

Abstract
We have previously cloned a cDNA encoding human prolylcarboxypeptidase (PRCP) and expressed the cDNA in the Schneider 2 (S2) drosophila cell line. Here, we further characterized this recombinant enzyme. Investigations were performed to determine whether recombinant PRCP (rPRCP) metabolizes kinins (BK 1–9 and BK 1–8). The metabolites of these kinins were identified by LC/MS. rPRCP metabolized BK 1–8 to BK 1–7, whereas rPRCP was ineffective in metabolizing BK 1–9. The hydrolysis of BK 1–8 by rPRCP was dose- and time-dependent. A homology model of PRCP was developed based upon the sequence of dipeptidyl-peptidase 7 (DPP7, PDB ID: 3JYH), and providentially, the structure of PRCP (PDB ID: 3N2Z) was characterized during the course of our investigation. Docking studies of bradykinin oligopeptides were performed both from the homology model, and from the crystal structure of PRCP. These docking studies may provide a better understanding of the contribution of specific residues involved in substrate selectivity of human PRCP.
INTRODUCTION
Emerging experimental evidence supports the existence of a complex interaction between the plasma kallikrein-kinin-system (KKS) and renin-angiotensin-system (RAS) via bond breaking mechanism studies[1;2]. An intriguing aspect of these studies is that the counteracting properties of the KKS and RAS results in blood pressure regulation as well as water and electrolyte balance. The upregulation of both the RAS and KKS leading to subsequent physiological and cellular responses such as inflammation, cell growth and differentiation, and angiogenesis following tissue injury, support the importance of these systems as potential targets for therapeutic intervention in a variety of diseases[3–5]. The activation of RAS results in the generation of bioactive angiotensin peptides with hypertensive[6], antihypertensive[7], immunostimulating[8;9] and cytomodulatory[10;11] activities. The major metabolic end products released by RAS that induce vasoconstriction and thrombotic events are angiotensin II (Ang II) and angiotensin III (Ang III)[12].
Prolylcarboxypeptidase (PRCP), a serine protease, is ubiquitously found in plasma and various human tissues[13;14]. However, PRCP is particularly abundant in lysosomes [14]. PRCP is one of the key enzymes in the RAS, which converts Ang II to angiotensin 1–7 (Ang 1–7) and Ang III angiotensin 2–7 (Ang 2–7)[15].
PRCP activates the zymogen prekallikrein (PK) to kallikrein irreversibly, most likely via cleavage of a single bond[16;17]. However, the mechanism by which PRCP activates PK is not well detailed. Kallikrein represents a key component of the KKS pathway in generating bioactive peptides with varying biological activities. The bioactive peptides of KKS include; cleaved high molecular weight kininogen (HKa)[18] and bradykinin (BK 1–9)[2]. While activated factor XII represents a potent activator of PK under pathophysiological conditions, PCRP activates PK to kallikrein at levels under plasma basal conditions[19;20]. Activation of this protease cascade leads to the generation of BK 1–9 in acute inflammation and des-Arg9-bradykinin (BK 1–8) in chronic inflammation[21]. PRCP-induced PK activation results in modulation of endothelial cell function through BK1–9 generation, a process which is involved in the induction of vasorelaxtion via nitric oxide and prostaglandin generation[22]. Thus, PRCP activates two distinct vasodilator pathways by activating bradykinin receptors and angiotensin 1–7 – mediating nitric oxide[23;24] as well as prostacyclin formation[25;26]. These studies suggest that PRCP is cardioprotective against thrombosis and hypertension by acting as a vasodilator [27;28].
Several laboratories independently have identified BK as a substrate of the same processing enzyme, PRCP[17;29]. Detailed metabolite studies of kinins (BK1–9 and BK1–8) by PRCP have not yet been undertaken. We have previously cloned a cDNA encoding human PRCP and expressed it in the Schneider 2 (S2) drosophila cell line[30]. Investigations were performed to determine whether rPRCP has the ability to metabolize kinins.
For the first time, a liquid chromatography-mass spectrometry (LC-MS) technique was used to analyze the metabolism of both BK1–9 and BK1–8 by human rPRCP. In the present study, we have also used a computational approach to predict the structure of human PRCP.
MATERIALS AND METHODS
Materials
HyQ SFX – Drosophila insect cells and insect cell culture medium were purchased from HyClone (Logan, UT), Hygromycin B was obtained from Invitrogen (Carlsbad, California). Human pulmonary artery endothelial cells (HPAEC) were obtained from ATCC. Bradykinin (1–9), Bradykinin (1–8) and Bradykinin (1–7) were bought from Sigma-Aldrich (St. Louis, MO).
Prekallikrein activation on endothelial cells
PK activation on confluent monolayers of HPAEC (4 × 104 cells density/well in microtiter plate cuvette wells) was determined according to the previously described method[31]. The substrate H-D-Pro-Phe-Arg-p-nitroanilide (S2302) (Dia-Pharma, Franklin, OH) was used to determine kallikrein activity. The rate of paranitraniline liberation from S2302 was determined by measuring the absorbance at 405 nm[31].
