

Abstract
Here, we demonstrate that in HeLa cells, Ser317 of Chk1 undergoes phosphorylation in response to replication stress induced by hydroxyurea. We also demonstrate the existence of constitutive (interphase and mitotic) Chk1 kinase phosphorylation, the translocation of its phosphorylated form from the nucleus to cytoplasm in prometaphase as well as strong labeling of apoptotic nuclei with α-Chk1S317 antibodies. Additionally, we show that caffeine, 2-aminopurine, staurosporine and sodium metavanadate can induce premature chromosome condensation (PCC) by the abrogation of the S-M checkpoint. Staurosporine appeared to be the most effective PCC inductor, and as in the case of the remaining inductors, the addition of hydroxyurea each time brought about an increase in the number of cells showing PCC symptoms (synergic effect). The forced premature mitosis was accompanied by an increasing index of double-strand breaks marked by the phosphorylation of histone H2AX on Ser139. Moreover, we found that the chemicals used brought about minor actin and tubulin network rearrangements that occurred following either replication stress or drug-induced cell cycle delay. At the same time, it was found that the extent of the cytoskeleton rearrangement did not hinder PCC in all its subperiods, i.e., from PCC-type prophase to PCC-type telophase.
Keywords: Checkpoint kinase 1, Cytoskeleton rearrangement, H2AX, Premature chromosome condensation, S-phase checkpoints
Introduction
Cell cycle checkpoints are the mechanisms by which cells respond to the presence of intrinsic (DNA replication disturbances, oxidative damage, DNA double-stranded breaks) and/or extrinsic (radiation, chemotherapeutic agents, chemicals) stress factors. The G1/S and G2/M checkpoints constitute only a part of a complex system of mechanisms serving to both prevent replication errors and inhibit abnormal mitosis. The mechanisms determining cell cycle development are functionally connected with the system of three S-phase checkpoints, which block mitosis initiation in the case of structural DNA damage (the intra-S-phase checkpoint or DSB-induced replication-independent intra-S-phase checkpoint) or inhibition of its biosynthesis (the replication checkpoint or replication-dependent intra-S-phase checkpoint). The S-M checkpoint (the third type of checkpoint monitoring the S-phase course) ensures that the G2 phase and mitosis can begin after complete genome replication (like the replication checkpoint, the S-M checkpoint depends on DNA replication) (Bartek et al. 2004). The S-phase checkpoints not only slow down or arrest replication forks, but also inhibit the late-firing origins (Alvino et al. 2007). However, if damage is still not repaired, cells can exit S phase and arrest later—at the G2/M transition (reviewed by Bartek et al. 2004).
The exposure of cells to genotoxic stress results in activation of DNA-damage checkpoints that induce delays in various cell cycle phases or cell cycle arrest (Gardner and Burke 2000). Checkpoint kinase 1 (Chk1)—one of distal transducer kinases—is the main component of DNA-damage response (DDR) and plays an essential role in G1/S, the S-phase, G2/M and mitotic spindle checkpoints (Dai and Grant 2010 and references therein). In addition, Chk1 regulates the timing and progression of DNA replication forks in unperturbed cell cycles (Maya-Mendoza et al. 2007), preserving the viability of stalled replication forks and—to perform ‘the S-phase checkpoint’ function—is able to prevent the firing of late origins when early-firing origins are stalled (Feijoo et al. 2001). Chk1 is activated by the proximal transducer kinases related to phosphoinositide 3-kinases (PIKKs): ataxia telangiectasia and Rad3-related kinase (ATR) and—in a supportive way—ataxia telangiectasia-mutated kinase (ATM). Activated by phosphorylation on serines 317 and 345, Chk1 phosphorylates the Cdc25 phosphatase family members facilitating cell cycle arrest (Liu et al. 2000; Löffler et al. 2006). Under physiological conditions, Chk1 shows either nuclear or centrosomal localization to prevent premature mitosis. Cytotoxic drug-induced autophosphorylation and changes in cellular location of Chk1 are essential for its full activation (Clarke and Clarke 2005; Löffler et al. 2007). Moreover, mitotic Chk1 phosphorylation is connected with translocation of Chk1 from the nucleus to the cytoplasm in prophase (Enomoto et al. 2009). In response to replication stress, however, Chk1 is not only activated, but it also triggers the ubiquitin-dependent destruction rendered therapy where the anticancer therapeutic agent proves inadequate (Merry et al. 2010; Zhang et al. 2005; Zhou and Bartek 2004).
In turn, DNA double-stranded breaks activate the molecular system, in which ATM kinase is a superior element. The effector factors of this kinase are also used in the second parallel pathway, in which ATR kinase fulfils sensory and transmitter functions. The pathway subject to ATR kinase shows first of all the ability to react to the replication fork function disturbances. These can result from endogenous interactions, chemotherapy or experimental treatments leading to the inhibiting or disturbance of replication processes by the inhibition effect of hydroxyurea (HU), ultraviolet radiation (UV) or DNA alkylating methylmethanesulfonate (MMS). Both biochemical pathways function according to the following schemes: DNA double-stranded breaks (DSB) → ATM → Chk2 and the replication block → ATR → Chk1. In each of these, Cdc25 phosphatase is the target substrate (Guo et al. 2000). Histone H2A variant H2AX is rapidly phosphorylated on serine 139 [by ATM protein kinase (Chanoux et al. 2009), by ATR protein kinase and/or by DNA-dependent protein kinase (DNA-PK; reviewed in Podhorecka et al. 2010)]. ATM phosphorylates H2AX in response to the induction of DNA double-strand breaks caused by ionizing radiation, UV-light and hydroxyurea-mediated replication arrest (Halicka et al. 2005; Rogakou et al. 1999). ATR phosphorylates H2AX in response to DNA single-stranded breaks and during replication fork arrest (Ward and Chen 2001). DNA-PK phosphorylates H2AX in response to apoptotic-like DNA fragmentation (Mukherjee et al. 2006). Within minutes following DSBs induction, Ser139-phosphorylated H2AX accumulates in the vicinity of the sites of DNA damage, forming fluorescent foci (Riesterer et al. 2009; Rogakou et al. 1999; Rybaczek and Maszewski 2007).
More recently, a cell wall morphology checkpoint and a morphogenesis checkpoint have been identified as checkpoints (activated during replication stress) which also monitor the perturbation of tubulin and actin. In budding yeast, the morphogenesis checkpoint and the DNA-damage checkpoint involve septins as GTP/GDP-binding proteins that sense and transmit the status of cytoskeleton organization to cell cycle connected proteins in the nucleus (Kinoshita and Takeda 2007; Petretti et al. 2006; and reviewed in Enserink et al. 2006). Moreover, the spindle checkpoint can be activated in response to extensive chromosome damage during mitosis (Rieder et al. 1995). Generally, various checkpoint mechanisms ensure the high fidelity of DNA replication, DNA repair, chromosome alignment and, finally, division of nucleus and cytoplasm.
Under physiological conditions, cells do not initiate mitosis until DNA synthesis has been completed (Hartwell and Weinert 1989). However, a number of inhibitors may induce premature chromosome condensation (PCC) in the presence of incompletely replicated or damaged DNA by overriding the S-M checkpoint. The prematurely condensed chromosomes show pulverization due to the presence of unreplicated regions of DNA.
The results of studies performed hitherto on initiation of premature mitosis have shown that depending on the course of this process, the structure and functions of the division spindle apparatus being formed can be subject to significant change. Rearrangement of cytoskeleton during PCC induction concerns mainly the organization of chromosomes and their relation with spindle microtubules. Depending on the research model and the type of PCC inducing factor, the following events were observed: (1) proper formation of spindle, with which chromosomes remained in a functional relation, while chromatin fibers fixed to centromeres migrated to poles (root meristems of Allium cepa; Sen and Ghosh 1998); (2) the displacement of dislodged kinetochores along the spindle filaments (division apparatus in CHO cells; Brinkley et al. 1998); (3) the formation of bipolar spindle deprived of interaction with chromosomes (BHK cells, PCC induction with the use of 2-aminopurine; Andreassen and Margolis 1991); (4) a lack of spindle formation in HeLa cells treated with okadaic acid (Ghosh et al. 1992) and also due to the fusion of interphase and mitotic HeLa cells (Ghosh and Paweletz 1987); (5) structurally and functionally abnormal spindle during PCC induction after the fusion of thymocyte nuclei (Szollosi et al. 1986).
In the present article, we describe Chk1 and H2AX phosphorylation after the use of both phosphatase and protein kinase inhibitors in HeLa cells arrested in the S-phase. We combined either the S-phase arrested cells or chemically induced premature chromosome condensation with serine 317 of Chk1 kinase phosphorylation status. Experiments were designed to facilitate investigation of the relationship between the quantities of HeLa cells induced to enter premature condensation of chromosomes after caffeine, 2-aminopurine, staurosporine and sodium metavanadate treatments and the extent of drug-induced phosphorylation of H2AX histones. HeLa cells were exposed to the above drugs alone or in combination with hydroxyurea. Furthermore, we decided to determine the action of these inhibitors on the cell cytoskeleton network by performing immunostaining of actin and tubulin.
Materials and methods
Cell lines
HeLa (human cervical epidermoid carcinoma) cells were grown as monolayers in RPMI1640 medium (Gibco BRL, Grand Island, NY) supplemented with 10% heat-inactivated FBS (Gibco, BRL, Paisley) and antibiotics (100 U/ml penicillin and 100 mg/ml streptomycin, Polfa, Tarchomin, Warsaw, Poland) at 37°C in a humidified atmosphere of 5% CO2. One hour before treatment, the medium was removed, and cells were washed once with phosphate-buffered saline (PBS), and then the cell medium was replaced with the one free of antibiotics.
Chemicals and drugs
Hydroxyurea (HU, 2.5 mM; 10 mM), sodium metavanadate (Van, 200 μM), and staurosporine (ST, 200 nM) were purchased from Sigma (St. Louis, MO, USA). Caffeine (CF, 5 mM) was supplied by Merck (Darmstadt, Germany), 2-aminopurine (2-AP, 10 mM) was obtained from ICN Biomedicals (Eschwege, Germany). HeLa cells were incubated with the above chemicals and drugs, which were dissolved in DMSO, but the final solvent concentration in the medium did not exceed 0.1%. For micronucleus assays some cells were incubated with DMSO at 0.1% as a control to assess the appearance of micronuclei in untreated (control) HeLa cells. Dimethyl sulfoxide (DMSO) was obtained from Sigma (St. Louis, MO, USA).
