

Abstract
Voltage-gated sodium channels are the primary target of pyrethroids, an important class of synthetic insecticides. Pyrethroids bind to a distinct receptor site on sodium channels and prolong the open state by inhibiting channel deactivation and inactivation. Recent studies have begun to reveal sodium channel residues important for pyrethroid binding. However, how pyrethroid binding leads to inhibition of sodium channel deactivation and inactivation remains elusive. In this study, we show that a negatively charged aspartic acid residue at position 802 (D802) located in the extracellular end of transmembrane segment 1 of domain II (IIS1) is critical for both the action of pyrethroids and the voltage dependence of channel activation. Charge-reversing or -neutralizing substitutions (K, G, or A) of D802 shifted the voltage dependence of activation in the depolarizing direction and reduced channel sensitivity to deltamethrin, a pyrethroid insecticide. The charge-reversing mutation D802K also accelerated open-state deactivation, which may have counteracted the inhibition of sodium channel deactivation by deltamethrin. In contrast, the D802G substitution slowed open-state deactivation, suggesting an additional mechanism for neutralizing the action of deltamethrin. Importantly, Schild analysis showed that D802 is not involved in pyrethroid binding. Thus, we have identified a sodium channel residue that is critical for regulating the action of pyrethroids on the sodium channel without affecting the receptor site of pyrethroids.
Keywords: Pyrethroid insecticide, Sodium channel, Activation, Deactivation
Introduction
Voltage-gated sodium channels are integral transmembrane proteins responsible for the rapidly rising phase of action potentials and are critical for electrical signaling in most excitable cells (Catterall, 2000). Because of their crucial role in membrane excitability, sodium channels are targeted by a variety of neurotoxins from animals and plants, therapeutic drugs, and synthetic chemical insecticides (Cestele and Catterall, 2000; Wang and Wang, 2003). The pore-forming α-subunit of the sodium channel contains four homologous domains (I–IV), each formed by six membrane spanning segments (S1–S6) connected by intracellular and extracellular loops. The S1–S4 segments serve as the voltage-sensing module, whereas the S5 and S6 segments and the loops connecting them function as the pore-forming module. In response to depolarization, S4s move outward across the membrane, which triggers a series of conformational changes in the sodium channel protein, resulting in opening of the sodium channel pore (i.e., activation) (Catterall, 1992). Upon repolarization, the S4 segments move inward and the channels return to the closed state, a process known as deactivation.
Pyrethroid insecticides are a large class of synthetic insecticides derived structurally from the natural pyrethrins extracted from flowers of certain Chrysanthemum species. They represent one of the most important classes of insecticides used to control medically and agriculturally important pests worldwide. Electrophysiological studies have long established that pyrethroids exert their insecticidal activity by inhibiting channel deactivation and inactivation, resulting in prolonged opening of sodium channels, as evident in the prominent tail currents observed upon repolarization of membrane potential (Narahashi, 1988, 2000). Salgado and Narahashi (1993) provided the initial electrophysiological evidence that pyrethroids trap sodium channels in the open state in crayfish giant axons. However, almost nothing is known about the molecular mechanism by which pyrethroids inhibit sodium channel deactivation at the molecular level. Furthermore, little is known about the molecular features critical for deactivation. Therefore, understanding the molecular mechanism underlying the action of pyrethroids not only is important from a practical standpoint but also provides insight into the fundamental gating mechanisms critical for sodium channel function. Here we report that an aspartic acid residue (D802) located in domain II transmembrane segment 1 (IIS1) of the cockroach sodium channel (BgNav) is critical for channel gating and the action of pyrethroids.
Materials and methods
Site-directed mutagenesis
Site-directed mutagenesis was performed by PCR using mutant primers and Pfu Turbo DNA polymerase (Stratagene, La Jolla, CA). All mutagenesis results were verified by DNA sequencing.
Expression of BgNav sodium channels in Xenopus oocytes
The procedures for oocyte preparation and cRNA injection are identical to those described previously (Tan et al., 2002). For robust expression of the BgNav sodium channels, cRNA was coinjected into oocytes with Drosophila melanogaster tipE cRNA (1:1 ratio), which enhances the expression of insect sodium channels in oocytes (Feng et al., 1995; Warmke et al., 1997).