Measuring the generation of bradykinin by PRCP-dependent PK activation pathway on endothelial cells
HPAEC (104 cells/well) were plated and cultured in 96 well plates. Triplicate samples of cells were incubated were incubated with 600 nM HK for 1 h at 37°C, as previously described[31]. After the incubation, cells were washed and treated with 600 nM PK. Afterward, supernatants were collected and concentrated from 300 μl to a final volume of 50 μl and frozen at −20 °C. The generation of BK 1–9, BK 1–8 and BK 1–7 in the samples was determined by LC/MS apparatus (details under LC-MS studies).
Expression, purification and characterization of rPRCP
rPRCP was prepared according to the previously described method[30].
Time course of metabolism of bradykinin 1–9 (BK 1–9) by rPRCP
A stock solution of 100 μL rPRCP, 150 μL of 10 mM sodium acetate (pH 4.8), 200μL of DPBS (pH 7.1) and 50 μL of bradykinin (1–9) was prepared. The pH of the mixture was determined to be 6.2. The activity of the enzyme was stopped at the following time points: 0, 1, 2, 5, 10, 15, 30, 60 and 120 minutes. This was quelled by reducing the pH of the mixture to 1.4 through transferring 50 μL of the mixture to centrifuge tubes with 10 μL of 1M phosphoric acid. Concentration of bradykinin (1–9) at the start of the reaction was 83.3 μM.
Time course of metabolism of des-Arg9-bradykinin (BK1–8) by rPRCP
100 μL rPRCP, 150 μL of 10 mM sodium acetate (pH 4.8), 200μL of DPBS (pH 7.1) and 50 μL of bradykinin (1–8) were mixed as described for bradykinin (1–9). The pH of the mixture was determined to be 6.2. The enzyme activity was stopped at same time points as for bradykinin (1–9) at 0, 1, 2, 5, 10, 15, 30, 60 and 120 minutes by reducing the pH of the mixture to 1.4 by means of transferring 50μL of the mixture to centrifuge tubes with 10 μL of 1 M phosphoric acid. The concentration of bradykinin (1–8) at the start of the reaction was 833.3 μM.
LC-MS studies
LC-MS experiments for the analysis of bradykinin and PRCP incubation mixtures were conducted based on the previously described method with modification[32]. Bradykinin m/z 354.3 [MH]+2; bradykinin 1–8 m/z 452.7 [MH]+2; bradykinin 1–7 m/z 379.2 [MH]+2. 5 μL of the incubation mixture containing bradykinin and rPRCP were diluted with water spiked with 0.1% formic acid (45 μL), and 10 μL of this solution was used for analysis. The rate constants for PRCP substrates (BK1–9, BK1–8, Ala-Pro-paranitraniline) were determined for rPRCP, as previously described[15].
Computational Studies
All IFD and MD studies were performed on a 2×4-core Intel Xeon L5420 2.5GHz SMP Linux Workstation.
Induced Fit Docking
The IFD workflow provided in the Maestro software environment (Schrodinger Suite 2009 IFD docking protocol; Glide version 5.5; Prime version 2.1, Schrodinger, New York, NY) was used to dock bradykinin oligopeptides into the lowest non-translational and non-rotational normal modes identified through NMA. The crystal structure of PRCP was treated to the Protein Preparation Wizard workflow (Version 9.1, Schrodinger, New York, NY). Preprocessing was performed to assign bond orders, add hydrogens, create disulfide bonds, delete waters beyond 5 Å from HET groups, and to fill in missing side chains and loops using Prime. Refinement was performed with automatic H-bond assignment using exhaustive sampling, including sampling of water orientations. Next minimization was performed using the OPLS2005 force field, with convergence set to an RMSD of 0.30 Å. Hydrogens were minimized initially followed by the entire protein.
After the protein preparation workflow, the sulfate ion present in the crystal structure was removed and IFD was performed on BK1–8 and BK1–9. Both peptides structures were built within the Maestro interface. The Glide grid was setup to use the centroid of residues 298, 348, 359, 371, 432, 455, 456, and 460. PRCP was prepared for initial Glide docking with receptor and ligand van der Waals scaling of 0.50 to generate 20 poses. Induced fit was then performed by refining residues within 5 Å of ligand poses, with optimization of side chains. Redocking was performed using the XP settings for structures within 30.0 kcal/mol of the best pose, which produced 14 surviving poses. The top ranked pose was selected for further analysis.
RESULTS
Catabolic metabolites of kinins by rPRCP
Bradykinin is a critical mediator in physiological and pathophysiological processes, many of which have been well-characterized in in-vitro and in-vivo assays. We have previously cloned a cDNA encoding human PRCP and expressed it in Schneider insect cells. Here, investigations were performed to determine whether rPRCP metabolizes bradykinin 1–9 [1Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg9 (BK1–9)] and bradykinin 1–8 [1Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe8 (des-Arg9-BK, BK1–8)].