HeLa cells treatments (replication stress, induction of premature chromosome condensation, and the micronucleus test)
HeLa cells were blocked with either 2.5 mM or 10 mM hydroxyurea (the S-phase arrest) for 24 h. For recovery test in the micronucleus assays, the above chemicals were replaced with a fresh medium for 9 h (in the case of 8 h recovery, a higher frequency of anaphase bridges was formed and this was the reason to repeat the experiments for a recovery time extended to 9 h). For the analyses connected with PCC induction series were selected, in which HeLa cells were blocked with the use of 2.5 mM hydroxyurea. The cells synchronized with the use of 2.5 mM hydroxyurea were then treated for additional 8 h with (1) 5 mM caffeine, (2) 10 mM 2-aminopurine, (3) 200 nM staurosporine, (4) 200 μM sodium metavanadate, and (5) either 2.5 mM hydroxyurea or 5 mM caffeine, (6) either 2.5 mM hydroxyurea or 10 mM 2-aminopurine, (7) either 2.5 mM hydroxyurea or 200 nM staurosporine, as well as (8) either 2.5 mM hydroxyurea or 200 μM sodium metavanadate. The quantification of premature mitosis and scoring of the MN frequency were determined by counterstaining with DAPI (0.1 mg/ml; 4′,6-diamidino-2-phenylindole; Sigma-Aldrich, Saint Quentin, France) for 5 min at room temperature. The cells were observed using an Optiphot-2 fluorescence microscope (Nikon, Warsaw, Poland) equipped with UV-2A filter (UV-light; λ = 518 nm). All images were recorded at exactly the same time of integration with a DXM 1200 CCD (Nikon, Warsaw, Poland) camera.
Antibodies
Immunocytochemical detection of both total and phosphorylated at Ser317 form of Chk1 was performed using rabbit polyclonal antibodies purchased from Cell Signaling Technology (Danvers, MA, USA). Rabbit polyclonal antibodies specific to phospho-H2AX(Ser139) were purchased from Upstate Biotechnology (Lake Placid, NY, USA). Bound primary antibodies were detected in both cases, with secondary goat anti-rabbit fluorescein isothiocyanate (FITC)-labeled antibody (ICN Biomedicals, Eschwege, Germany). Anti-rabbit IgG (AP-linked) antibody was used for immunoblotting (Cell Signaling Technology Danvers, MA, USA). Mouse monoclonal antibody to β-tubulin and secondary goat anti-mouse FITC-conjugated antibody were from Sigma-Aldrich (Saint Quentin, France). Actin was stained by phalloidin coupled to rhodamine (mixed isomers from Amanita phalloides from Sigma, St. Louis, MO, USA) at 50 μg/ml, after fixation in 2.5% glutaraldehyde buffered with PBS.
Western blot analysis
Proteins were extracted using TriPure Isolation Reagent (Roche Diagnostics Corporation, Indianapolis, IN, USA) according to the instructions of the manufacturer. Total protein concentrations in the cell lysates were determined using Ultrospec 110 pro (Amersham Biosciences, Austria). Western blot analysis was carried out by separating protein extracts on 7% polyacrylamide-SDS gel and blotting onto a nitrocellulose membrane (φ 0.45 μm, Schleicher & Schüell, Germany). Signals were visualized with of NBT/BCIP (Nitro blue tetrazolium chloride/5-bromo-4-chloro-3-indolyl phosphate, toluidine salt, Sigma-Aldrich, Saint Quentin, France) as substrates. The β-tubulin protein was used as an internal control.
Immunofluorescence
HeLa cells were grown in 8-well tissue culture plates containing sterile coverslips and treated as indicated in the figure legends. For immunocytochemical detection of phospho-Chk1(Ser317), phospho-H2AX(Ser139), and β-tubulin, HeLa cells were fixed for 15 min in 4% formaldehyde buffered with PBS. The cells were pre-treated in a blocking buffer (10% horse serum, 1% bovine serum albumin; BSA, 0.02% NaN3, 1× PBS) for 1 h at room temperature to minimize the non-specific adsorption of the antibodies to the coverslip, and were incubated overnight in a humidified atmosphere (4°C) with primary antibody. Polyclonal antibody to phospho-Chk1 (Ser317) was used at 1:500, polyclonal antibody to phospho-H2AX (Ser139) was used at 1:750, and β-tubulin was detected with a monoclonal antibody at a 300-fold dilution. The cells were washed three times (5 min each) with PBS/0.2% Triton X-100 (PBT). Secondary antibodies, including FITC-conjugated goat anti-rabbit (for Chk1 and H2AX), and goat anti-mouse antibodies (anti-β-tubulin), were used at 1:1000 for 1 h at room temperature in the dark. Next, they were washed three times with PBT (5 min each) and then for 5 min in PBS. DNA was detected by incubation with 0.2 μg/ml propidium iodide (PI; Sigma, St. Louis, MO, USA) in PBS for 5 min. The cells were embedded in PBS/glycerol mixture (9:1) with 2.3% DABCO (diazabicyclo [2.2.2] octane; Sigma-Aldrich, Saint Quentin, France). Observations were made using an Optiphot-2 fluorescence microscope (Nikon, Warsaw, Poland) equipped with B-2A filter (λ = 450–490 nm) for FITC, and with G-2A filter (λ = 518 nm) for propidium iodide. Phase-contrast images of cells grown inside a 37°C, 5% CO2 chamber were obtained using an Optiphot-2 fluorescence microscope (Nikon, Warsaw, Poland). Negative control sections incubated with non-immune serum in place of primary antibodies were free from immunostaining (data not shown; explanation: these negative control sections gave bright propidium iodide signals but completely lacked fluorescence in the wavelength corresponding either to anti-H2AX or anti-Chk1, both FITC-conjugated). Image data were collected at exactly the same time of integration with a DXM 1200 CCD (Nikon, Warsaw, Poland) camera. Images were processed and arranged using Adobe Photoshop CS5 (Adobe, San Jose, CA).
Statistical analysis
Statistical analyses were performed by means of STATISTICA 8.0 PL software (StatSoft INC, Tulsa, Oklahoma). All data were expressed as mean ± SD. Differences between groups were assessed by the non-parametric Mann–Whitney U test (for impaired data) and Kruskal–Wallis test; t Student test was used for data normally distributed. A probability P < 0.05 was considered statistically significant. Means values of the number of micronucleus (MN) per 1000 cells were calculated for significance among both HU concentration tested and the DMSO control.
Results
Caffeine, 2-aminopurine, staurosporine and sodium metavanadate can induce premature chromosome condensation in HeLa cells: addition of HU enhances this phenomenon
The HeLa-MN test (determining the frequencies of micronuclei in interphase HeLa cells after treatment with 2.5 or 10 mM HU, and 9 h post-treatment with fresh medium) was used for the identification of appropriate HU concentration for further investigations (which concerned the effectiveness of the selected HU concentration that blocked replication and increased the effectiveness of PCC induction with minimal side effects). We presented the results of our observations as the number of micronuclei per 1,000 cells. The scoring criteria for MN were developed for experiments with mammalian cells as described by Tolbert et al. (1992). A positive result occurred when the chromatin intensity and staining pattern were judged similar to that of the main nucleus, and when the MN was contained within the same cytoplasm as the main nucleus (Fig. 1A[f]). In a control series, micronuclei in a quantity of 1.1 ± 0.56 were observed. After 24 h incubation in 2.5 mM HU and 9 h post-incubation in a medium containing no HU, the number of micronuclei increased to 3.4 ± 1.16. Incubation in HU solution with a higher concentration (10 mM) significantly increased the formation of micronuclei in terms of 1,000 cells (4.5 ± 1.42). Finally, it was decided that in the study on PCC induction, 2.5 mM HU would be used in HeLa cells; among other reasons this was because 2.5 mM HU appeared to be effective in blocking cells in the S-phase (mitotic index: 0.3%, Fig. 1B) with only a relatively small adverse effect on the DNA structure, examined via light phase-contrast microscopy as well as under a fluorescence microscope after staining with DAPI and/or propidium iodide (numerical data are shown in Fig. 1C).
Fig. 1.

A Normal mitotic HeLa cell (a). The major categories of PCC phenotype: S-PCC (b), G2-PCC (c), and chromosome segregation defects (d). Representative nucleus displaying signs of apoptosis (e). HeLa cells treated with hydroxyurea were stained with DAPI dye for DNA. Micronucleus formation (f; arrows), were found significantly increased in comparison with the control. Scale bars = 10 μm. (B) Mitotic indices (%) and % of cell nuclei displaying signs of apoptosis evaluated for HeLa cells (the cells were treated as described in the figure legend). (C) Quantification of PCC phenotypes in HeLa cells
During the main series of tests concerned with PCC induction, HeLa cells were subjected to 24 h of HU followed by 8 h of PCC inductors as the mechanism of action of the chemical used allowed some of the blocked cells to override the S-phase checkpoints and, thus, to advance aberrant mitotic division. The following agents appeared to be PCC inductors: caffeine (CF, methyloxantine disturbing the molecular organization of the S-M checkpoint, which is known as a broad-spectrum nonspecific inhibitor of ATR/ATM); 2-aminopurine (2-AP), an inhibitor of cyclin-dependent kinases (CDK); staurosporine (ST) an ATP-competitive kinase inhibitor, related to protein C kinase; as well as sodium metavanadate (Van), an agent retarding the catalytic properties of phosphatases, including phosphatase of the cdc25 type as a main CDK activator (Bezrookove et al. 2003; Ghosh et al. 1998; Gotoh and Durante 2006; Laredo et al. 1994; Steinmann et al. 1991; Wang et al. 1999; Zhang et al. 2001).
