|---|
| Negative control (n = 4) | 0.09 ± 0.005 | 6 ± 0.7 |
| Positive control (n = 6) | 0.84 ± 0.03 | 207 ± 7.6 |
| Tac LD (n = 6) | 0.85 ± 0.04b | 205 ± 5.5 |
| Hir LD (n = 6) | 0.75 ± 0.06b | 207 ± 10 |
| Tac HD (n = 4) | 0.33 ± 0.02c | 111 ± 6c |
| Hir HD (n = 4) | 0.51 ± 0.01c | 135 ± 5c |
| Combo (n = 6) | 0.25 ± 0.02c | 85 ± 4.8c |
Values are mean ± standard error of the mean.
p = 0.2 compared with positive controls.
p < 0.001 compared with positive controls.
BAL. bronchoalveolar lavage; combo. hirudin 0.125 mg/kg IV and tacrolimus 0.0125 mg/kg intratracheal; Hir HD. hirudin 1 mg/kg IV; Hir LD. hirudin 0.125 mg/kg IV; Tac HD. tacrolimus 0.2 mg/kg intratracheal; Tac LD. tacrolimus 0.0125 mg/kg intratracheal.
Table 2.
Left Lung Bronchoalveolar Lavage Cytokine-Chemokine Contenta
| Experimental Group | CINC (pg/mL) | IL-1β (pg/mL) | TNF-α (pg/mL) |
|---|---|---|---|
| Negative control (n = 4) | 51 ± 12 | 37 ± 5 | 22 ± 5.6 |
| Positive control (n = 6) | 2,772 ± 268 | 587 ± 44 | 275 ± 32 |
| Tac LD (n = 6) | 2,435 ± 287b | 591 ± 54b | 278 ± 28b |
| Hir LD (n = 6) | 2,325 ± 324b | 534 ± 52b | 314 ± 14b |
| Tac HD (n = 4) | 686 ± 23c | 437 ± 12b | 106 ± 12d |
| Hir HD (n = 4) | 759 ± 42c | 161 ± 46c | 227 ± 22b |
| Combo (n = 8) | 275 ± 31c | 134 ± 18c | 64 ± 8c |
Values are mean ± standard error of the mean.
p 0.2 compared with positive controls.
p < 0.001 compared with positive controls.
p = 0.007 compared with positive controls.
CINC = cytokine-induced neutrophil chemoattractant; Combo = hirudin 0.125 mg/kg IV and tacrolimus 0.0125 mg/kg intratracheal; Hir HD = hirudin 1 mg/kg IV; Hir LD = hirudin 0.125 mg/kg IV; IL-1β = interleukin-1 beta; Tac HD = tacrolimus 0.2 mg/kg intratracheal; Tac LD = tacrolimus 0.0125 mg/kg intratracheal; TNF-α = tumor necrosis factor alpha.
Permeability Index
Positive control animals demonstrated a greater than ninefold increase in permeability index compared with unmanipulated negative controls (p < 0.001). Both high-dose monotherapy with hirudin and tacrolimus, provided significant protection versus positive controls, reducing permeability index 39% and 61%, respectively (p < 0.001).
Conversely, low-dose therapy with either agent was not protective (p = 0.2 and p = 0.2, respectively). Treatment with a combination of low-dose hirudin and tacrolimus reduced vascular permeability by 70% (p < 0.001).
Bronchoalveolar Lavage Inflammatory Cell Counts
Compared with negative control animals, positive controls animals had a greater than 34-fold increase in inflammatory cells in left lung lavage fluid (p < 0.001). High-dose hirudin and high-dose tacrolimus decreased BAL inflammatory cell counts compared with positive controls by 35% and 46%, respectively (p < 0.001). Low-dose therapy with either drug did not reduce inflammatory cell counts compared with positive controls (p 0.2 and p 0.2, respectively). Combined low-dose therapy was profoundly protective, reducing cell counts by 59% (p < 0.001).
Left Lung Bronchoalveolar Lavage Cytokine-Chemokine Content
The CINC, IL-1β, and TNF-α contents within the left lung BAL fluid was significantly increased in positive controls compared with negative controls (53-, 15-, and 12 fold increase, respectively, p <0.001). High-dose hirudin therapy resulted in a significant decrease in both CINC and IL-1β production by 72% (p <0.001) but did not alter TNF-α production (p = 0.42), whereas high-dose tacrolimus therapy resulted in a significant decrease in CINC and TNF-α production by 75% and 61%, respectively (p < 0.001 and p = 0.007, respectively). Low-dose hirudin or tacrolimus monotherapy did not alter CINC, IL-1β, or TNF-α production significantly. However, low-dose combination therapy resulted in a significant decrease in CINC, IL-1β, and TNF-α production by 90%, 77%, and 77%, respectively (p < 0.001, p < 0.001, p = 0.005, respectively).