Electrophysiological recording and analysis
The voltage dependence of activation and inactivation was measured using the two-electrode voltage clamp technique. Methods for two-electrode recording and data analysis were similar to those described previously (Tan et al., 2005). Sodium currents were measured with a Warner OC725C oocyte clamp (Warner Instrument, Hamden, CT) and processed with a Digidata 1322A interface (Axon Instruments Inc., Foster City, CA). Data were sampled at 50 kHz and filtered at 2 kHz. Leak currents were corrected by p/4 subtraction. pClamp 8.2 software (Axon Instruments Inc., CA) was used for data acquisition and analysis. The maximal peak sodium current was limited to <2.0 μA to achieve optimal voltage control by adjusting the amount of cRNA and the incubation time after injection.
The voltage dependence of sodium channel conductance (G) was calculated by measuring the peak current at test potentials ranging from −80 mV to +65 mV in 5-mV increments and divided by (V−Vrev), where V is the test potential and Vrev is the reversal potential for sodium ion. Peak conductance values were normalized to the maximal peak conductance (Gmax) and fitted with a two-state Boltzmann equation of the form G/Gmax=[1+exp (V−V1/2)/k]−1, in which V is the potential of the voltage pulse, V1/2 is the voltage for half-maximal activation, and k is the slope factor.
The voltage dependence of sodium channel inactivation was determined by using 100-ms inactivating prepulses ranging from −120 mV to 0 mV in 5-mV increments from a holding potential of −120 mV, followed by test pulses to −10 mV for 20 ms. The peak current amplitude during the test depolarization was normalized to the maximum current amplitude and plotted as a function of the prepulse potential. Data were fitted with a two-state Boltzmann equation of the form I/Imax=[1+(exp(V−V1/2)/k)]−1, in which I is the peak sodium current, Imax is the maximal current evoked, V is the potential of the voltage prepulse, V1/2 is the half-maximal voltage for inactivation, and k is the slope factor.
Cell-attached macropatch recording and analysis
Methods for cell-attached macropatch recordings and data analysis are similar to those described in Groome et al. (1999). For cell-attached macropatch recordings, voltage clamping and data acquisition were done using an EPC-9 patch-clamp amplifier (HEKA, Lambrecht, Germany) controlled via Pulse 8.67 software (HEKA). Data were acquired at 10 μs per point and low-pass-filtered at 3 kHz during acquisition. All experiments were run at 20 °C. Voltage clamp protocols were run from a holding potential of −150 mV. Leak subtraction was performed automatically by the software using a p/4 protocol. Subsequent analyses and graphing were done using PulseFit (HEKA) and Igor Pro (Wavemetrics, Lake Oswego, OR). Activation kinetics were measured from the rising phase (10% to 90% of peak current) of sodium current during 20-ms step commands to voltages ranging from −45 to +30 mV in 5-mV increments. To measure open-state deactivation, channels were opened with brief depolarization with (0 mV, 0.2 ms), and tail currents were elicited by hyperpolarizing commands to voltages ranging from −180 to −40 mV. Deactivation time constants (τD) were derived from the monoexponential decay of tail currents according to the function: I(t)=offset+a1exp(−t/τD), where I(t) is current amplitude as a function of time, offset is the plateau amplitude (asymptote), a1 is current amplitude at time 0, and ττD is the time constant.
Measurement of tail currents induced by pyrethroids
The method for application of pyrethroids in the recording system was identical to that described by Tan et al. (2002). The effects of pyrethroids were measured 10 min after toxin application. The pyrethroid-induced tail current was recorded during a 100-pulse train of 5-ms step depolarizations from −120 to 0 mV with 5-ms interpulse intervals (Vais et al., 2000). The percentage of channels modified by pyrethroids was calculated using the equation M={[Itail/(Eh −ENa)]/[INa/(Et−ENa)]}×100 (Tatebayashi and Narahashi, 1994), where Itail is the maximal tail current amplitude, Eh is the potential to which the membrane is repolarized, ENa is the reversal potential for sodium current determined from the current–voltage curve, INa is the amplitude of the peak current during depolarization before pyrethroid exposure, and Et is the potential of step depolarization. Dose–response curves were fitted to the Hill equation: M=Mmax/{1+(Kd/[P])n}, in which [P] represents the concentration of pyrethroid, Kd represents the concentration of pyrethroid that produced the half-maximal effect, n represents the Hill coefficient, and Mmax is the maximal percentage of sodium channel modified.