The following PCC symptoms were observed: (1) irregular condensation of chromatin in prophase, (2) fragmentation of chromosomes, (3) disturbances in their metaphase system, (4) lost and lagging chromatids and chromosomes during anaphase, and (5) segregation defects observed later at telophase (comp. Ghosh et al. 1996; Krause et al. 2001; Nghiem et al. 2001; Riesterer et al. 2009; Rybaczek et al. 2008). Chemically induced PCC comprised only a small portion of the population of cells blocked previously in S-phase by the 24 h effect of 2.5 mM HU (Fig. 1). The causes of selective PCC induction are unknown but a similar phenomenon has also been observed in HeLa cells undergoing PCC under the influence of okadaic acid (Ghosh et al. 1998) as well as in plant cells (Allium cepa, Sen and Ghosh 1998; and Vicia faba meristems, Rybaczek et al. 2008). Depending on the fragmentation degree of chromosomes forced to undergo premature mitosis, two types of PCC phenotypes were distinguished for prophase, prometaphase and metaphase figures: (1) S-PCC (with numerous fragmentations without chromatid-like pairs elements, Fig. 1A[b]) and (2) G2-PCC (with a relatively small number of breakpoints: <20, leading to the losses of relatively large fragments of chromosomes during anaphase, Fig. 1A[c]). Additionally, we observed chromosome segregation defects (chromosomal bridges and lagging chromosomes, Fig. 1A[d]) during anaphase (comp. Krause et al. 2001; Nghiem et al. 2001; Rybaczek et al. 2008). Not surprisingly, micronuclei were observed as a consequence of incorrect chromosome segregation (Fig. 1A[f]). On the other hand, for nuclei, where the damage appeared to be widespread (probably also from the part of PCC population of the S-PCC phenotype), apoptotic type changes were initiated (Fig. 1A[e]); see also the list of numerical data from Fig. 1B (black bars, % of apoptosis) and from Fig. 1C (black rectangles, % of nuclei of S-PCC phenotype). The effectiveness of each of the inhibitors used as PCC inductors is not identical (Fig. 1C). Taking into account the percentage of cell numbers showing PCC symptoms (calculated as the sum: S-PCC + G2-PCC + segregation defects), their effectiveness can be presented in a diminishing series: HU/ST (9.7%) > HU/Van (9.5%) > HU/CF (7.9%) > HU/2-AP (7.2%) > ST (5.1%) > Van (3.7%) > CF (3.2%) > 2-AP (2.1%). This series indicates that the strongest synergic influence on PCC induction is exerted by the combination of staurosporine (ST), an inhibitor of protein kinases, with hydroxyurea (HU), which inhibits DNA replication by depleting dNTP pools. The comparison of the effects of individual drugs and their combination with HU shows that the addition of HU considerably enhances PCC symptoms (Fig. 1C). These results are consistent with previous reports on the effects of HU with both phosphatase and protein kinase inhibitors on the PCC induction in Vicia faba cells (Rybaczek et al. 2008).
A suggested model for the hydroxyurea-induced checkpoint signaling and modulation by caffeine, 2-aminopurine, staurosporine, and sodium metavanadate is presented in Fig. 2.
Fig. 2.

A model for the hydroxyurea-induced checkpoint signaling and modulation by caffeine, 2-aminopurine, staurosporine and sodium metavanadate. The mechanisms connected with the S-phase checkpoints are set in motion under the conditions of replication stress [under the influence of hydroxyurea (HU), an inhibitor of ribonucleotide reductase (RNR)], which results in the phosphorylation of Chk1 kinase by superior kinases: ATM and ATR. The activated Chk1 can inactivate Cdc25C phosphatase by phosphorylation on Ser216, which results in the inhibition of Cdc2 activation and G2/M passages (orange pathway). Caffeine (CF), 2-aminopurine (2-AP), staurosporine (ST) and sodium metavanadate (Van) induce the premature condensation of chromosomes (PCC), most probably by omitting the mechanism that blocks Cdc25 phosphatase and Cdk1/cyclin B complex (red and blue pathways)
Both S-phase arrest and loss of S-M dependency are related to the changes in phosphorylation of Chk1S317
The results of Western blot test with the use of antibodies recognizing total protein and Chk1 kinase phosphorylated on serine 317 showed a strong increase in the amount of total Chk1 and twofold increase (in comparison with the control band on the same blot) of the active form of this enzyme in protein extracts obtained from HeLa cells blocked in S-phase under the influence of hydroxyurea (Fig. 3a, b). The analyses performed (ANOVA calculated by means of Kruskal–Wallis’ test) point to the statistical significance of the results obtained [Chk1 total: H = 60.2, P < 0.001; control/2.5 mM HU P < 0.001; control/10 mM HU P < 0.001; Chk1-P (Ser317): H = 51.9, P < 0.001; control/2.5 mM HU P < 0.001; control/10 mM HU P = 0.000001] (Fig. 3b).
Fig. 3.

Expression levels of the total and phosphorylated Chk1 protein kinase at Ser317 by Western blot analysis (a). Data shown are representative of three independent experiments. The relative levels of Chk1 (total) and Chk1-P (Ser317) after normalization for β-tubulin, as determined by densitometry analysis of the bands, are shown in the histogram (b; the pixel values [pv; 1–255] categorized according to densitometry analysis of the bands intensities and expressed in arbitrary units [a.u.]). Columns, mean from three independent experiments; bars, SD. * P < 0.001, compared with control (Mann–Whitney U test; U = 0); filled diamond, P < 0.001, compared with control (Mann–Whitney U test; U = 18.5); filled square, P < 0.001, compared control (Mann–Whitney U test; U = 71.5)
These results support a model in which, under normal cell cycle conditions and in the absence of DNA damage, HeLa cells have constitutively phosphorylated Chk1 preventing premature mitosis. Our results show that 9% of control HeLa cells had a basal level of Chk1S317 immunofluorescence (Fig. 4a–a′ and Fig. 5a). However, the intensity of this phosphorylation was weak (about 8%; Figs. 4a–a′ and 5b; comp. Schmitt et al. 2006).
Fig. 4.

Micrographic pictures showing the intranuclear localization of phospho-Chk1S317 in HeLa cells; control (a–a′), a negative control section incubated with secondary antibodies only (b–b′), cells incubated for 24 h with 2.5 mM hydroxyurea (c–c′), cells incubated for 24 h with 10 mM hydroxyurea (d–d′), cells incubated for 24 h with 2.5 mM hydroxyurea and post-treated with 5 mM caffeine (e–e′), cells incubated for 24 h with 2.5 mM hydroxyurea and post-treated with the mixture of 2.5 mM hydroxyurea and 5 mM caffeine (f–f′), cells incubated for 24 h with 2.5 mM hydroxyurea and post-treated with 10 mM 2-aminopurine (g–g′), cells incubated for 24 h with 2.5 mM hydroxyurea and post-treated with the mixture of 2.5 mM hydroxyurea and 10 mM 2-aminopurine (h–h′), cells incubated for 24 h with 2.5 mM hydroxyurea and post-treated with 200 nM staurosporine (i–i′), cells incubated for 24 h with 2.5 mM hydroxyurea and post-treated with the mixture of 2.5 mM hydroxyurea and 200 nM staurosporine (j–j′), cells incubated for 24 h with 2.5 mM hydroxyurea and post-treated with 200 μM sodium metavanadate (k–k′), cells incubated for 24 h with 2.5 mM hydroxyurea and post-treated with the mixture of 2.5 mM hydroxyurea and 200 μM sodium metavanadate (l–l′). Scale bar = 10 μm
Fig. 5.

a Immunolabeling indices (%) estimated for HeLa cells stained with anti-Chk1S317 antibodies. Columns, mean from five independent experiments; bars, SD. * P = 0.009, compared with control (Mann–Whitney tests); # P = 0.009, compared with 2.5 mM HU (Mann–Whitney tests); & P = 0.03, compared with 2.5 mM HU (Mann–Whitney tests). b Quantification of anti-Chk1S317 pixel intensity per cell. Columns, mean from at least 108 cells per sample taken from five independent experiments; bars, SD. * P < 0.01, compared with control (Mann–Whitney tests); + P < 0.001, compared with control (Mann–Whitney tests); # P < 0.01 compared with 2.5 mM HU (Mann–Whitney tests); & P = 0.000215, compared with 2.5 mM HU (Mann–Whitney tests); $ P = 0.000038 compared with 2.5 mM HU (Mann–Whitney tests)
Here, we also report that Chk1 was phosphorylated at Ser317 during normal mitosis (Fig. 6). The fraction of control cells undergoing mitotic division constitutes about 2.5% (Fig. 1B). Since all mitotic cells in the control are marked with antibodies that recognize Chk1 kinase phosphorylated on Ser317, the constitutive phosphorylation level of Chk1 kinase, denoted with a white bar in Fig. 5a, combines the population of interphase and mitotic nuclei. Mitotic phosphorylation of Chk1S317 is associated with the translocation of phosphorylated Chk1 from the nucleus to the cytoplasm during prometaphase to anaphase (Fig. 6a–c′′; comp. Enomoto et al. 2009; Peddibhotla et al. 2009). The labeling pattern in the form of large brightly fluorescing foci is characteristic of telophase nuclei and nuclei from the post-telophase [early G1 phase (Fig. 6d–d′′)]. Surprisingly, not only foci size but also their number increases in telophase in comparison with the nuclei of prophase, metaphase and anaphase cells (Fig. 6). Also a strong but homogeneous labeling is shown by the total population of apoptotic nuclei (Fig. 6e–e′′).
Fig. 6.