We initially determined the rate of Ala-Pro-pNA hydrolysis by rPRCP. rPRCP (50 ng) liberated paranitroanilide from Ala-Pro-pNA, which was detected at 405 nm. A hyperbolic dependence was observed for the rPRCP, consistent with Henri-Michaelis–Menten behavior. rPRCP metabolized Ala-Pro-pNA in a dose-dependent fashion, Figure 1A. Next, investigations were performed to determine the metabolism of BK1–9 and BK1–8 by rPRCP. rPRCP (50 ng) was incubated with the increasing concentrations of BK1–8 (ranging from 0.08 to 1000 μM) and incubated at 37 °C, Figure 1B. BK1–8 and its metabolites were detected by LC-MS. rPRCP metabolized BK 1–8 in a dose-dependent manner.
Figure 1.
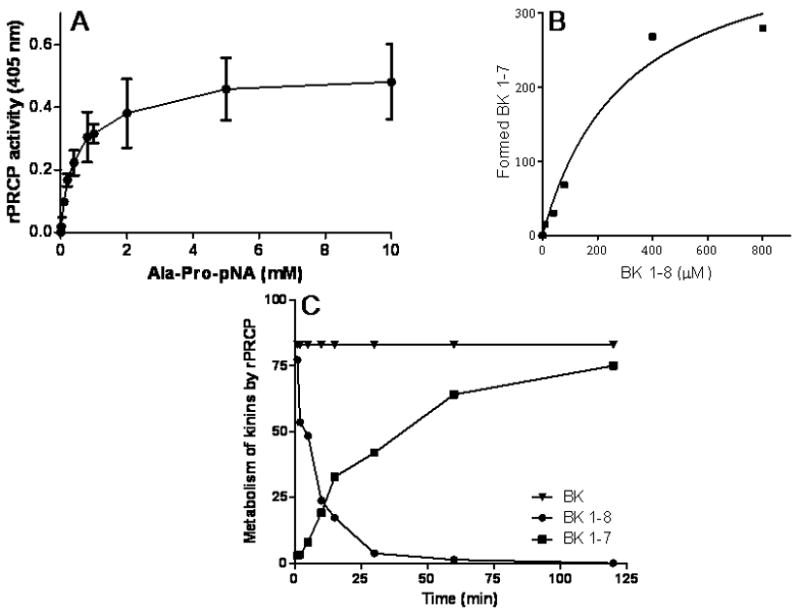
The correlation between the reaction rate and the substrate concentration. Panel A. rPRCP was incubated with the increasing concentrations of Ala-Pro-p nitroanilide (Ala-Pro-pNA) for 1 hour at 37 °C. The hydrolysis of Ala-Pro-pNA was detected at 405 nm. The slope of the dose-response curve is linear from 0.1 mM to 1 mM. Panel B. Dose-response curve for bradykinin 1–8 (BK 1–8) doses. rPRCP was incubated with the increasing concentrations of BK 1–8 as indicated in the figure. The generation of BK 1–7 was quantified by LC-MS. Panel C. Time-course of BK 1–8 metabolism and BK 1–7 formation. rPRCP was incubated with BK 1–8 at various time interval at 37 °C. Using BK 1–7 as a standard, the generation of BK 1–7 from BK 1–8 by rPRCP was determined by LC/MS apparatus.
Further studies determined the time-dependant BK 1–8 metabolism by rPRCP using LC/MS. Samples were then collected at different time points. Bradykinin 1–7 [1Arg-Pro-Pro-GlyPhe-Ser-Pro7 (BK1–7)] was used as a standard. BK1–7 was detected in the sample within 2 min following incubation of BK1–8 with rPRCP along with a concomitant reduction of the amount of BK1–8, Figure 1C. rPRCP generated a rapid burst of BK1–7 production, and peak effect occurred within 40 to 60 minutes. Further studies were performed to determine the products of BK 1–9 metabolism by rPRCP using LC/MS. rPRCP was ineffective in metabolizing BK 1–9. Taken together, metabolism of BK 1–8 by rPRCP was time-dependent.
Kinetic constants (KM, kcat) for the hydrolysis of BK1–8, and Ala-Pro-pNA are tabulated in Table 1. The study revealed that the KM values for the hydrolysis of Ala-Pro-pNA and BK 1–8 are 476 μM and 306 μM respectively. The value of kcat for BK1–8 (622 min−1) was not significantly larger than that for Ala-Pro-pNA (521± 53 min−1). However, rPRCP was two-fold more efficient at the liberating Phe residue from BK 1–8 than at the splitting off the paranitroaniline from Ala-Pro-pNA.