Transcription Factor Nuclear Translocation
Electrophoretic mobility shift assays for NFκB and AP-1 were performed on lungs harvested after 15 minutes of reperfusion to assess the nuclear translocation of these relevant transcription factors.
Representative shift assays are shown for NFκB(Fig 1) and AP-1 (Fig 2) with corresponding graphs depicting their relative optical densitometry data (Figs 3 and 4, respectively). High-dose tacrolimus therapy reduced NFκB but not AP-1 translocation compared with positive controls. High-dose hirudin inhibited AP-1 translocation but not NFκB. Individual low-dose inhibition with either hirudin or tacrolimus did not significantly reduce either NFκB or AP-1. In contrast, combination low-dose therapy markedly reduced transactivation of both NFκB and AP-1.
Fig 1.
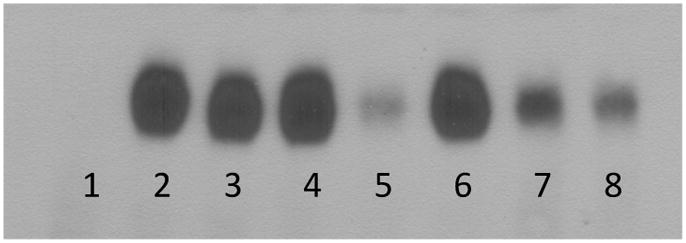
Representative electromobility shift assay for nuclear factor kappa B (NFκB). Unmanipulated negative controls (lane 1, n = 4) demonstrate minimal NFκB nuclear translocation compared with positive controls (lane 2, n = 6) (p = 0.009). Low-dose tacrolimus (lane 3, n = 6), low-dose hirudin (lane 4, n = 6), and high-dose hirudin therapy (lane 6, n = 6) did not significantly reduce NFκB translocation compared with positive controls (p = 0.2). High-dose tacrolimus (lane 5, n = 6) reduced NFκB translocation by 66% (p = 0.024) compared with positive controls. Low-dose combination therapy (lanes 7 and 8, n = 6) reduced NFκB translocation by 83% (p = 0.007) compared with positive controls.
Fig 2.
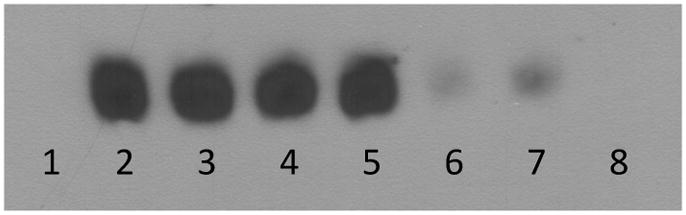
Representative electromobility shift assay for activator protein-1 (AP-1). Unmanipulated negative controls (lane 1, n. 4) demonstrate minimal AP-1 nuclear translocation compared with positive controls (lane 2, n = 6) (p < 0.001). Low-dose tacrolimus (lane 3, n = 6), low-dose hirudin (lane 4, n = 6), and high-dose tacrolimus therapy (lane 5, n = 6) did not significantly reduce AP-1 translocation compared with positive controls (p = 0.02). High-dose hirudin (lane 6, n = 6) significantly reduced AP-1 translocation by 80% (p < 0.001) compared with positive controls. Low-dose combination therapy (lanes 7 and 8, n = 6) reduced AP-1 translocation by 86% (p = 0.001) compared with positive controls.
Fig 3.
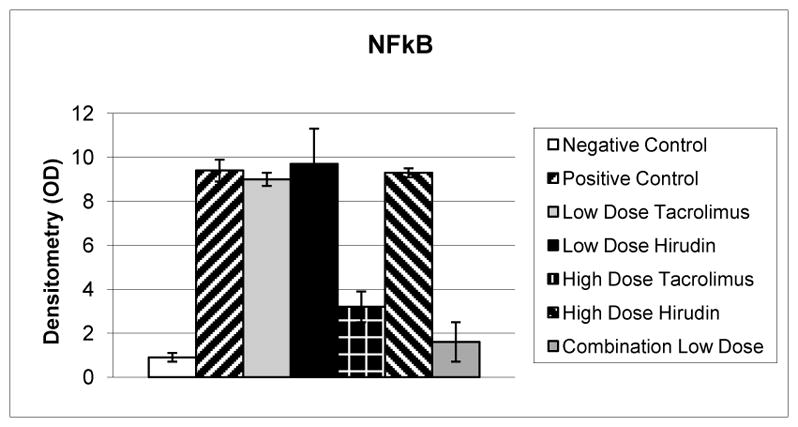
Relative optical densitometry (OD) of electromobility shift assay for nuclear factor kappa B (NFκB). Positive controls (n = 6) demonstrate a greater than tenfold increase in NFκB nuclear translocation compared with negative controls (p = 0.009, n = 4). Low-dose tacrolimus, low-dose hirudin, and high-dose hirudin therapy (n = 6 per group) did not significantly reduce NFκB translocation compared with positive controls (p = 0.2). High-dose tacrolimus therapy (n. 6) reduced NFκB translocation by 66% (p = 0.024) compared with positive controls. Low-dose combination therapy (n. 6) reduced NFκB translocation by 83% (p = 0.007) compared with positive controls.