Schild analysis
Schild analysis is similar to that described in Tan et al. (2005). Kd values were determined from the dose–response curves of the active 1R-cis-isomer on sodium channels by measuring the amplitude of the tail current induced by the 1R-cis-isomer and calculating the percentage of channel modification as described above. A series of Kd values (denoted as Kd′) of the active 1R-cis-isomer were determined in the presence of increasing concentrations of the inactive 1S-cis-isomer. Schild analysis was used to determine the affinity of the inactive isomer, calculated from the equation log (dose ratio−1)=logKB−log[B], where dose ratio=Kd′/Kd, [B] is the molar concentration of the inactive 1S-cis-isomer, and KB is the dissociation constant of the inactive 1S-cis-isomer. Log (dose ratio −1) was plotted as a function of −log[B]. The data were fitted with a linear regression, generating the Schild plot slope and the x-intercept, pA2, which equals −logKB.
Results are reported as mean±SEM. Statistical significance was determined by using one-way ANOVA with Scheffe’s post hoc analysis, and significant values were set at p<0.05 or as indicated in the table and figure legends.
Results
The D802G substitution in IIS1 reduces BgNav channel sensitivity to deltamethrin
In a previous study, we identified two cockroach sodium channel variants, BgNav1-1 and BgNav2-1, with distinct functional and pharmacological properties (Tan et al., 2002). Specifically, BgNav2-1 is 100-fold more resistant to the pyrethroid insecticide deltamethrin than BgNav1-1. The differential pyrethroid sensitivities between BgNav1-1 and BgNav2-1 channels are partially attributable to the presence of exon G1 and G2, respectively (Tan et al., 2002). A V1356A change in exon G2 reduces the sensitivity of BgNav2-1 channels to pyrethroids (Du et al., 2006). In addition to the selective inclusion of exon G1 or G2, BgNav1-1 and BgNav2-1 also contain nine amino acid differences scattered throughout the protein. To determine whether any of these amino acid differences are involved in the differential sensitivity of BgNav1-1 and BgNav2-1 to deltamethrin, reciprocal site-directed mutagenesis was performed in the two channels. The sensitivities of the resulting recombinant channels to deltamethrin were evaluated by measuring deltamethrin-induced tail currents and determining the percentages of channel modification by deltamethrin. These analyses revealed that of the nine amino acid substitutions, only the D (in BgNav1-1) to G (in BgNav2-1) change at position 802, contributed significantly to the reduced sensitivity of the BgNav2-1 channel to deltamethrin (Fig. 1). The values of EC20 are 0.04 μM, 0.48 μM, 0.51 μM, and 12.4 μM for BgNav1-1, BgNav1-1D802G, BgNav2-1G802D, and BgNav2-1, channels, respectively. BgNav1-1D802G was 10-fold more resistant to deltamethrin than BgNav1-1. Conversely, the G-to-D substitution in BgNav2-1 channels (i.e., BgNav2-1G802D) increased channel sensitivity to deltamethrin by 20-fold (Fig. 1B). The D802G substitution also reduced BgNav1-1 channel sensitivity to permethrin, a type I pyrethroid (Fig. 1C).
Fig. 1.

The D802G change in the BgNav2-1 channel is partially responsible for the resistance of BgNav2-1 to deltamethrin. A, Tail currents induced by deltamethrin in BgNav2-1G802D, BgNav2-1, BgNav1-1, and BgNav1-1D802G channels. The tail current was elicited by a 67-Hz train of 100 5-ms depolarization from −120 to 0 mV. B and C, Percentage of channel modification by deltamethrin (B) and permethrin (C). Percentages of channels modified by pyrethroids was calculated using the equation M={[Itail/(Eh−ENa)]/[INa/(Et−ENa)]}×100 (Tatebayashi and Narahashi, 1994),where Itail is the maximal tail current amplitude, Eh is the potential to which the membrane is repolarized, ENa is the reversal potential for sodium current determined from the current–voltage curve, INa is the amplitude of the peak current during depolarization before pyrethroid exposure, and Et is the potential of step depolarization. The values of EC20 for deltamethrin are 0.04 μM, 0.48 μM, 0.51 μM, and 12.4 μM for BgNav1-1, BgNav1-1D802G, BgNav2-1G802D, BgNav2-1 channels, respectively. The values of EC20 for permethrin are 0.03 μM and 0.25 μM for BgNav1-1 and BgNav1-1D802G, respectively.
The decay of the tail current from oocytes expressing BgNav1-1 channels was slow and biphasic, which is typical of type II-pyrethroid induced tail currents. In contrast, the deltamethrin-induced tail current of BgNav2-1 channels exhibited a very rapid and monophasic decay (Fig. 1A and Table 1). This striking difference in tail current kinetics can largely be explained by the difference (D or G) in amino acid at position 802 between the two variants. The rate of the decay of the deltamethrin-induced tail current of BgNav2-1G802D was significantly slower than BgNav2-1 and exhibited two components. However, the decay rate for BgNav1-1D802G is not significantly different from that of BgNav1-1 and maintained as two components (Fig. 1A and Table 1).