Table 1.
rPRCP substrate specificity
| Substrate | Km (μmol/L) | Kcat (min−1) | Kcat/Km (L.μmol−1.min−1) |
|---|---|---|---|
| BK1–9 | ns | - | - |
| BK1–8 | 306 | 622 | 2.04 |
| Ala-Pro-p-nitroanilide | 476 ± 40 | 521± 53 | 1.09 ± 0.03 |
Bradykinin generation on HPAEC
Studies were performed to determine the activation of the bradykinin-forming pathway on HPAEC. As shown in Figure 2A, the activation of the complex of HK/PK on endothelium is dose-dependent, confirming a previously described observation[33]. Next, we determined whether the human anti-PRCP antibody would block this bradykinin-forming pathway on cells. The human anti-PRCP antibody strongly inhibited kallikrein generation on HPAEC, suggesting that PRCP activates PK to kallikrein, Figure 2B.
Figure 2.
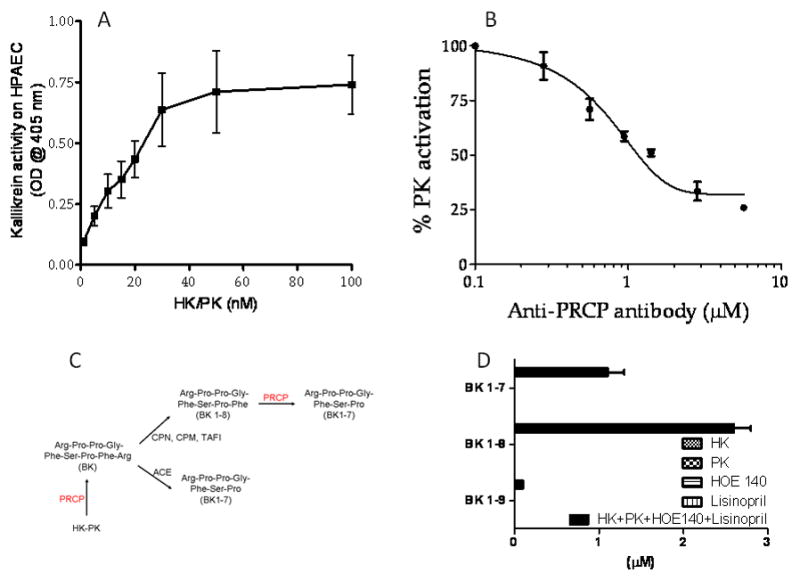
Panel A. Prekallikrein activation on endothelial cells. Pulmonary artery endothelial cells (HPAEC, 3×104 cells/well) were platted and cultured in 96 well plates. Cells were incubated with increasing concentrations of HK and PK. Panel B. Effect of anti-PRCP antibody on PK activation on HPAEC. Triplicate samples of cells were incubated with increasing concentrations of anti-PRCP antibody in the absence or presence of the complex of HK and PK. The control was without anti-PRCP antibody. Panel C. Schematic illustration of the pathways involved in the conversion of bradykinin to its respective metabolites. Panel D. The generation of BK 1–9, BK 1–8, and BK 1–7 were determined on HPAEC by LC/MS. The negative control was HK, a BK precursor.
Since BK generation is mediated by kallikrein, an approach to the determination of BK levels would be to assess PRCP-dependent PK activation at the cellular level. We determined the generation of kinins (BK 1–9, BK 1–8, BK 1–7) after the activation of the HK/PK complex on endothelial cells. To improve detection of kinins on cells, we selectively blocked degradation of bradykinin with an angiotensin converting enzyme (ACE) inhibitor and a specific inhibitor of the bradykinin B2 receptor (B2), Figure 2C. HPAECs were incubated with the complex of HK-PK (600 nM each) in the presence of lisinopril (30 μM, an ACE inhibitor) and HOE 140 (10 μM, a B2 antagonist) for 1h at 37 °C. For the first time, the level of kinins in the concentrated supernatant (six-fold) from three wells of the cultured HPAEC was monitored and quantified by LC-MS. As shown in Figure 2D, the assembly and activation of complex of HK/PK on HPAEC leads to generation of BK 1–9, BK 1–8, and BK 1–7 into the supernatant of HPAEC. No kinin was generated when cells were treated only with HK (600 nM) or PK (600 nM) alone. Similarly, the chemical signature of HOE 140 (10 μM) or lisinopril (30 μM) were also determined by LC-MS. The retention times of BK 1–8 and its metabolites were unaffected by HOE 140 or lisinopril. Together these novel data indicate that PRCP converts BK 1–8 to BK 1–7 on the cell surface.
Docking of Bradykinin Peptides
BK1–9 and BK 1–8 were treated with the IFD workflow provided by Maestro. The putative loop comprising residues E348–S353 presents a dilemma when assessing docking modes. The loop may be oriented so that the cavity leading to the active site residues is aligned roughly parallel to the plane of the β-sheets. Alternatively, the loop could also be oriented so that access to the active site residues must occur through an axis perpendicular to the β-sheets. An additional consideration is that any proteolytic mechanism requires that the scissile bond be located proximal to the active site.
Substrate access to the active site
Notably, none of the BK1–9 structures were successfully docked. While there are two potential ways to access the active site residues, BK1–8 appears to bind through the long groove between the SKS and hydrolase domains[34].