Fig 4.
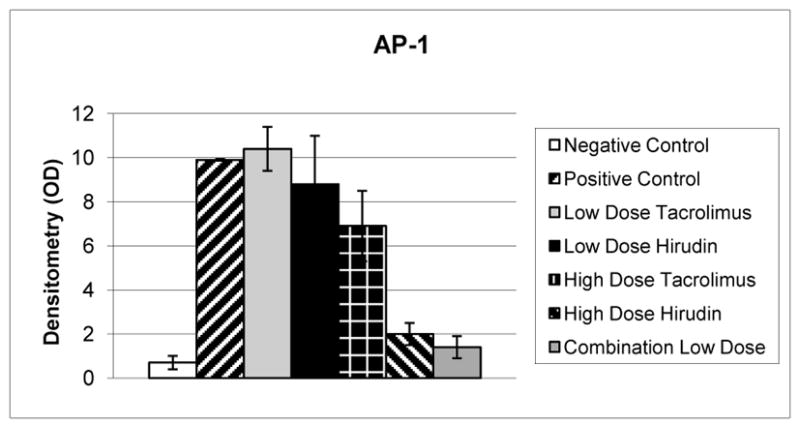
Relative optical densitometry (OD) of electromobility shift assay activator protein-1 (AP-1). Positive controls (n = 6) demonstrate a greater than 14-fold increase in AP-1 nuclear translocation compared with negative controls (p = 0.001, n. 4). Low-dose tacrolimus, low-dose hirudin, and high-dose tacrolimus therapy (n = 6 per group) did not significantly reduce AP-1 translocation compared with positive controls (p = 0.02). High-dose hirudin therapy (n = 6) significantly reduced AP-1 translocation by 80% (p = 0.001) compared with positive controls. Low-dose combination therapy (n = 6) reduced AP-1 translocation by 86% (p < 0.001) compared with positive controls.
Activated Partial Thromboplastin Time
High-dose hirudin resulted in an elevated aPTT (51 seconds) compared with untreated animals (21 seconds). Low-dose hirudin therapy did not cause an increase in aPTT (22.7 seconds).
Comment
In response to oxidative stress, transcriptional upregulation and subsequent secretion of proinflammatory mediators including CINC, IL-1β, and TNF-α, leads to lung injury. Previous work has demonstrated this to be specifically dependent on the nuclear translocation of the transcription factors NFκB and AP-1 [6, 10]. The present study confirms that calcineurin inhibition with tacrolimus, and thrombin inhibition with hirudin, effectively reduce NFκB and AP-1 translocation, respectively, leading to a reduction in reperfusion injury severity. Using monotherapy, high doses are required to exert the protective effects; however, when animals are treated with low-dose tacrolimus and hirudin simultaneously synergistic protection was achieved. We found the individual protective effects of both hirudin and tacrolimus to occur only with utilization of high doses. Protection from reperfusion injury, as measured by permeability index and BAL cell count, was lost when dosing of each agent was reduced by almost 90%. Similarly, low-dose therapy with either hirudin or tacrolimus did not decrease the transactivation of NFκBor AP-1 or alter the production of proinflammatory mediators. Interestingly, when such low-dose therapy was used in a combined fashion, a dramatic reduction in nuclear translocation of both transcription factors and proinflammatory mediator production was seen as well as a corresponding reduction in lung injury as assessed by multiple parameters.
Full injury manifestation in LIRI is dependent on the activation of multiple proinflammatory signaling cascades. Redundancy in signaling is evident at both the pretranscriptional and post-transcriptional levels. Inhibition targeting post-transcriptional events, as demonstrated by individual cytokine inhibition, is only modestly effective at diminishing LIRI severity [9]. Our results indicate a similar redundancy in pretranscriptional signaling. Here we have demonstrated two discrete but interrelated pathways involved in transcription factor activation in our in vivo model. High-dose monotherapy (either hirudin or tacrolimus) appears to effectively inhibit a high percentage of its available target ligand, resulting in a significant reduction in activation of the corresponding transcription factor. Low-dose mono-therapy provides incomplete inhibition, thus allowing redundant pretranscriptional pathways to promote enough NFκB and AP-1 translocation to initiate production of critical proinflammatory mediators.