Table 1.
Time constants of decay of tail currents induced by deltamethrin.
| Na+ channel type |
Deltamethrina |
||
|---|---|---|---|
| τ1 (s) | τ2 (s) | Nb | |
| BgNav2-1 | 0.88±0.17 | 25 | |
| BgNav2-1G802D | 1.33±0.31* | 0.35±0.07 | 8 |
| BgNav1-1 | 1.56±0.41 | 0.35±0.08 | 15 |
| BgNav1-1D802G | 1.37±0.16 | 0.26±0.06 | 10 |
Deltamethrin-induced tail currents were fit with double exponential equations except for tail currents of BgNav2-1 which were fit with single exponential. Each value represents the mean±SEM.
Number of oocytes.
Significantly different from the BgNav2-1 channel using one-way ANOVA with Scheffe’s post hoc analysis (p<0.05).
D802G does not alter pyrethroid binding to the BgNav1-1 channel
The effect of D802G on sensitivity to pyrethroids could be the result of an effect either at the binding site or during a binding-independent stage of pyrethroid action. To distinguish between the two mechanisms, we examined whether the D802G substitution alters pyrethroid binding in BgNav1-1 channels using Schild analysis. To this end, we examined the competition for the pyrethroid binding site between the inactive isomer of perthmethrin, 1S-cis, with the active 1R-cis permethrin (Lund and Narahashi, 1982). This method was used previously on the pyrethroid resistance mutations, L993F and F1519I, as described in Tan et al. (2005). Consistent with earlier findings, the modification of sodium channels by the active isomer was reduced in the presence of the inactive isomer on BgNav1-1 channels (Fig. 2A). However, unlike the L993F and F1519I mutations, the D802G substitution did not alter the affinity of the inactive isomer for BgNav1-1 channels (Figs. 2C and D). These results suggest that the D802G substitution reduces the channel sensitivity to pyrethroids via a mechanism that is independent of pyrethroid binding.
Fig. 2.

D802G does not reduce the binding of 1S-cis-permethrin to the BgNav1-1 channel. A, The inactive isomer 1S-cis-permethrin reduces the amplitude of the tail current induced by the active isomer, but by itself does not elicit a tail current. B and C, Percentages of channel modification by 1R-cis-permethrin in the presence of a series of concentrations of 1S-cis-permethrin in BgNav1-1 (B) and BgNav1-1D802G (C) channels. The modification curves were linearized by a logarithm of modification percentage at the Y-axis to estimate the EC20 value of each line. D, Schild plots (i.e., pA2 plots) showing generally similar binding affinities of 1S-cis-permethrin to the BgNav1-1 and BgNav1-1D802G channels. Each point represents the average of four oocytes. The affinity of inactive isomer (KB) was calculated from the equation log(A′/A−1)=logKB−log[B], where A represents EC20 without inactive isomer, A′ represents EC20 in the presence of inactive isomer 1S-cis-permethrin, KB is the dissociation constant of inactive isomer, and B is inactive isomer concentration. The data were fitted with a linear regression, generating x-intercept, pA2, which equals −logKB. The values of KB are 1.6 μM and 2.1 μM for BgNav1-1 and BgNav1-1D802G channels, respectively.
A negatively charged residue at 802 is critical for pyrethroid action and voltage-dependent activation of the sodium channel
We investigated the effect of the charge of the amino acid located at position 802 in BgNav1-1 on channel gating and sensitivity to pyrethroids by replacing D802 with positively charged K, negatively charged E, or another neutral residue A. K, G, or A substitutions significantly reduced channel sensitivity to deltamethrin compared with BgNav1-1, with K802 being the most resistant channel (Fig. 3). In contrast, the BgNav1-1D802E channel was as sensitive as the wild-type BgNav1-1 channel (Fig. 3). Thus, a negatively charged residue at 802 appears to be essential for channel sensitivity to deltamethrin.
Fig. 3.