Hydrogen bonding interactions
1Arg of docked BK1–8 interacts with the acid side chain of D277 via the Nω (1.768 Å) of the guanidinium group, while the N-terminus appears to interact with the side chain acid of D393 (1.905 Å). Additional interactions occur between the backbone nitrogen of 4Phe and the side-chain of T356 (1.934 Å), and between the 5Ser β-OH and the backbone carbonyl of E93 (1.912 Å). The C-terminal carboxyl group of BK1–8 interacts with the Nω of R460 (1.889 Å), and with Nτ of H455 (1.517 Å) and H456 (1.802 Å). R460 and H456 hydrogen-bond to a common oxygen on the acid, whereas H455 hydrogen-bonds to the remaining oxygen. The presence of the basic R460 in the S1’ subsite may help to explain the substrate selectivity of BK1–8 over BK1–9, due to the potential coulombic repulsion of this residue with the C-terminal arginine of BK1–9. The scissile carbonyl lies 4.456 Å from the β-OH of S179, which suggests that the proteolytic mechanism is mediated through the activation of a water molecule. A listing of potential van der Waals contacts is provided in Table 2.
Table 2.
Van der Waals contacts of BK1–8 with PRCP.
| Subsite | Residues |
|---|---|
| S1' | L298, F297, V289, M288, W98, H455, R460, H456, S179 |
| S1 | M288, S179, W432, W359 |
| S2 | W356, E93, G94, G355 |
| S3 | L298, T356, W359 |
| S4 | T356 |
| S5 | S360, A363, L390 |
| S6 | S360, Q397, C364, C394, L390, D393 |
| S7 | D277 |
Discussion
Prolylcarboxypeptidase (PRCP, EC 3.4.16.2), a serine protease, liberates an amino acid residue at the C-terminus of relatively small peptides[17;35]. PRCP metabolizes peptides such as angiotensin II, angiotensin III, and alpha-melanocyte stimulating hormone (α-MSH). PRCP also activates the cell matrix-associated prekallikrein[36]. Thus, PRCP has mixed substrate specificity, suggesting that PRCP is able to generate several distinct and physiologically important products.
The present study determined: 1) the kinetic parameters of the kinin hydrolysis reaction by LC-MS, 2) the correlation between KM and kcat values and the substrate structure, and 3) the IC50 for the inhibition of rPRCP by the specific anti-PRCP antibody. 4) We developed a simple and an accurate functional assay to measure BK and its metabolites along the surface of endothelial cells as well as in the presence of rPRCP. 5) An induced-fit docking study of BK peptides with the crystal structure of PRCP provided several poses of BK1–8 which putatively explains the selectivity for BK1–8 over BK1–9.
Studies determined whether rPRCP would have the ability to enrich specific markers, namely, BK 1–7 from either BK 1–9 or BK 1–8. It was observed that rPRCP metabolizes BK 1–8, whereas the metabolism of BK 1–9 was unaffected by the enzyme. The inability of rPRCP to metabolize BK 1–9 is remarkable. This observation indicates that PRCP has a narrow substrate selectivity profile. We propose that the potential coulombic repulsion of the basic R460 in the S1’ subsite with the C-terminal arginine of BK1–9 may explain the selectivity of PRCP to its substrates. rPRCP converted BK 1–8 to BK 1–7. The increase in rPRCP concentration correlated well with the increase in the generation of BK 1–7. The hydrolysis of BK 1–8 by rPRCP was dose - and time-dependent. Notably, PRCP not only activates PK to kallikrein but also metabolizes BK 1–8 to BK 1–7 on HPAEC. The results suggest that PRCP has both proinflammatory and anti-inflammatory actions.
In the present study, we have also used a computational approach to predict the structure of human PRCP, and serendipitously, a crystal structure of PRCP was disclosed. Due to the initial failure of the homology model and induced fit docking (IFD) to provide a docked structure of BK1–8, molecular dynamics (MD) was used to relax the structure for further normal mode analysis (NMA) and induced fit docking of bradykinin oligopeptides (see Supplementary Data). We initially used DPP7 as a structural template for homology modeling. In theory, this approach would result in a valid structural model because PRCP has 40% amino-acid primary sequence identity to DPP7, and the crystal structure of DPP7 was previously disclosed[37;38]. Following the release of the PRCP crystal structure, the docking studies were performed again following a much simpler protocol. The discrepancies between the initial and follow-up studies may serve as a pedagogical case-study into the suitability of homology modeling, with and without refinement, for docking oligopeptides into uncharacterized protein structures.
The proteolytic mechanism could involve either the classical acylation–deacylation mechanism of the Ser-His-Asp triad, or possibly through a mechanism involving Ser-His-His,[39] given that two histidines are found neighboring each other in PRCP. Oxyanion stabilization of the tetrahedral intermediate could involve the backbone of E93, along with either the side chain carboxyl of E93, or potentially the 4-OH of Y180, in a manner analogous to prolyl oligopeptidase[40]. The structural studies presented here enabled us to predict a putative BK 1–8 docking mode in the active site of PRCP. Moreover, these structure-function studies provide insights into the framework for the design and interpretation of ongoing studies on homodimerization, substrate specificities, identification of structurally and functionally important amino acid residues, and its post-translational regulation.