Though the exact mechanism is unclear, the dramatic decrease in transcription factor activation, proinflammatory mediator production, and injury severity observed with combined pathway inhibition suggests significant interaction between the calcineurin and thrombin dependent signaling pathways at the pretranscriptional level. Even incomplete inhibition of multiple proximal signaling steps was demonstrated to be just as effective as more complete, targeted inhibition of only one pathway, suggesting the potential for synergistic inhibition of redundant, interactive pretranscriptional inflammatory signaling.
There are several advantages to the use of low-dose combination therapy over high-dose monotherapy. At the low dose employed, hirudin caused no alteration in the aPTT. Previous work has also determined that low-dose intratracheal tacrolimus is undetectable in peripheral blood samples four hours after administration [8]. Therefore, combination low-dose therapy offers the ability to minimize the deleterious side effects of these medications while providing clinically significant protection from lung reperfusion injury. Though this warm ischemia model may not recapitulate the complex intracellular signaling response in LIRI, it focuses on the aspect of lung transplantation that is most injurious to the recipient. Identifying ways to ameliorate the cellular response to warm ischemia, perhaps with low-dose combination therapy administered to donors prior to harvesting, may reduce the incidence and severity of LIRI as well as the associated morbidities. Additionally, given the commonality of downstream signaling pathways in acute lung injury and LIRI, low-dose combination therapy may be protective in other types of acute lung injury, such as in acute respiratory distress syndrome or secondary to sepsis, though we currently lack data to support this. Further studies evaluating the optimal dose and timing of administration and confirming this protection are warranted.
Acknowledgments
This research was funded by the University of Washington, Department of Surgery, Cardiothoracic Surgery Division. The authors of this research had full control of the design of the study, methods used, outcome parameters, analysis of data, and production of the written report.
References
- 1.King RC, Binns OA, Rodriguez F, et al. Reperfusion injury significantly impacts clinical outcome after pulmonary transplantation. Ann Thorac Surg. 2000;69:1681–5. doi: 10.1016/s0003-4975(00)01425-9. [DOI] [PubMed] [Google Scholar]
- 2.Farivar AS, Woolley SM, Fraga CH, Byrne K, Mulligan MS. Proinflammatory response of alveolar type II pneumocytes to in vitro hypoxia and reoxygenation. Am J Transplant. 2004;4:346–51. doi: 10.1111/j.1600-6143.2004.00352.x. [DOI] [PubMed] [Google Scholar]
- 3.Naidu BV, Farivar AS, Woolley SM, Byrne K, Mulligan MS. Chemokine response of pulmonary artery endothelial cells to hypoxia and reoxygenation. J Surg Res. 2003;114:163–71. doi: 10.1016/s0022-4804(03)00330-5. [DOI] [PubMed] [Google Scholar]
- 4.Naidu BV, Krishnadasan B, Farivar AS, et al. Early activation of the alveolar macrophage is critical to the development of lung ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 2003;126:200–7. doi: 10.1016/s0022-5223(03)00390-8. [DOI] [PubMed] [Google Scholar]
- 5.Ramachandran R, Hollenberg MD. Proteinases and signaling: pathophysiological and therapeutic implications via PARs and more. Br J Pharmacol. 2008;153 (suppl 1):S263–82. doi: 10.1038/sj.bjp.0707507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farivar AS, Delgado MF, McCourtie AS, Barnes AD, Verrier ED, Mulligan MS. Crosstalk between thrombosis and inflammation in lung reperfusion injury. Ann Thorac Surg. 2006;81:1061–7. doi: 10.1016/j.athoracsur.2005.09.047. [DOI] [PubMed] [Google Scholar]
- 7.Krishnadasan B, Naidu B, Rosengart M, et al. Decreased lung ischemia reperfusion injury in rats after preoperative administration of cyclosporine and tacrolimus. J Thorac Cardiovasc Surg. 2002;123:756–67. doi: 10.1067/mtc.2002.120351. [DOI] [PubMed] [Google Scholar]
- 8.Woolley SM, Farivar AS, Naidu BV, et al. Endotracheal calcineurin inhibition ameliorates injury in an experimental model of lung ischemia reperfusion. J Thorac Cardiovasc Surg. 2004;127:376–84. doi: 10.1016/j.jtcvs.2003.09.034. [DOI] [PubMed] [Google Scholar]
- 9.Krishnadasan B, Naidu BV, Byrne K, Fraga C, Verrier ED, Mulligan MS. The role of proinflammatory cytokines in lung ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 2003;125:261–72. doi: 10.1067/mtc.2003.16. [DOI] [PubMed] [Google Scholar]
- 10.Naidu BV, Krishnadasan B, Byrne K, et al. Regulation of chemokine expression by cyclosporine A in alveolar macro-phages exposed to hypoxia and reoxygenation. Ann Thorac Surg. 2002;74:899–905. doi: 10.1016/s0003-4975(02)03746-3. [DOI] [PubMed] [Google Scholar]