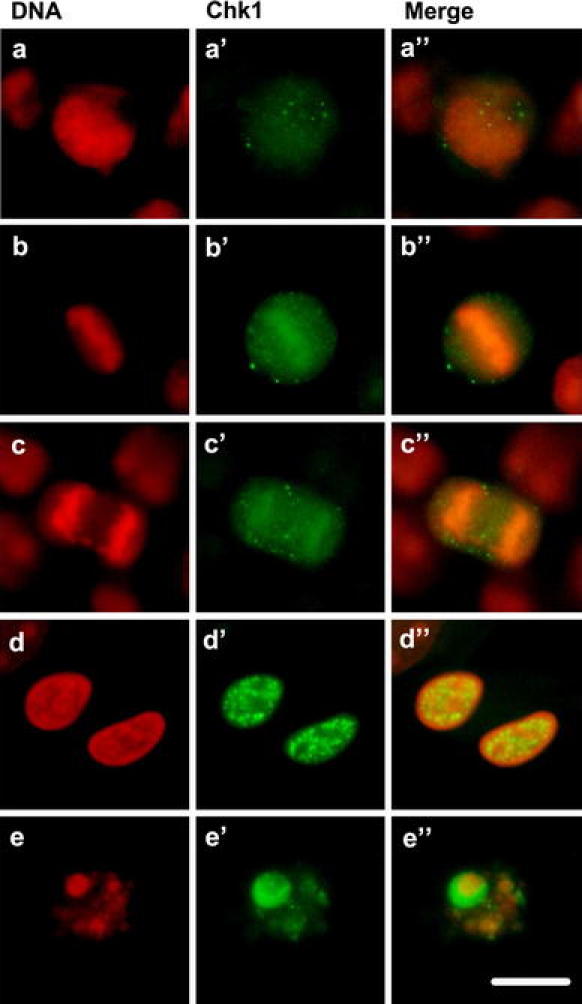
The mitotic, post-mitotic and apoptotic Chk1S317 phosphorylation patterns in HeLa cells after caffeine-induced PCC. Prophase (a–a′′), metaphase (b–b′′), anaphase (c–c′′), post-telophase (d–d′′), apoptosis (e–e′′). Scale bar = 10 μm
HeLa cells were collected in the S-phase by exposure to hydroxyurea (HU), an inhibitor of ribonucleotide reductase. The cell cycle arrest in response to HU was documented by using a fluorescence microscope which showed that arrested nuclei revealed Chk1S317-positive signals (above 60% and 80% after 24 h incubation-period with 2.5 mM and 10 mM HU, respectively). The effect of HU was dose-dependent (Fig. 4c–c′, d–d′ and Fig. 5a). We observed rapidly enhanced fluorescence intensity of Chk1 after HU treatment (Fig. 5b), in comparison with control. This result suggests that the greater activation of Chk1 in HU-treated HeLa cells may play an essential role in response to S-phase checkpoint activation.
The purpose of the present study was to examine the effect of both protein kinase and protein phosphatase inhibitors on the S-M checkpoint abrogation in HeLa cells. In this way, we examined the functional importance of S-M dependency. We found that HeLa cells were able to override the S-M checkpoint, related to the observed decrease in Chk1S317 phosphorylation (Figs. 4e–l′, 5a). This phenomenon of breaking the S-M dependency was induced after treatment of HeLa cells with CF, 2-AP, ST, Van and, also, after combined treatment of the above chemicals with 2.5 mM HU (Fig. 5a). In HeLa cells treated for 24 h with 2.5 mM HU and then post-incubated in all the chemical solutions described above, the number of Chk1S317-positive cells (Fig. 5a) and the intensity of fluorescence of Chk1S317 (Fig. 5b) were significantly higher compared to the control. As shown in Fig. 5, an association was found between the control and the following experimental series: HU → CF, HU → 2-AP, HU → ST, HU → Van (Kruskal–Wallis H test signed rank: P = 0.01) and for HU → HU + CF, HU → HU + 2-AP, HU → HU + ST, HU → HU + Van (P = 0.0028). It is noteworthy that sodium metavanadate (acts as alone, i.e. HU → Van) and either 2-AP or ST, (i.e. HU → HU + 2-AP, HU → HU + ST) were the most effective for inducing Chk1 phosphorylation (for Van: P = 0.01; for 2-AP: P = 0.006; for ST: P = 0.05). An association was also found between the 2.5 mM HU and the following experimental series HU → CF, HU → 2-AP, HU → ST, HU → Van (P = 0.017) and for HU → HU + CF, HU → HU + 2-AP, HU → HU + ST, HU → HU + Van (P = 0.01). Caffeine was the most effective agent for reducing of Chk1S317 phosphorylation (P = 0.023; see Fig. 5a). All correlations pertaining to Chk1 intensity per cell (showed in Fig. 5b) were significant (P < 0.001 for all experimental series reported herein).
During chemically induced checkpoint overriding, the cell nuclei were unable either to finish the S-phase or to execute DNA repair following G2 phase, resulting in PCC induction and commencement of the earliest stages of apoptosis (Fig. 1B and Fig. 6e-e′′). We observed that CF, ST and Van were highly potent inhibitors frequently leading to apoptosis after PCC induction. Similar results were obtained by Niida and Nakanishi (2006) and Shimizu and et al. (1995). In this context, Chk1 has been shown to inactivate the Cdc25C and Cdc25A phosphatases by phosphorylation, resulting in the accumulation of tyrosine 15-phosphorylated Cdc2 (Niida and Nakanishi 2006) followed then by “inappropriate” activation of Cdc2-promoted apoptosis (Shimizu et al. 1995).
Treatment with HU leads to increased H2AX phosphorylation, addition of PCC inductors suppresses the formation of phospho-H2AX foci
HeLa cells exposed to the long-lasting prolonged incubation with either 2.5 mM HU or 10 mM HU exhibited phospho-H2AX fluorescence (Figs. 7, 8a–c). Generally, HU-induced fluorescent foci spread throughout the whole area of nucleoplasm, but phospho-H2AX immunostaining after 10 mM HU treatment was additionally represented by nuclear pan-staining (Fig. 7). Either labeling indices and mean number of fluorescent foci or the intensity of fluorescence were significantly elevated in a dose-dependent manner in the HU-treated cells compared to control (Fig. 8a–c). Compared to untreated HeLa cells, in the case of the fraction of H2AX-positive cells in series exposed for 24 h to 2.5 mM/10 mM HU, increased nearly 60/70 times (labeling index), 40/60 times (mean number of foci) and above 100 times (H2AX intensity per cell), respectively (Fig. 8a–c). However, about 1% of control cells also revealed weak phospho-H2AX fluorescence (Fig. 7). Our studies showed that a significant level of Ser139 phosphorylation in histone H2AX occurred in HeLa cells treated for 24 h with 2.5 mM HU and then transferred either to water-based solutions inducing PCC (e.g. HU → CF, HU → 2-AP, HU → ST, HU → Van) or into the mixtures of 2.5 mM HU and above regulators of key enzymes of the cell cycle (e.g. HU → HU + CF, HU → HU + 2-AP, HU → HU + ST, HU → HU + Van). In all the above-described experimental sequences, the number of H2AX-positive cells (Fig. 8a) and the mean number of H2AX foci (Fig. 8b) were found to be significantly higher compared to the control. As shown in Fig. 8a, an association was found between the control and the following experimental series: HU → CF, HU → 2-AP, HU → ST, HU → Van (Kruskal–Wallis H test signed rank: P = 0.0196) and for HU → HU + CF, HU → HU + 2-AP, HU → HU + ST, HU → HU + Van (P = 0.009). It is noteworthy that sodium metavanadate (alone or in combination with hydroxyurea) was most effective for inducing the formation of phosphorylated H2AX (P = 0.01). An association also was found between the 2.5 mM HU and the following experimental series HU → CF, HU → 2-AP, HU → ST, HU → Van (P = 0.0196) and for HU → HU + CF, HU → HU + 2-AP, HU → HU + ST, HU → HU + Van (P = 0.01; see Fig. 8a). The quantity of foci was significantly higher than those in the control (Fig. 8b). An association was found between the control and the following experimental series: HU → CF, HU → 2-AP, HU → ST, HU → Van (P = 0.024) and for HU → HU + CF, HU → HU + 2-AP, HU → HU + ST, HU → HU + Van (P = 0.01). Most effective for the production of H2AX-foci was either sodium metavanadate (P = 0.01 for: HU → Van; and P = 0.046 for HU → HU + Van) or staurosporine (P = 0.03 for: HU → HU + ST; see Fig. 8). An association was also found between the 2.5 mM HU and the following experimental series HU → CF, HU → 2-AP, HU → ST, HU → Van (P = 0.024). Kruskal–Wallis H test signed rank for HU → HU + CF, HU → HU + 2-AP, HU → HU + ST, HU → HU + Van was at P = 0.017 (Fig. 8b). All the other correlations (showed in Fig. 8c) were significant (P < 0.001 for all experimental series reported herein).
Fig. 7.

Micrographic pictures showing the intranuclear localization of phospho-H2AXS139 in HeLa cells. Scale bar = 10 μm
Fig. 8.
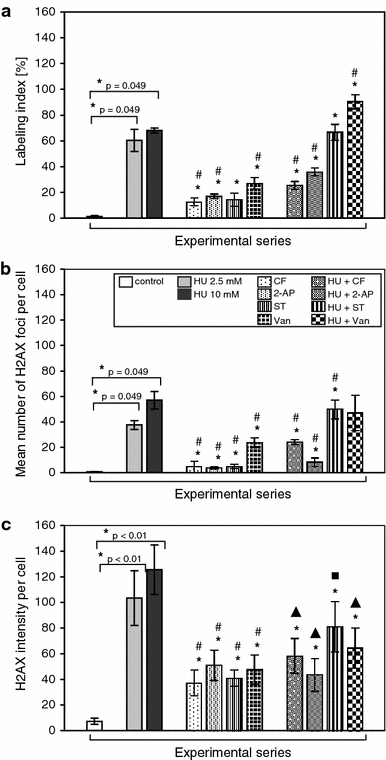
a Immunolabeling indices (%) estimated for HeLa cells stained with anti-H2AXS139 antibodies. Columns, mean from three independent experiments; bars, SD. * P = 0.049, compared with control (Mann–Whitney tests); # P = 0.049, compared with 2.5 mM HU (Mann–Whitney tests). b Mean number of intranuclear phospho-H2AX foci generated in HeLa cells. Columns, mean from at least 300 cells per sample taken from three independent experiments; bars, SD. * P = 0.049, compared with control (Mann–Whitney tests); # P = 0.049, compared with 2.5 mM HU (Mann–Whitney tests). c Quantification of anti-H2AXS139 pixel intensity per cell. Columns, mean from at least 106 cells per sample taken from three independent experiments; bars, SD. * P < 0.01, compared with control (Mann–Whitney tests); # P < 0.01, compared with 2.5 mM HU (Mann–Whitney tests); filled diamond, P < 0.01, compared with 2.5 mM HU (t tests with Cochran-Cox correction); filled square, P < 0.001 compared with 2.5 mM HU (t tests on log-transformed values, which were normally distributed)
The level of H2AX phosphorylation as well as the mean number of H2AX foci were reduced in HeLa cells incubated with the above chemicals without HU (i.e. HU → CF, HU → 2-AP, HU → ST, HU → Van), as compared with the cells treated with 2.5 mM HU for 24 h. We also observed a significant decrease in labeling indices and the mean number of foci during simultaneous treatment with CF (HU → HU + CF) and 2-AP (HU → HU + 2-AP) in comparison with that treated with 2.5 mM HU for 24 h (Fig. 8a–b), which was due to the mechanism of action of these inhibitors [e.g. caffeine inhibits the checkpoint kinase ATM (ataxia telangiectasia mutated) that phosphorylates H2AX in response to DNA damage; Rybaczek et al. 2008 and references therein].