In conclusion, our data presented here strongly indicate that PRCP targets BK1–8 but not BK1–9 and implies a key regulatory role of PRCP for the kinin-mediated process. One interpretation of these data is that PRCP protects chronically inflamed tissues by inactivating BK1–8. PRCP appears to have a critical role in the inflammatory cascade. Further study on the elucidation of the role of PRCP in vivo in patients with chronic inflammation is of great importance.
Supplementary Material
Figure 3.
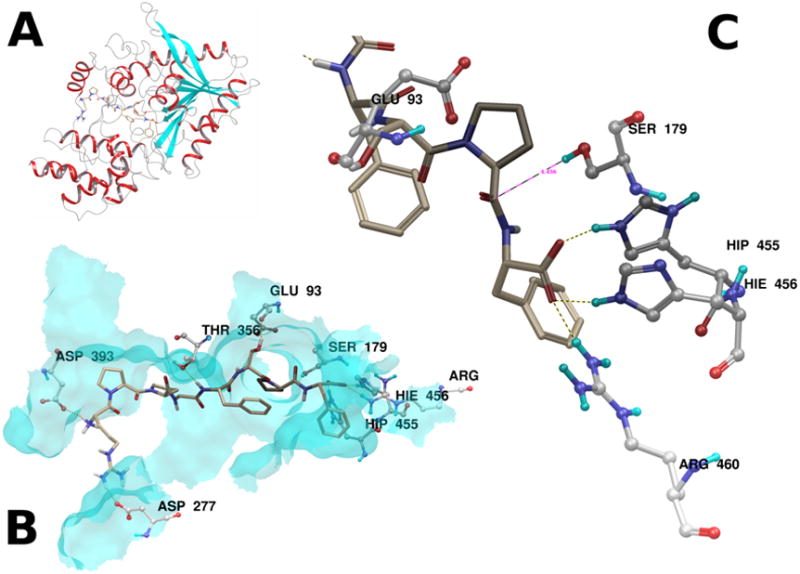
Panel A. PRCP is shown as a ribbon diagram, with the top ranked pose of BK1–8 docked into the active site groove. Panel B. The major hydrogen bonding interactions between BK1–8 and the top ranked pose are illustrated. Panel C. The scissile Pro-Phe amide bond lies 4.456 Å from the side chain oxygen of S179. Hydrogen bonding interactions from the active site histidines H455 and H456, as well as R460 stabilize the C-terminal acid. The proximity of the side chain acid of G93 suggests a possible role in the proteolytic mechanism.
Acknowledgments
This work was supported by National Science Foundation [MRI 0619774] and National Institute of Health [NCRR/NIH P20RR021929] to ZSM
Abbreviations
- PRCP
Prolylcarboxypeptidase
- DPP7
dipeptidyl-peptidase 7
- BK 1–8
Bradykinin 1–8
- HK
high molecular weight kininogen
- PK
prekallikrein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Wilkinson-Berka JL, Fletcher EL. Angiotensin and bradykinin: targets for the treatment of vascular and neuro-glial pathology in diabetic retinopathy. Curr Pharm Des. 2004;10:3313–3330. doi: 10.2174/1381612043383179. [DOI] [PubMed] [Google Scholar]
- 2.Campbell DJ. The renin-angiotensin and the kallikrein-kinin systems. Int J Biochem Cell Biol. 2003;35:784–791. doi: 10.1016/s1357-2725(02)00262-5. [DOI] [PubMed] [Google Scholar]
- 3.Wilkinson-Berka JL, Fletcher EL. Angiotensin and bradykinin: targets for the treatment of vascular and neuro-glial pathology in diabetic retinopathy. Curr Pharm Des. 2004;10:3313–3330. doi: 10.2174/1381612043383179. [DOI] [PubMed] [Google Scholar]
- 4.Fabiani ME, Dinh DT, Story DF. Interaction of the renin-angiotensin system, bradykinin and sympathetic nerves with cholinergic transmission in the rat isolated trachea. Br J Pharmacol. 1997;122:1089–1098. doi: 10.1038/sj.bjp.0701499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campbell DJ. The kallikrein-kinin system in humans. Clin Exp Pharmacol Physiol. 2001;28:1060–1065. doi: 10.1046/j.1440-1681.2001.03564.x. [DOI] [PubMed] [Google Scholar]
- 6.Allikmets K, Parik T, Viigimaa M. The renin-angiotensin system in essential hypertension: associations with cardiovascular risk. Blood Press. 1999;8:70–78. doi: 10.1080/080370599438239. [DOI] [PubMed] [Google Scholar]
- 7.Castro-Chaves P, Pintalhao M, Fontes-Carvalho R, Cerqueira R, Leite-Moreira AF. Acute modulation of myocardial function by angiotensin 1–7. Peptides. 2009;30:1714–1719. doi: 10.1016/j.peptides.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 8.Hisada Y, Sugaya T, Yamanouchi M, Uchida H, Fujimura H, Sakurai H, Fukamizu A, Murakami K. Angiotensin II plays a pathogenic role in immune-mediated renal injury in mice. J Clin Invest. 1999;103:627–635. doi: 10.1172/JCI2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geara AS, Azzi J, Jurewicz M, Abdi R. The renin-angiotensin system: an old, newly discovered player in immunoregulation. Transplant Rev (Orlando ) 2009;23:151–158. doi: 10.1016/j.trre.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 10.Ager EI, Neo J, Christophi C. The renin-angiotensin system and malignancy. Carcinogenesis. 2008;29:1675–1684. doi: 10.1093/carcin/bgn171. [DOI] [PubMed] [Google Scholar]