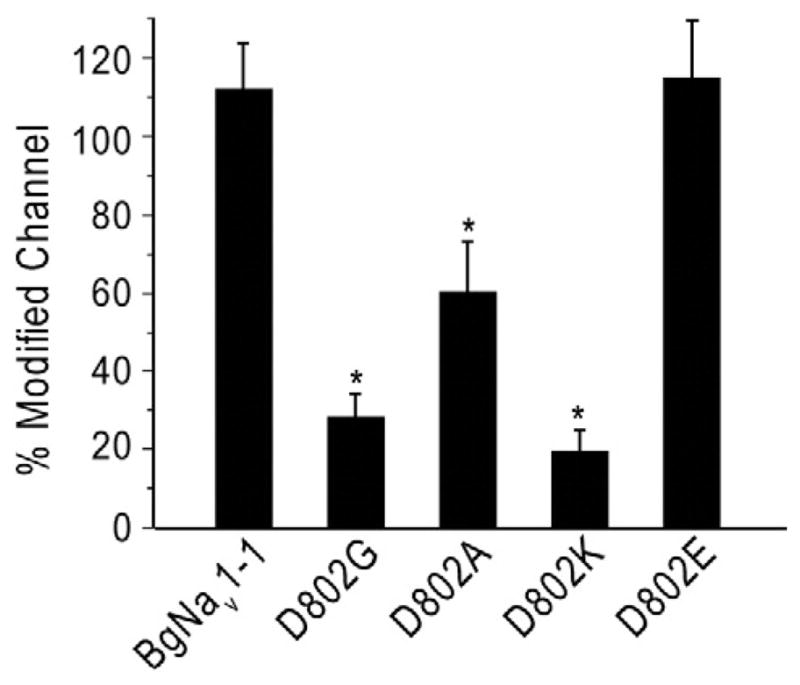
Effects of amino acid substitutions at D802 of the BgNav1-1 channel on channel sensitivity to deltamethrin. Percentage of channel modification by deltamethrin (1 μM) was determined using the method described in Materials and methods, and also in Fig. 1B. * indicates a significant difference compared to the BgNav1-1 channel (p<0.05).
We found that the D802G substitution in BgNav1-1 shifted the voltage dependence of activation by 10 mV in the depolarizing direction (Fig. 4A and Table 2). Conversely, when the reverse G802D substitution was introduced into BgNav2-1, the voltage dependence of activation was shifted in the hyperpolarizing direction by 18 mV (Fig. 4A). Furthermore, A or K substitution at 802 also shifted the voltage dependence of activation of BgNav1-1 channels in the depolarizing direction by 14 and 20 mV, respectively (Fig. 4B). In contrast, the E substitution shifted the voltage dependence of activation by only 4 mV in the hyperpolarizing direction (Fig. 4B and Table 2). These results suggest a correlation between pyrethroid resistance and depolarized voltage dependences of channel activation.
Fig. 4.

Effects of amino acid substitutions at D802 on channel gating properties. A, Voltage dependence of activation of BgNav1-1D802G and BgNav2-1G802D channels. B, Voltage dependence of activation of recombinant channels with substitutions at 802 other than D or G. The sodium currents were measured using a 20-ms test pulse ranging from −80 mV to+65 mV in 5-mV increments in BgNav1-1, BgNav2-1, and mutant channels. The values of half-maximal voltage for activation (V1/2) and slope factor (k) of BgNav1-1, BgNav2-1, and mutant channels are shown in Table 2. The normalized peak conductance was plotted against the potential of test pulses.
Table 2.
Voltage dependences of activation and inactivation of BgNav2-1, BgNav1-1, and their mutants.
| Na+ channel type | Activation |
Inactivation |
n | ||
|---|---|---|---|---|---|
| V1/2 (mV) | K (mV) | V1/2 (mV) | K (mV) | ||
| BgNav2-1 | −19.2±0.6 | 4.2±0.1 | −45.6±0.6 | 5.4±0.3 | 25 |
| BgNav2-1G802D | −37.5±1.1* | 4.1±0.4 | −55.1±0.9* | 6.1±0.1 | 8 |
| BgNav1-1 | −24.1±0.7 | 5.5±0.3 | −44.1±0.7 | 5.1±0.2 | 15 |
| BgNav1-1D802G | −14.8±1.0* | 6.0±0.4 | −42.9±0.2 | 6.1±0.5 | 10 |
| BgNav1-1D802A | −10.5±1.2* | 6.7±0.6 | −45.8±0.5 | 6.2±0.2 | 8 |
| BgNav1-1D802K | −4.2±1.5* | 5.7±0.9 | −40.0±0.1 | 6.3±0.3 | 12 |
| BgNav1-1D802E | −28.8±0.1 | 4.2±0.4 | −47.2±1.1 | 5.7±0.3 | 9 |
Significantly different from the parental channels using one-way NOVA with Scheffe’s post hoc analysis (p<0.05).
In contrast, there was no correlation between pyrethroid resistance and the voltage dependence of channel inactivation. Specifically, the D802K/G/A/E substitutions in BgNav1-1 did not significantly affect the voltage dependence of inactivation, whereas the G802D substitution in BgNav2-1 caused a 10-mV hyperpolarizing shift (Table 2).