We therefore assessed the effect of CF, 2-AP, ST and Van on the induction of phospho-H2AX foci in interphase HeLa cells or in HeLa cells committed to enter PCC. Immunocytochemistry using anti-phospho-H2AX antibodies shows that the above-mentioned chemicals may promote the formation of phospho-H2AX foci in a population of interphase cells (Fig. 7) and in mitotic cells (the mean number of phospho-H2AX foci observed in PCC-like anaphases was scored only in those series in which PCC was induced exclusively by the influence of PCC inductor, i.e., in the absence of HU. The following numerical values were obtained: 4.4 ± 1.01 for 2-AP; 2.8 ± 0.56 for CF; 2.4 ± 0.29 for Van; 1.5 ± 1.1 for ST). In interphase HeLa cells, phospho-H2AX accumulated at discrete fluorescent foci spread throughout the whole area of nucleoplasm, without height tendency at the region of euchromatin or heterochromatin (Fig. 7). The presence of phospho-H2AX foci in the mitotic figures (PCC-like anaphases) indicated that cells were proliferating despite the presence of double-stranded breaks within DNA molecules (An et al. 2010; Ha et al. 2009; Huang et al. 2008; Rybaczek et al. 2007).
Chemically induced tubulin and actin rearrangements concerning mainly the fraction of interphase nuclei: PCC-like mitoses was observed
Immunocytochemical tests of the microtubule system with the use of antibodies directed against β-tubulin and staining actin filaments with rhodamine-labeled phalloidin were performed to determine the character of changes in cytoskeleton under the conditions of replication stress and after the use of various PCC inductors. The tests showed that, as a result of PCC induction, the organization of cytoskeleton structures in HeLa cells did not significantly change, while the division spindle being formed was functional, which was also confirmed indirectly by the observation of all the stages of mitotic division (from prophase through prometaphase and metaphase until the division of chromosomes in anaphase and their decondensation in telophase; Fig. 1A). The use of fluorescein-marked antibodies directed against β-tubulin revealed that the number of microtubules constituting the main element of the structural–functional organization of division spindle was only slightly lower in PCC-type cells than that of control cells (data not shown).
Microtubules (MTs) form a dynamic network that plays a crucial role in all of the cellular changes, cell polarity and intracellular traffic. When cycling cells enter mitosis, the interphase microtubules rearrange into a mitotic spindle that is responsible for proper chromosome segregation during cell division (Wittmann et al. 2001). We used immunofluorescence tests to localize β-tubulin (Fig. 9A). In interphase control cells, MTs emanate from one point and are distributed throughout the entire cytoplasm (Fig. 9A[a]). This pattern was not dramatically altered upon incubation with inhibitors: CF, 2-AP and ST (Fig. 9[c–e]). We observed the extension of MTs and unusual MT-curling following HU treatment (Fig. 9A[b]). However, the most visible changes within the interphase microtubule architecture was revealed using Van: the bundling-type organization of MTs could then be seen (Fig. 9[f]). Our results are in agreement with the data presented by Meyer and et al. (1981) and Schepis et al. (2006) who reported that during the course of Vaccinia Virus infection actin underwent microtubule-dependent rearrangements. The results of the analyses described here show that regulation of both actin and tubulin morphology is an essential part of the DNA-damage stress response pathway. The tests performed by Ghosh et al. (1998) with the help of a HeLa cell model revealed that cells simultaneously treated with okadaic acid (OA) and staurosporine did not show a cytoskeletal rearrangement typical of normal mitotic cells approaching prophase, and thus these cells did not exhibit premature mitosis. However, HeLa cells treated only with OA or only with fostriecin seemed to induce either the cytoskeletal rearrangement or premature mitosis.
Fig. 9.

A Organization of the cellular microtubule network in HeLa cells; control (a), cells incubated for 24 h with 2.5 mM hydroxyurea or with 10 mM hydroxyurea (b), cells incubated for 24 h with 2.5 mM hydroxyurea and post-treated either with 5 mM caffeine or with the mixture of 2.5 mM hydroxyurea and 5 mM caffeine (c), cells incubated for 24 h with 2.5 mM hydroxyurea and post-treated either with 10 mM 2-aminopurine or with the mixture of 2.5 mM hydroxyurea and 10 mM 2-aminopurine (d), cells incubated for 24 h with 2.5 mM hydroxyurea and post-treated either with 200 nM staurosporine or with the mixture of 2.5 mM hydroxyurea and 200 nM staurosporine (e), cells incubated for 24 h with 2.5 mM hydroxyurea and post-treated either with 200 μM sodium metavanadate or with the mixture of 2.5 mM hydroxyurea and 200 μM sodium metavanadate (f). Scale bar = 10 μm. B Actin rearrangements occurred in HeLa cells during hydroxyurea-induced arrest and following PCC induction; control (a), cells incubated for 24 h with 2.5 mM hydroxyurea or with 10 mM hydroxyurea (b), cells incubated for 24 h with 2.5 mM hydroxyurea and post-treated either with 5 mM caffeine or with the mixture of 2.5 mM hydroxyurea and 5 mM caffeine (c), cells incubated for 24 h with 2.5 mM hydroxyurea and post-treated either with 10 mM 2-aminopurine or with the mixture of 2.5 mM hydroxyurea and 10 mM 2-aminopurine (d), cells incubated for 24 h with 2.5 mM hydroxyurea and post-treated either with 200 nM staurosporine or with the mixture of 2.5 mM hydroxyurea and 200 nM staurosporine (e), cells incubated for 24 h with 2.5 mM hydroxyurea and post-treated either with 200 μM sodium metavanadate or with the mixture of 2.5 mM hydroxyurea and 200 μM sodium metavanadate (f). Scale bar = 10 μm
Actin is a cytoskeletal component of the microfilament that provides the structural basis for cell morphogenesis (Bretscher 1991). In the present study, fluorescent phalloidin was used to investigate the actin cytoskeleton of human HeLa cells, which revealed that the actin network underwent several rearrangements occurring during replication stress and drug-induced delay (Fig. 9B). As shown in Fig. 9B[a], in control HeLa cells, the actin network appeared as thin fibers that crossed the cytoplasm. Upon HU treatment the actin patterns had disappeared in the central part of the cell, and instead, long fibers clearly outlined the cell borders (Fig. 9B[b]). The induction of actin stress fibers identified mainly at the peripheral rims of labeled HeLa cells, with no F-actin staining in the central region of the cell, appeared to be due to CF action (Fig. 9B[c]). Amiri et al. (2007) also showed that, in response to the Hsp90 inhibitor geldanamycin, HeLa cells generated cytoplasmic actin stress fibers that span the cells’ length. Cells treated with 2-AP formed many protrusions (Fig. 9B[d]). Minimal actin reorganization was revealed following treatment with ST (Fig. 9B[e]). Actin filaments in Van-treated cells appeared as fibers that crossed the entire cytoplasm. Moreover, lamellipodia-like structures were observed around the cell following application of Van (Fig. 9B[f]; comp. Schepis et al. 2006).
Discussion
Interest in factors releasing cells from S-M dependence is connected with the search of effective chemotherapeutic agents or stimulators of suicidal death of malignant cells. Beside mutations that either eliminate particular biochemical elements of the S-phase checkpoints or block their biological activity, many chemical substances can disturb the effectiveness of the system monitoring the course of DNA replication. Their action involves an overriding or breakage of the mechanism function of genome integrity supervision and the ordinary course of DNA biosynthesis and, as a direct consequence of this, cancellation of S-M dependence and “unauthorized”, premature mitosis initiation (PCC).
Staurosporine-induced morphological changes typical either of apoptosis (internucleosomal DNA fragmentation) or premature mitosis and the two staurosporine-induced phenomena are biochemically different (in their requirement for p34cdc2 kinase activation; Yoshida et al. 1997).
Caffeine blocks the activity of ATR/ATM kinases in vitro (Cortez 2003), and the PCC process in vivo induced by it is the result of putting into inertia the whole biochemical pathway, which is responsible for cascade transfer of the signal of replication apparatus dysfunction. The inhibition action of caffeine in relation to ATR/ATM kinases blocks their ability to phosphorylate Chk1/Chk2 kinases thereby preserving the catalytic activity of Cdc25 phosphatases, which fulfill the role of inductors of the Cdk1/cyclin B complexes and release mitotic phosphorylations (Fig. 2; Savitsky and Finkel 2002). Partly inconsistent with this hypothetical model of PCC induction are the results of Cortez’ studies (published in 2003) on the ATR dependent phenomena in HCT116 cells, which suggest that although caffeine fails to decrease the phosphorylation level of Chk1 and Chk2 kinases, it blocks the function of checkpoint induced by HU and ionizing radiation. Thus, it seems that at least some of the numerous effects connected with caffeine interference in the functional effectiveness of the S-phase checkpoints are connected with mechanisms independent of its inhibition influence on the ATM and ATR kinases.
The inhibitory influence of 2-aminopurine on the activity of the main mitosis promote, MPF, being incompatible with the PCC induction interaction, constituted a source of various hypotheses concerning the mechanisms of inducing suicidal cell divisions (Ghosh et al. 1998; Sen and Ghosh 1998). About the same time, however, publication of the first study suggesting clearly that the activation of Chk1 kinase at the S-M checkpoint, resulting from the replication block was sensitive to caffeine (Kumagai et al. 1998) occurred. Successive years witnessed papers reporting the analogues of the action mechanism of caffeine and 2-AP. These showed that target factors susceptible to the action of such inhibitors were ATM and ATR kinases (Dimitrova and Gilbert 2000). The results of these observations can explain, at least partly, the reduction in the p53 phosphorylation level by 2-AP in the cells subjected to the action of adriamycin, and either γ or UV radiation. However, the activity of p53 blocked under the influence of 2-AP takes place also in cells deprived of ATM, ATR or DNA-PK (Huang et al. 2003).