- 11.Daniels MD, Hyland KV, Engman DM. Treatment of experimental myocarditis via modulation of the renin-angiotensin system. Curr Pharm Des. 2007;13:1299–1305. doi: 10.2174/138161207780618812. [DOI] [PubMed] [Google Scholar]
- 12.Duke LM, Eppel GA, Widdop RE, Evans RG. Disparate roles of AT2 receptors in the renal cortical and medullary circulations of anesthetized rabbits. Hypertension. 2003;42:200–205. doi: 10.1161/01.HYP.0000083341.64034.00. [DOI] [PubMed] [Google Scholar]
- 13.Sleat DE, Wang Y, Sohar I, Lackland H, Li Y, Li H, Zheng H, Lobel P. Identification and validation of mannose 6-phosphate glycoproteins in human plasma reveal a wide range of lysosomal and non-lysosomal proteins. Mol Cell Proteomics. 2006;5:1942–1956. doi: 10.1074/mcp.M600030-MCP200. [DOI] [PubMed] [Google Scholar]
- 14.Skidgel RA, Erdos EG. Cellular carboxypeptidases. Immunol Rev. 1998;161:129–141. doi: 10.1111/j.1600-065x.1998.tb01577.x. [DOI] [PubMed] [Google Scholar]
- 15.Mallela J, Perkins R, Yang J, Pedigo S, Rimoldi JM, Shariat-Madar Z. The functional importance of the N-terminal region of human prolylcarboxypeptidase. Biochem Biophys Res Commun. 2008;374:635–640. doi: 10.1016/j.bbrc.2008.07.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hooley E, McEwan PA, Emsley J. Molecular modeling of the prekallikrein structure provides insights into high-molecular-weight kininogen binding and zymogen activation. J Thromb Haemost. 2007;5:2461–2466. doi: 10.1111/j.1538-7836.2007.02792.x. [DOI] [PubMed] [Google Scholar]
- 17.Shariat-Madar Z, Mahdi F, Schmaier AH. Identification and characterization of prolylcarboxypeptidase as an endothelial cell prekallikrein activator. J Biol Chem. 2002;277:17962–17969. doi: 10.1074/jbc.M106101200. [DOI] [PubMed] [Google Scholar]
- 18.Wu Y, Dai J, Schmuckler NG, Bakdash N, Yoder MC, Overall CM, Colman RW. Cleaved high molecular weight kininogen inhibits tube formation of endothelial progenitor cells via suppression of matrix metalloproteinase 2. J Thromb Haemost. 2009 doi: 10.1111/j.1538-7836.2009.03662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwaki T, Castellino FJ. Plasma levels of bradykinin are suppressed in factor XII-deficient mice. Thromb Haemost. 2006;95:1003–1010. doi: 10.1160/TH06-03-0128. [DOI] [PubMed] [Google Scholar]
- 20.Zhu L, Carretero OA, Liao TD, Harding P, Li H, Sumners C, Yang XP. Role of prolylcarboxypeptidase in angiotensin II type 2 receptor-mediated bradykinin release in mouse coronary artery endothelial cells. Hypertension. 2010;56:384–390. doi: 10.1161/HYPERTENSIONAHA.110.155051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Isordia-Salas I, Sainz IM, Pixley RA, Martinez-Murillo C, Colman RW. High molecular weight kininogen in inflammation and angiogenesis: a review of its properties and therapeutic applications. Rev Invest Clin. 2005;57:802–813. [PubMed] [Google Scholar]
- 22.Sharma JN, Al-Sherif GJ. Pharmacologic targets and prototype therapeutics in the kallikrein-kinin system: bradykinin receptor agonists or antagonists. ScientificWorldJournal. 2006;6:1247–1261. doi: 10.1100/tsw.2006.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feterik K, Smith L, Katusic ZS. Angiotensin-(1–7) causes endothelium-dependent relaxation in canine middle cerebral artery. Brain Res. 2000;873:75–82. doi: 10.1016/s0006-8993(00)02482-3. [DOI] [PubMed] [Google Scholar]
- 24.Sharma JN. Hypertension and the bradykinin system. Curr Hypertens Rep. 2009;11:178–181. doi: 10.1007/s11906-009-0032-7. [DOI] [PubMed] [Google Scholar]
- 25.Dharmani M, Mustafa MR, Achike FI, Sim MK. Effects of angiotensin 1–7 on the actions of angiotensin II in the renal and mesenteric vasculature of hypertensive and streptozotocin-induced diabetic rats. Eur J Pharmacol. 2007;561:144–150. doi: 10.1016/j.ejphar.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 26.Hoffmann G, Dusing R. ACE-inhibition, kinins, and vascular PGI2 synthesis. Eicosanoids. 1992;5(Suppl):S60–S62. [PubMed] [Google Scholar]