The D802K substitution, but not the D802G substitution, accelerates open state deactivation
The molecular determinants involved in voltage-dependent activation also regulate channel deactivation (Kontis et al., 1997; Groome et al., 1999). Therefore, we compared the kinetics of activation and deactivation in D802K and D802G channel mutants using the cell-attached macropatch technique. In this set of experiments, we introduced these mutations into another channel, BgNav1-1a, which gives more robust expression in Xenopus oocytes than BgNav1-1 and was derived from BgNav1-1 by reverting four unique amino acid changes in BgNav1-1 to the sequence in GenBank (accession number: U73583) (Song et al., 2004). D802K significantly accelerated open state deactivation nearly over the entire range of command voltages (between −160 mV and −40 mV), as shown by the decrease in deactivation time constant (τD) of compared to the BgNav1-1a channel (Fig. 5A). In contrast, D802G slowed deactivation kinetics (Fig. 5A). For the kinetics of activation, D802K slowed the rise time of peak inward sodium current during activation, but D802G had no effect on the kinetics of activation (Fig. 5B).
Fig. 5.

Effects of D802K and/D802G on sodium channel gating kinetics. A, Open-state deactivation. Channels were opened with 0 mV, 0.5-ms depolarizing pulses followed by 20-ms hyperpolarizing commands to the voltages shown. Values shown are mean±SEM of time constants for the monoexponential decay in tail currents elicited during command hyperpolarization. B, Activation kinetics. Channels were opened with 20-ms depolarizing commands to the voltages shown. Values shown represent the mean±SEM for 10–90% of the rise time towards peak inward current during step depolarization.
Discussion
This study illustrates the utility of studying naturally occurring sodium channel variants (e.g., BgNav1-1 and BgNav2-1 channels) to seek a better understanding of the molecular mechanisms underlying pyrethroid action. Previously, we showed that the BgNav2-1 channel is more resistant to pyrethroids than the BgNav1-1 channel (Tan et al., 2005). Following this lead, we demonstrated that this difference in sensitivity can be traced to a highly conserved amino acid residue at position 802 in IIS1 that is critical for mediating pyrethroid action on voltage-sensitive sodium channels through a mechanism that is independent of the pyrethroid receptor.
Computer modeling of the house fly sodium channel based on the crystal structure of mammalian Kv1.2 predicts that IIS5, IIS6, the IIS4-S5 linker, and IIIS6 form a hydrophobic cavity that serves as the pyrethroid receptor (O’Reilly et al., 2006). Recently, systematic site-directed mutagenesis of residues in these regions has provided further experimental support for this model (Usherwood et al., 2007; Du et al., 2009). Schild analysis conducted in this study showed that D802, located in IIS1, is not part of the pyrethroid receptor site. Instead, owing to the importance of D802 to activation and deactivation, the D802G/K/A substitutions likely induced a conformational state of the sodium channel that counteracts the action of pyrethroids on channel deactivation and/or inactivation.
How does a negatively charged residue in IIS1 modulate the voltage-dependent gating of sodium channels? D802 is located at the extracellular end of IIS1. Interestingly, a negatively charged residue (D or E) is present at this position in all known vertebrate and invertebrate sodium channels (Borjesson and Elinder, 2008). Neutralization of an E residue in the mammalian Nav1.2 channel (equivalent to D802 in BgNav) also caused a depolarizing shift in the voltage dependence of activation (Cestele et al., 2006). Thus, the role of this negatively charged residue in sodium channel activation and deactivation is likely conserved. It is well established that positively charged residues in S4s are essential for voltage sensing (Catterall, 2000). Substantial experimental data show that negatively charged residues in S2–S3 make contact with the positively charged residues in S4s by electrostatic interactions (Borjesson and Elinder, 2008, reference therein; e.g., Papazian et al., 1995; Tiwari-Woodruff et al., 1999). For example, the electrostatic interactions between IIS4 and IIS2/IIS3 have been shown to be important for the voltage dependence of activation and the action of a scorpion β-toxin on the rat Nav1.2a sodium channel (Mantegazza and Cestele, 2005). A recent study showed that sequential ion pair formation between negative charges in S2 and S3 and positive charges in S4 are predicted to catalyze the transmembrane movement of S4 in NaChBac, a bacterial sodium channel (DeCaen et al., 2008, 2009). Computer modeling also predicts that several negatively charged residues including D802 in S1 electrostatically interact with positive charges in S4 during voltage-gated transmembrane movement of S4s in potassium, calcium, and sodium channels (Borjesson and Elinder, 2008). Therefore, it is possible that disruption of these electrostatic interactions could cause an alteration of S4 transmembrane movement leading to modification of channel gating and pyrethroid sensitivity. At the position corresponding to D802, mammalian sodium channels also have a conserved negatively charged residue (D or E). A similar mechanism might also underlie the interaction of mammalian sodium channels with pyrethroids.