Sodium metavanadate shows a strong inhibitory action in relation to the activity of tyrosine phosphatases and p34cdc2/H1 kinase (phosphorylation of Tyr15); it also brings about the dephosphorylation of H1 and H3 and decondensation of chromosomes (citation of Ajiro et al. 1996). In malignant epithelial lung cells (of A549 line), Van induces increases in the level of reactive oxygen forms and the inhibition of the G2/M passage by influencing components of the antioxidative system. These processes are accompanied by the intensive expression of p21 and Chk1 kinase, a slight increase in B1 cyclin level, degradation of Cdc25C phosphatase, and activation of MAPK and p38 kinases (Zhang et al. 2001; Zhang et al. 2003). As the general character of PCC-type changes induced in HeLa cells treated with sodium metavanadate resembles the modifications brought about by the action of CDK inhibitors, it seems probable that the chemical base of these analogues is a common factor functioning at the molecular level (stimulation of tyrosine kinases, blockage of ATPase and consequently the effect on cyclin B/Cdc2 complex or on the system of ubiquitin-dependent destruction of cyclin B; Ajiro et al. 1996). Taking into account the above facts, the possibility arises that no matter whether HeLa cells are subjected to the action of either protein kinase inhibitors or sodium metavanadate, the final effect of these interactions is the same. According to this hypothesis, one can assume that the ability of cells to initiate PCC arises not only as the consequence of direct inhibition of CDK activity, but also as a secondary effect connected with the reduction in the activity of protein Cdc25 phosphatase, i.e., an enzyme indispensable for the proper formation of the catalytic region in the cyclin B/CDK complex. Explaining the action mechanism of sodium metavanadate as a PCC inductor is not easy. The investigations performed by Ghosh et al. (1998) show that the percentage of HeLa cells (unsubjected to the replication stress) prematurely initiating mitosis under the influence of okadaic acid (inhibitor of phosphatase 1 and 2A; PP-1, PP-2A), decrease steadily after 3-h action of Van, but clearly increase after the incubation prolonged by successive 2-h periods. The simulation of PCC with a mixture of okadaic acid and sodium metavanadate is accompanied by the total disappearance of cyclin B. These results seem to indicate the possibility of PCC induction independent of the MPF activation process. It seems that the factors causing premature mitosis initiation may be, in this case, members of the Nek (NIMA-related kinase) family of serine/threonine protein kinases (Noguchi et al. 2002), being the homologues of NIMA kinase (Never-In-Mitosis, gene A) in Aspergillus nidulans. Production of the stable form of this protein in HeLa cells can induce PCC without the contribution of Cdk1/cyclin B complexes (O’Connell et al. 1994). It seems that an alternative pathway of mitosis activation related to MPF is now the only possible explanation the PCC induction under the influence of Van in HeLa cells subjected to replication stress.
Chemically induced premature mitosis is a phenomenon seen in many species (from plants to mammals; Gotoh and Durante 2006; Nghiem et al. 2001; Rybaczek et al. 2008). In our studies, we also observed the induction of premature mitosis in HeLa cells treated with ST, CF, 2-AP and Van. Neither the three protein kinase inhibitors: caffeine, 2-aminopurine and staurosporine nor sodium metavanadate, a potent protein phosphatase inhibitor, abrogate the S-M dependency to a large extent (no more than 10% in the case of the most effective, staurosporine). Steinmann et al. (1991) found that the HeLa cells arrested in the S-phase for 3–5 h with 2.5 mM HU and then treated with CF, 6-DMAP, or 2-AP for 8 h in the presence of nocodazole (or treated with okadaic acid for 6 h) displayed little or no response to any of the compounds (% of premature mitosis <1). They reported that DNA damage observed in HeLa cells often resulted in a delay in the G2 → M progression, and that suppression of DNA damage-induced G2 delay due to CF, 2-AP, and 6-DMAP action also was a form of premature mitosis. In the present work, apart from the above-mentioned suppression of G2 delay, the induction of PCC from S- and G2-phase was investigated (through the joint treatment involving two kinds of inhibitor: hydroxyurea, which slows down S-phase progression, combined with the others, which affect the activity of protein kinases or protein phosphatases). We affirm that the frequency of premature cell divisions was then higher than in the case of solutions without HU. Thus, the tests performed clearly indicate the synergic effect of hydroxyurea upon the PCC inductors used. Although each of these compounds is characterized by different biological activity and interaction with various target protein molecules (Fig. 2), the end result of their combined action during PCC induction shows a cumulative effect. Despite the fact that the interpretation of cumulative effects is not easy in the light of current information about biochemical pathways connected with PCC induction, it seems possible that one cannot exclude the separate, but facilitating action of each of the two inhibitors used in the mixture: hydroxyurea would play then the role of a classical suppressor of replication activity, while the PCC inductor would interfere to inhibit stimulation of biochemical pathways leading to initiation of PCC despite its failure to realize the replication process. The results of the tests permitted a penetrating analysis of the available scientific literature allowing us to state that the effectiveness of PCC induction depends not only on the biological material subjected to tests (e.g., cell line), but also on the type of PCC inductor, its concentration and, to a great extent, on the experimental methodology used.
Premature entry into mitosis often is associated with the formation of micronuclei, multi- or unipolar mitotic spindles, lagging chromosomes and subsequently apoptosis. The staining of tubulin subunits in HT-29 cells (colorectal adenocarcinoma) undergoing mitotic catastrophe showed an increase in the number of cells with atypically distributed tubulin subunits (Riesterer et al. 2009 and references therein).
Our immunofluorescence observations show that the action of the PCC inductors used in this study results in a clear decrease in the content of active form of Chk1 kinase in comparison with the material subjected to the action of 2.5 mM HU. The immunofluorescence analysis of cell nuclei following the use of antibodies recognizing the phosphorylation of Chk1 kinase on Ser317 shows that this reduction constitutes not only the effect of lower number of intranuclear focuses concentrating the modified molecules of this kinase, but above all, also that of the relative number of cells, in which its activation process took place. The presence of sodium metavanadate decreases the phosphorylation of this kinase to a lesser extent, but in spite of this one cannot assume that sodium metavanadate shows the properties of a phosphorylation inductor of Chk1 kinase (Fig. 4; comp. Zhang et al. 2001, 2003). Such an effect can result from the intensified expression (and activation) of Chk1 kinase, the excitation of catalytic functions superior when compared to sensory kinases or from the increased number of double-stranded breaks in DNA molecules induced by the genotoxic influence of sodium metavanadate (Fig. 8a, b; Ivancsits et al. 2002). We presume (according to Huang et al. 2006) that H2AX phosphorylation is not an ever-present feature of mitotic cells. However, in our opinion, during chemically induced premature mitosis HeLa cells acquire double-stranded DNA breaks (DSBs) and Chk1 phosphorylation at Ser317 is an essential part of the response to DNA damage produced during the S-phase. Ewald et al. (2007) have shown that H2AX histone is phosphorylated on Ser139 in response to the inhibition of DNA replication by gemcitabine (a concentration- and time-dependent phosphorylation), and consequently caused by the activation of the S-phase checkpoint pathway (Chk1/Cdc25A/cyclin-dependent kinase 2). Moreover, testing of the cells with Ml-1 and OCI-AML3 (human adult myelogenous leukemia cell lines) against the Chk1 inhibitor 7-hydroxystaurosporine (UCN-01), after previous pretreatment with gemcitabine, caused a tenfold increase in the phosphorylation of Ser139 histone H2AX and even the appearance of a strong staining of the pan-nuclear type (comp. Fig. 7; image after incubation in 10 mM HU). S23906, a novel acronycine derivative, induces mitotic catastrophe in the human cell lines HT-29 and HeLa, which is accompanied by both the induction of Ser139 histone H2AX phosphorylation and the phosphorylation-activating Chk1 kinase (Cahuzac et al. 2010).
The beginning of mitosis is correlated with the change in location of Cdc25C phosphatase, a major MPF activator. Despite some controversy, it is generally accepted that as with Cdk1/cyclin B1 complex, whilst Cdc25C phosphatase is present on the site of cytoplasm during interphase, during prophase it displaces to the cell nucleus (Kumagai and Dunphy 1999). The basic mitotic processes proceeding in the cell nucleus such as the breakdown of nuclear envelope are connected with the phosphorylations of specified substrates (e.g., nuclear lamins) catalyzed by Cdk1/cyclin B1 complexes. Their migration from the cytoplasm area, preceding the induction of many phenomena taking place within the area of nucleoplasm seems then to be an equally obvious and necessary event. The sequence of events, commencing with the translocation of Cdk1/cyclin B1 complexes resulting in the first symptoms of mitosis can be observed in numerous experiments. The breakdown of nuclear envelope in frog oocytes is inhibited when cyclins cannot migrate to the nucleus due to mutation within the CRS sequence (Walsh et al. 2003). Genetic modifications leading to the accumulation of considerable quantities of dephosphorylated active Cdk1 molecules, whose location is restricted to the area of cytoplasm, do not cause any symptoms typical of the premature condensation of chromosomes and do not initiate the breakdown of nuclear envelope. The expression of mutated Cdk1AF molecules, incapable of phosphorylation within the inhibition regions, leads to a considerable increase in the cytoplasmic activity of Cdk1 in mammalian cells. The characteristic phenomenon of premature mitosis is, however, not observed, which might explain the relatively small population of HeLa cells inducing PCC.
From these data we conclude that (1) caffeine, 2-aminopurine, staurosporine and sodium metavanadate abrogate the hydroxyurea-induced S-phase arrest in HeLa cells and induce premature chromosome condensation, suggesting the following sequence of effectiveness (from most to least effective): staurosporine > sodium metavanadate > caffeine > 2-aminopurine; (2) induction of DSB-like DNA damage associated with either inhibition of DNA replication by hydroxyurea or overriding of the S-phase checkpoints occurs leading to induction of PCC; (3) the actin and tubulin network undergoes several rearrangements either following replication stress or via drug-induced delay but in a manner that does not permit successive subperiods of the chemically induced premature mitosis. This being so, including elucidation of the chemical mechanisms involved in the induction of PCC, better understanding of the purpose and function of the Chk1, H2AX, or other proteins in the S-phase checkpoint response will be essential if practical applications of such scientific studies are to follow.