- 27.Mallela J, Yang J, Shariat-Madar Z. Prolylcarboxypeptidase: a cardioprotective enzyme. Int J Biochem Cell Biol. 2009;41:477–481. doi: 10.1016/j.biocel.2008.02.022. [DOI] [PubMed] [Google Scholar]
- 28.Schmaier AH. The plasma kallikrein-kinin system counterbalances the renin-angiotensin system. J Clin Invest. 2002;109:1007–1009. doi: 10.1172/JCI15490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deddish PA, Skidgel RA, Kriho VB, Li XY, Becker RP, Erdos EG. Carboxypeptidase M in Madin-Darby canine kidney cells. Evidence that carboxypeptidase M has a phosphatidylinositol glycan anchor. J Biol Chem. 1990;265:15083–15089. [PubMed] [Google Scholar]
- 30.Shariat-Madar Z, Mahdi F, Schmaier AH. Recombinant prolylcarboxypeptidase activates plasma prekallikrein. Blood. 2004;103:4554–4561. doi: 10.1182/blood-2003-07-2510. [DOI] [PubMed] [Google Scholar]
- 31.Ngo ML, Mahdi F, Kolte D, Shariat-Madar Z. Upregulation of prolylcarboxypeptidase (PRCP) in lipopolysaccharide (LPS) treated endothelium promotes inflammation. J Inflamm (Lond) 2009;6:3. doi: 10.1186/1476-9255-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cui L, Nithipatikom K, Campbell WB. Simultaneous analysis of angiotensin peptides by LC-MS and LC-MS/MS: metabolism by bovine adrenal endothelial cells. Anal Biochem. 2007;369:27–33. doi: 10.1016/j.ab.2007.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao Y, Qiu Q, Mahdi F, Shariat-Madar Z, Rojkjaer R, Schmaier AH. Assembly and activation of HK-PK complex on endothelial cells results in bradykinin liberation and NO formation. Am J Physiol Heart Circ Physiol. 2001;280:H1821–H1829. doi: 10.1152/ajpheart.2001.280.4.H1821. [DOI] [PubMed] [Google Scholar]
- 34.Soisson SM, Patel SB, Abeywickrema PD, Byrne NJ, Diehl RE, Hall DL, Ford RE, Reid JC, Rickert KW, Shipman JM, Sharma S, Lumb KJ. Structural definition and substrate specificity of the S28 protease family: the crystal structure of human prolylcarboxypeptidase. BMC Struct Biol. 2010;10:16. doi: 10.1186/1472-6807-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao X, Southwick K, Cardasis HL, Du Y, Lassman ME, Xie D, El-Sherbeini M, Geissler WM, Pryor KD, Verras A, Garcia-Calvo M, Shen DM, Yates NA, Pinto S, Hendrickon RC. Peptidomic profiling of human cerebrospinal fluid identifies YPRPIHPA as a novel substrate for prolylcarboxypeptidase. Proteomics. 2010 doi: 10.1002/pmic.201000145. [DOI] [PubMed] [Google Scholar]
- 36.Moreira CR, Schmaier AH, Mahdi F, da MG, Nader HB, Shariat-Madar Z. Identification of prolylcarboxypeptidase as the cell matrix-associated prekallikrein activator. FEBS Lett. 2002;523:167–170. doi: 10.1016/s0014-5793(02)02980-0. [DOI] [PubMed] [Google Scholar]
- 37.Burley SK. An overview of structural genomics. Nat Struct Biol. 2000;7(Suppl):932–934. doi: 10.1038/80697. [DOI] [PubMed] [Google Scholar]
- 38.Ginalski K, Grishin NV, Godzik A, Rychlewski L. Practical lessons from protein structure prediction. Nucleic Acids Res. 2005;33:1874–1891. doi: 10.1093/nar/gki327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu M, Bayjanov JR, Renckens B, Nauta A, Siezen RJ. The proteolytic system of lactic acid bacteria revisited: a genomic comparison. BMC Genomics. 2010;11:36. doi: 10.1186/1471-2164-11-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fulop V, Bocskei Z, Polgar L. Prolyl oligopeptidase: an unusual beta-propeller domain regulates proteolysis. Cell. 1998;94:161–170. doi: 10.1016/s0092-8674(00)81416-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.