It is important to point out that the D802G/A/K-mediated pyrethroid resistance is not exclusively correlated with the depolarizing shift in channel activation or accelerated channel deactivation. Although a depolarizing shift in the voltage dependence of activation by D802K/G/A was observed, the degree of the depolarizing shift by different substitutions is not directly correlated with the level of resistance to deltamethrin (Fig. 3). In theory, accelerated deactivation kinetics by the D802K mutation could counteract the inhibition of deactivation by pyrethroids and render the sodium channel pyrethroid-resistant. However, the D802G mutation slowed the deactivation kinetics, suggesting that there is a deactivation-independent mechanism. We hypothesize that D802 likely interacts with negative charges in IIS4 and modulates the transmembrane movement of IIS4. Disruption of such interactions may lead to altered channel gating which is unfavorable for the action of pyrethroids. Identification of alterations in the interactions between a negative residue in IIS1 and positive residues in IIS4 and their effect on pyrethroid activity should help uncover the gating state(s) that promotes pyrethroid action.
Acknowledgments
We thank Dr. Kris Silver for critical review of the manuscript; and thank Drs. Michael Gurevitz and Dalia Gordon for reviewing an earlier version of the manuscript. The work was supported by National Institutes of Health grant GM057440 to K.D.
Abbreviations
- IIS1
segment 1 in domain II
References
- Catterall WA. Cellular and molecular biology of voltage-gated sodium channels. Physiol Rev. 1992;72:S15–S48. doi: 10.1152/physrev.1992.72.suppl_4.S15. [DOI] [PubMed] [Google Scholar]
- Catterall WA. The structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- Cestele S, Catterall WA. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie. 2000;82:883–892. doi: 10.1016/s0300-9084(00)01174-3. [DOI] [PubMed] [Google Scholar]
- Cestele S, Yarov-Yarovoy V, Qu Y, Sampieri F, Scheuer T, Catterall WA. Structure and function of the voltage sensor of sodium channels probed by a β-scorpion toxin. J Biol Chem. 2006;281:21332–21344. doi: 10.1074/jbc.M603814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaen PG, Yarov-Yarovoy V, Zhao Y, Scheuer T, Catterall WA. Disulfide locking a sodium channel voltage sensor reveals ion pair formation during activation. Proc Natl Acad Sci USA. 2008;105:15142–15147. doi: 10.1073/pnas.0806486105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaen PG, Yarov-Yarovoy V, Sharp EM, Scheuer T, Catterall WA. Sequential formation of ion pairs during activation of a sodium channel voltage sensor. Proc Natl Acad Sci USA. 2009;106:22498–22503. doi: 10.1073/pnas.0912307106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Liu Z, Nomura Y, Bhambay B, Dong K. An alanine in segment 3 of domain III (IIIS3) of the cockroach sodium channel contributes to the low pyrethroid sensitivity of an alternative splice variant. Insect Biochem Mol Biol. 2006;36:161–168. doi: 10.1016/j.ibmb.2005.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Lee JE, Nomura Y, Zhang T, Zhorov BS, Dong K. Identification of a cluster of residues in transmembrane segment 6 of domain III of the cockroach sodium channel essential for the action of pyrethroid insecticides. Biochem J. 2009;419:377–385. doi: 10.1042/BJ20082082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borjesson SI, Elinder F. Structure, function and modification of the voltage sensor in voltage-gated ion channels. Cell Biochem Biophys. 2008;52:149–174. doi: 10.1007/s12013-008-9032-5. [DOI] [PubMed] [Google Scholar]
- Feng G, Deak P, Chopra M, Hall LM. Cloning and functional analysis of TipE, a novel membrane protein that enhances Drosophila Para sodium channel function. Cell. 1995;82:1001–1011. doi: 10.1016/0092-8674(95)90279-1. [DOI] [PubMed] [Google Scholar]