Acknowledgments
This work was supported by the Ministry of Science and Higher Education, grant number: No. N303 355935 (the contract no. 3559/B/P01/2008/35).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- 2-AP
2-Aminopurine
- ATM
Ataxia telangiectasia-mutated
- ATR
ATM- and Rad3-related
- CF
Caffeine
- Chk1
Checkpoint kinase 1
- HU
Hydroxyurea
- MN
Micronucleus
- Van
Sodium metavanadate
- PCC
Premature chromosome condensation
- RNR
Ribonucleotide reductase
- ST
Staurosporine
Contributor Information
Dorota Rybaczek, Phone: +48-42-6354514, FAX: +48-42-6354514, Email: doryb@biol.uni.lodz.pl, Email: dorota.rybaczek@gmail.com.
Magdalena Kowalewicz-Kulbat, Email: mkow@biol.uni.lodz.pl.
References
- Ajiro K, Yasuda H, Tsuji H. Vanadate triggers the transition from chromosome condensation to decondensation in a mitotic mutant (tsTM13) inactivation of p34cdc2/H1 kinase and dephosphorylation of mitosis-specific histone H3. Eur J Biochem. 1996;241:923–930. doi: 10.1111/j.1432-1033.1996.00923.x. [DOI] [PubMed] [Google Scholar]
- Alvino GM, Collingwood D, Murphy JM, Delrow J, Brewer BJ, Raghuraman MK. Replication in hydroxyurea: it’s a matter of time. Mol Cell Biol. 2007;27:6396–6406. doi: 10.1128/MCB.00719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiri A, Noei F, Feroz T, Lee JM. Geldanamycin anisimycins activate Rho and stimulate Rho- and ROCK-dependent actin stress fiber formation. Mol Cancer Res. 2007;5:933–942. doi: 10.1158/1541-7786.MCR-06-0362. [DOI] [PubMed] [Google Scholar]
- An J, Huang YC, Xu QZ, Zhou LJ, Shang ZF, Huang B, Wang Y, Liu XD, Wu DC, Zhou PK (2010) DNA-PKcs plays a dominant role in the regulation of H2AX phosphorylation in response to DNA damage and cell cycle progression. BMC Mol Biol. doi:10.1186/1471-2199-11-18 [DOI] [PMC free article] [PubMed]
- Andreassen PR, Margolis RL. Induction of partial mitosis in BHK cells by 2-aminopurine. J Cell Sci. 1991;100:290–310. doi: 10.1242/jcs.100.2.299. [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- Bezrookove V, Smits R, Moeslein G, Fodde R, Tanke HJ, Raap AK, Darroudi F. Premature chromosome condensation revisited: a novel chemical approach permits efficient cytogenetic analysis of cancers. Genes Chromosomes Cancer. 2003;38:177–186. doi: 10.1002/gcc.10268. [DOI] [PubMed] [Google Scholar]
- Bretscher A. Microfilament structure and function in the cortical cytoskeleton. Annu Rev Cell Biol. 1991;7:337–374. doi: 10.1146/annurev.cb.07.110191.002005. [DOI] [PubMed] [Google Scholar]
- Brinkley BR, Zinkowski RP, Mollon WL, Davis FM, Pisegna MA, Penhouse M, Rao PN. Movement and segregation of kinetochores experimentally detached from mammalian chromosomes. Nature. 1998;336:251–254. doi: 10.1038/336251a0. [DOI] [PubMed] [Google Scholar]
- Cahuzac N, Studény A, Marshall K, Versteege I, Wetenhall K, Pfeiffer B, Léonce S, Hickman JA, Pierré A, Glosteyn RM. An unusual DNA binding compound, S23906, induces mitotic catastrophe in cultured human cells. Cancer Lett. 2010;289:178–197. doi: 10.1016/j.canlet.2009.08.014. [DOI] [PubMed] [Google Scholar]
- Chanoux RA, Yin B, Urtishak KA, Asare A, Bassing CH, Brown EJ. ATR and H2AX cooperate in maintaining genome stability under replication stress. J Biol Chem. 2009;284:5994–6003. doi: 10.1074/jbc.M806739200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke CAL, Clarke PR. DNA-dependent phosphorylation of Chk1 and Claspin in a human cell-free system. Biochem J. 2005;388:705–712. doi: 10.1042/BJ20041966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D. Caffeine inhibits checkpoint responses without inhibiting the ataxia-telangiectasia-mutated (ATM) and ATM- and Rad3-related (ATR) protein kinases. J Biol Chem. 2003;278:37139–37145. doi: 10.1074/jbc.M307088200. [DOI] [PubMed] [Google Scholar]
- Dai Y, Grant S. New insight into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res. 2010;16:376–383. doi: 10.1158/1078-0432.CCR-09-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova DS, Gilbert DM. Temporally coordinated assembly and disassembly of replication factories in the absence of DNA synthesis. Nat Cell Biol. 2000;2:686–694. doi: 10.1038/35036309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto M, Goto H, Tomono Y, Kasahara K, Tsujimura K, Kiyono T, Inagaki M. Novel positive feedback loop between Cdk1 and Chk1 in the nucleus during G2/M transition. J Biol Chem. 2009;284:34223–34230. doi: 10.1074/jbc.C109.051540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enserink JM, Smolka MB, Zhou H, Kolodner RD. Checkpoint proteins control morphogenetic events during DNA replication stress in Saccharomyces cerevisae. J Cell Biol. 2006;157:729–741. doi: 10.1083/jcb.200605080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald B, Sampath D, Plunkett W. H2AX phosphorylation marks gemcitabine-induced stalled replication forks and their collapse upon S-phase checkpoint abrogation. Mol Cancer Ther. 2007;6:1239–1248. doi: 10.1158/1535-7163.MCT-06-0633. [DOI] [PubMed] [Google Scholar]
- Feijoo C, Hall-Jackson C, Wu R, Jenkins D, Leitch J, Gilbert DM, Smythe C. Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J Cell Biol. 2001;154:913–923. doi: 10.1083/jcb.200104099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RD, Burke DJ. The spindle checkpoint: two transitions, two pathways. Trends Cell Biol. 2000;10:154–158. doi: 10.1016/S0962-8924(00)01727-X. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Paweletz N. Centrosome-kinetochore interaction in multinucleate cells. Chromosoma. 1987;95:136–143. doi: 10.1007/BF00332186. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Paweletz N, Schroter D. Effects of okadaic acid on mitotic HeLa cells. J Cell Sci. 1992;103:117–124. doi: 10.1242/jcs.103.1.117. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Schroeter D, Paweletz N. Okadaic acid overrides the check point and accelerates progression of G2 phase to induce premature mitosis in HeLa cells. Exp Cell Res. 1996;227:165–169. doi: 10.1006/excr.1996.0262. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Paweletz N, Schroeter D. Cdc2-independent induction of premature mitosis by okadaic acid in HeLa cells. Exp Cell Res. 1998;242:1–9. doi: 10.1006/excr.1998.4115. [DOI] [PubMed] [Google Scholar]
- Gotoh E, Durante M. Chromosome condensation outside of mitosis: mechanisms and new tools. J Cell Physiol. 2006;209:297–304. doi: 10.1002/jcp.20720. [DOI] [PubMed] [Google Scholar]
- Guo Z, Kumagai A, Wang SX, Dunphy WG. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 2000;14:2745–2756. doi: 10.1101/gad.842500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha G-H, Kim H-S, Lee CG, Park H-Y, Kim E-J, Shin H-J, Lee J-C, Lee K-W, Lee C-W. Mitotic catastrophe is the predominant response to histone acetyltransferase depletion. Cell Death Differ. 2009;16:483–497. doi: 10.1038/cdd.2008.182. [DOI] [PubMed] [Google Scholar]
- Halicka HD, Huang X, Traganos F, King MA, Dai W, Darzynkiewicz Z. Histone H2AX phosphorylation after cell irradiation with UV-B. Cell Cycle. 2005;4:339–345. doi: 10.4161/cc.4.2.1486. [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- Huang S, Qu LK, Cuddihy AR, Ragheb R, Taya Y, Koromilas AE. Protein kinase inhibitor 2-aminopurine overrides multiple genotoxic stress-induced cellular pathways to promote cell survival. Oncogene. 2003;22:3721–3733. doi: 10.1038/sj.onc.1206490. [DOI] [PubMed] [Google Scholar]
- Huang X, Kurose A, Tanaka T, Traganos F, Dai W, Darzynkiewicz Z. Sequential phosphorylation of Ser-10 on histone H3 and Ser-139 on histone H2AX and ATM activation during premature chromosome condensation: relationship to cell-cycle phase and apoptosis. Cytometry A. 2006;69:222–229. doi: 10.1002/cyto.a.20257. [DOI] [PubMed] [Google Scholar]
- Huang H, Fletcher L, Beeharry N, Daniel R, Kao G, Yen TJ, Muschel RJ. Abnormal cytokinesis after X-irradiation in tumor cells that override the G2 DNA damage checkpoint. Cancer Res. 2008;68:3724–3732. doi: 10.1158/0008-5472.CAN-08-0479. [DOI] [PubMed] [Google Scholar]
- Ivancsits S, Pilger A, Diem E, Schaffer A, Rudiger HW. Vanadate induces DNA strand breaks in cultured human fibroblasts at doses relevant to occupational exposure. Mutat Res. 2002;519:25–35. doi: 10.1016/s1383-5718(02)00138-9. [DOI] [PubMed] [Google Scholar]
- Kinoshita M, Takeda S. Connecting the dots between septins and the DNA damage checkpoint. Cell. 2007;7:777–779. doi: 10.1016/j.cell.2007.08.022. [DOI] [PubMed] [Google Scholar]