- Groome JR, Fujimoto E, George AL, Ruben PC. Differential effects of homologous S4 mutations in human skeletal muscle sodium channels on deactivation gating from open and inactivated states. J Physiol. 1999;516:687–698. doi: 10.1111/j.1469-7793.1999.0687u.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontis KJ, Rounaghi A, Goldin AL. Sodium channel activation gating is affected by substitutions of voltage sensor positive charges in all four domains. J Gen Physiol. 1997;110:391–401. doi: 10.1085/jgp.110.4.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund AE, Narahashi T. Dose-dependent interaction of the pyrethroid isomers with sodium channels of squid axon membranes. NeuroTox. 1982;3:11–24. [PubMed] [Google Scholar]
- Mantegazza M, Cestele S. Beta-scorpion toxin effects suggest electrostatic interactions in domain II of voltage-dependent sodium channels. J Physiol. 2005;568:13–30. doi: 10.1113/jphysiol.2005.093484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narahashi T. Molecular and cellular approaches to neurotoxicology: past, present and future. In: Lunt GG, editor. Neurotox ‘88: Molecular Basis of Drug & Pesticide Action. Elsevier; New York: 1988. pp. 563–582. [Google Scholar]
- Narahashi T. Neuroreceptors and ion channels as the basis for drug action: past, present, and future. J Pharmacol Exp Ther. 2000;294:1–26. [PubMed] [Google Scholar]
- O’Reilly AQ, Khambay BPS, Williamson MS, Field LM, Wallace BA, Davies TGE. Modeling insecticide binding sites at the voltage-gated sodium channel. Biochem J. 2006;396:255–263. doi: 10.1042/BJ20051925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papazian DM, Shao XM, Seoh SA, Mock AF, Huang Y, Wainstock H. Electrostatic interactions of S4 voltage sensor in Shaker K+ channel. Neuron. 1995;14:1293–1301. doi: 10.1016/0896-6273(95)90276-7. [DOI] [PubMed] [Google Scholar]
- Salgado VL, Narahashi T. Immobilization of sodium channel gating charge in crayfish giant axons by the insecticide fenvalerate. Mol Pharmacol. 1993;43:626–634. [PubMed] [Google Scholar]
- Song W, Liu Z, Tan J, Yoshiko N, Dong K. RNA editing generates tissue-specific sodium channels with distinct gating properties. J Biol Chem. 2004;279:32554–32561. doi: 10.1074/jbc.M402392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Liu Z, Nomura Y, Goldin AL, Dong K. Alternative splicing of an insect sodium channel gene generates pharmacologically distinct sodium channels. J Neurosci. 2002;22:5300–5309. doi: 10.1523/JNEUROSCI.22-13-05300.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Liu Z, Wang R, Huang ZY, Chen AC, Gurevitz M, Dong K. Identification of amino acid residues in the insect sodium channel critical for pyrethroid binding. Mol Pharmacol. 2005;67:513–522. doi: 10.1124/mol.104.006205. [DOI] [PubMed] [Google Scholar]
- Tatebayashi H, Narahashi T. Differential mechanism of action of the pyrethroid tetramethrin on tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels. J Pharmacol Exp Ther. 1994;270:595–603. [PubMed] [Google Scholar]
- Tiwari-Woodruff SK, Schulteis CT, Mock AF, Papazian DM. Biophys J. 1999;72:1489–1500. doi: 10.1016/S0006-3495(97)78797-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usherwood PN, Davies TG, Mellor IR, O’Reilly AO, Peng F, Vais H, Khambay BP, Field LM, Williamson MS. Mutations in DIIS5 and the DIIS4–S5 linker of Drosophila melanogaster sodium channel define binding domains for pyrethroids and DDT. FEBS Lett. 2007;581:5485–5492. doi: 10.1016/j.febslet.2007.10.057. [DOI] [PubMed] [Google Scholar]
- Vais H, Williamson MS, Goodson SJ, Devonshire AL, Warmke JW, Usherwood PNR, Cohen C. Activation of Drosophila sodium channels promotes modification by deltamethrin: reductions in affinity caused by knock-down resistance mutations. J Gen Physiol. 2000;115:305–318. doi: 10.1085/jgp.115.3.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SY, Wang GK. Voltage-gated sodium channels as primary targets of diverse lipid-soluble neurotoxins. Cell Signal. 2003;15:151–159. doi: 10.1016/s0898-6568(02)00085-2. [DOI] [PubMed] [Google Scholar]
- Warmke JW, Reenan RAG, Wang P, Qian S, Arena JP, Wang J, Wunderler D, Liu K, Kaczorowski GJ, Van Der Ploeg LHT, Ganetzky B, Cohen CJ. Functional expression of Drosophila para sodium channels: modulation by the membrane protein tipE and toxin pharmacology. J Gen Physiol. 1997;110:119–133. doi: 10.1085/jgp.110.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]