- Krause SA, Loupert M-L, Vass S, Schoefelder S, Harrison S, Heck MMS. Loss of cell cycle checkpoint control in Drosophila RFC4 mutants. Mol Cell Biol. 2001;21:5156–5168. doi: 10.1128/MCB.21.15.5156-5168.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Dunphy WG. Binding of 14–3-3 proteins and nuclear export control the intracellular localization of the mitotic inducer Cdc25. Genes Dev. 1999;13:1067–1072. doi: 10.1101/gad.13.9.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Guo Z, Emami KH, Wang SX, Dunphy WG. The Xenopus Chk1 protein kinase mediates a caffeine-sensitive pathway of checkpoint control in cell-free extracts. J Cell Biol. 1998;142:1559–1569. doi: 10.1083/jcb.142.6.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laredo J, Huynh A, Muller C, Jaffrézou J-P, Bailly J-D, Cassar G, Laurent G, Demur C. Effect of the protein kinase C inhibitor staurosporine on chemosensitivity to daunorubicin of normal and leukemic fresh myeloid cells. Blood. 1994;84:229–237. [PubMed] [Google Scholar]
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. doi: 10.1101/gad.840500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löffler H, Rebacz B, Ho AD, Lukas J, Bartek J, Krämer A. Chk1-dependent regulation of Cdc25B functions to coordinate mitotic events. Cell Cycle. 2006;5:2543–2547. doi: 10.4161/cc.5.21.3435. [DOI] [PubMed] [Google Scholar]
- Löffler H, Bochtler T, Fritz B, Tews B, Ho AD, Lukas J, Bartek J, Krämer A. DNA damage-induced accumulation of centrosomal Chk1 contributes to its checkpoint function. Cell Cycle. 2007;6:2541–2548. doi: 10.4161/cc.6.20.4810. [DOI] [PubMed] [Google Scholar]
- Maya-Mendoza A, Petermann E, Gillespie DA, Caldecott KW, Jackson DA. Chk1 regulates the density of active replication origins during the vertebrate S phase. EMBO J. 2007;26:2719–2731. doi: 10.1038/sj.emboj.7601714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merry C, Fu K, Wang J, Yeh IJ, Zhang Y. Targeting the checkpoint kinase Chk1 in cancer therapy. Cell Cycle. 2010;9:279–283. doi: 10.4161/cc.9.2.10445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer RK, Burger MM, Tschannen R, Schafer R. Actin filaments bundles in Vaccinia Virus infected fibroblasts. Arch Virol. 1981;67:11–18. doi: 10.1007/BF01314597. [DOI] [PubMed] [Google Scholar]
- Mukherjee B, Kessinger C, Kobayashi J, Chen BP, Chen DJ, Chatterjee A, Burma S. DNA-PK phosphorylates histone H2AX during apoptotic DNA fragmentation in mammalian cells. DNA Repair. 2006;5:575–590. doi: 10.1016/j.dnarep.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Nghiem P, Park PK, Kim Y, Vaziri C, Schreiber SL. ATR inhibition selectively sensitizes G1 checkpoint-deficient cells to lethal premature chromatin condensation. Proc Natl Acad Sci U S A. 2001;98:9092–9097. doi: 10.1073/pnas.161281798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niida H, Nakanishi M. DNA damage checkpoints in mammals. Mutagenesis. 2006;21:3–9. doi: 10.1093/mutage/gei063. [DOI] [PubMed] [Google Scholar]
- Noguchi K, Fukazawa H, Murakami Y, Uehara Y. Nek11, a new member of the NIMA family of kinases, involved in DNA replication and genotoxic stress responses. J Biol Chem. 2002;277:39655–39665. doi: 10.1074/jbc.M204599200. [DOI] [PubMed] [Google Scholar]
- O’Connell MJ, Norbury C, Nurse P. Premature chromatin condensation upon accumulation of NIMA. EMBO J. 1994;13:4926–4937. doi: 10.1002/j.1460-2075.1994.tb06820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peddibhotla S, Lam MH, Gonzalez-Rimbau M, Rosen JM. The DNA-damage effector checkpoint kinase 1 is essential for chromosome segregation and cytokinesis. Proc Natl Acad Sci U S A. 2009;106:5159–5164. doi: 10.1073/pnas.0806671106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petretti C, Savoian M, Montembault E, Glover DM, Prigent C, Giet R. The PITSLRE/CDK11p58 protein kinase promotes centrosome maturation and bipolar spindle formation. EMBO Rep. 2006;7:418–424. doi: 10.1038/sj.embor.7400639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podhorecka M, Składanowski A, Bozko P (2010) H2AX phosphorylation: its role in DNA damage response and cancer therapy. J Nucl Acids. doi:10.4061/2011/920161 [DOI] [PMC free article] [PubMed]
- Rieder CL, Cole RW, Khodjakov A, Sluder G. The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitor signal produced by unattached kinetochores. J Cell Biol. 1995;130:941–948. doi: 10.1083/jcb.130.4.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesterer O, Matsumoto F, Wang L, Pickett J, Molkentine D, Giri U, Milas L, Raju U (2009) A novel Chk inhibitor, XL-844, increases human cancer cell radiosensitivity through promotion of mitotic catastrophe. Invest New Drugs. doi:10.1007/s10637-009-9361-2 [DOI] [PubMed]
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand break in vivo. J Cell Biol. 1999;146:905–915. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybaczek D, Maszewski J. Induction of foci of phosphorylated H2AX histones and premature chromosome condensation after DNA damage in Vicia faba root meristem. Biol Plant. 2007;51:443–450. doi: 10.1007/s10535-007-0094-9. [DOI] [Google Scholar]
- Rybaczek D, Bodys A, Maszewski J. H2AX foci in late S/G2- and M-phase cells after hydroxyurea- and aphidicolin-induced DNA replication stress in Vicia. Histochem Cell Biol. 2007;128:227–241. doi: 10.1007/s00418-007-0311-9. [DOI] [PubMed] [Google Scholar]
- Rybaczek D, Żabka A, Pastucha A, Maszewski J. Various chemical agents can induce premature chromosome condensation in Vicia faba. Acta Physiol Plant. 2008;30:663–672. doi: 10.1007/s11738-008-0163-5. [DOI] [Google Scholar]
- Savitsky PA, Finkel T. Redox regulation of Cdc25C. J Biol Chem. 2002;277:20535–20540. doi: 10.1074/jbc.M201589200. [DOI] [PubMed] [Google Scholar]
- Schepis A, Schramm B, de Haan CAM, Locker JK. Vaccinia virus-induced microtubule-dependent cellular rearrangements. Traffic. 2006;7:308–323. doi: 10.1111/j.1600-0854.2005.00381.x. [DOI] [PubMed] [Google Scholar]
- Schmitt E, Boutros R, Froment C, Monsarrat B, Ducommun B, Dozier C. CHK1 phosphorylates CDC25B during the cell cycle in the absence of DNA damage. J Cell Sci. 2006;119:4269–4275. doi: 10.1242/jcs.03200. [DOI] [PubMed] [Google Scholar]
- Sen R, Ghosh S. Induction of premature mitosis in S-blocked onion cells. Cell Biol Int. 1998;22:867–874. doi: 10.1006/cbir.1998.0315. [DOI] [PubMed] [Google Scholar]
- Shimizu T, O’Connor PM, Kohn KW, Pommier Y. Unscheduled activation of cyclin B1/Cdc2 kinase in human promyelocytic leukemia cell line HL60 cells undergoing apoptosis induced by DNA damage. Cancer Res. 1995;55:228–231. [PubMed] [Google Scholar]
- Steinmann KE, Belinsky GS, Lee D, Schlegel R. Chemically induced premature mitosis: differential response in rodent and human cells and the relationship to cyclin B synthesis and p34cdc2/cyclin B complex formation. Proc Natl Acad Sci U S A. 1991;88:6843–6847. doi: 10.1073/pnas.88.15.6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szollosi D, Czolowska R, Soltynska MS, Tarkowski AK. Ultrastructure of cell fusion and premature chromosome condensation (PCC) of thymocyte nuclei in metaphase II mouse oocytes. Biol Cell. 1986;56:239–250. doi: 10.1111/j.1768-322x.1986.tb00455.x. [DOI] [PubMed] [Google Scholar]
- Tolbert PE, Shy CM, Allen JW. Micronuclei and other nuclear anomalies in buccal smears: methods development. Mutat Res. 1992;271:69–77. doi: 10.1016/0165-1161(92)90033-i. [DOI] [PubMed] [Google Scholar]
- Walsh S, Margolis SS, Kornbluth S. Phosphorylation of the cyclin B1 cytoplasmic retention sequence by mitogen-activated protein kinase and Plx. Mol Cancer Res. 2003;1:280–298. [PubMed] [Google Scholar]
- Wang S-W, Norbury C, Harris AL, Toda T. Caffeine can override the S-M checkpoint in fission yeast. J Cell Sci. 1999;112:927–937. doi: 10.1242/jcs.112.6.927. [DOI] [PubMed] [Google Scholar]
- Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;51:47759–47762. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- Wittmann T, Hyman A, Desai A. The spindle: a dynamic assembly of microtubules and motors. Nat Cell Biol. 2001;3:E28–E34. doi: 10.1038/35050669. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Usui T, Tsujimura K, Inagaki M, Beppu T, Horinouchi S. Biochemical differences between staurosporine-induced apoptosis and premature mitosis. Exp Cell Res. 1997;232:225–239. doi: 10.1006/excr.1997.3538. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Huang C, Li J, Leonard SS, Lanciotti R, Butterworth L, Shi X. Vanadate-induced cell growth regulation and the role of reactive oxygen species. Arch Biochem Biophys. 2001;392:311–320. doi: 10.1006/abbi.2001.2464. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Leonard SS, Huang C, Vallyathan V, Castranova V, Shi X. Role of reactive oxygen species and MAPKs in vanadated-induced G2/M phase arrest. Free Radic Biol Med. 2003;34:1333–1342. doi: 10.1016/S0891-5849(03)00145-X. [DOI] [PubMed] [Google Scholar]
- Zhang YW, Otterness DM, Chiang GG, Xie W, Liu YC, Mercurio F, Abraham RT. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol Cell. 2005;19:607–618. doi: 10.1016/j.molcel.2005.07.019. [DOI] [PubMed] [Google Scholar]
- Zhou BB, Bartek J. Targeting the checkpoint kinases: chemosensitization versus chemoprotection. Nat Rev Cancer. 2004;4:216–225. doi: 10.1038/nrc1296. [DOI] [PubMed] [Google Scholar]