

Abstract
Influenza virus non-structural protein 1 (NS1) is the centrepiece of the viral response to the host interferon (IFN) system. NS1 has been demonstrated previously to be a potential therapeutic target for antiviral therapy by identification of specific small-molecule inhibitors. This study demonstrated the biological mechanism for a potent new NS1 antagonist. Compound JJ3297 inhibited virus replication by more than three orders of magnitude without affecting cell viability. Importantly, it efficiently reversed NS1-induced inhibition of IFN mRNA production. The hypothesis was tested that JJ3297 facilitates IFN production in infected cells, leading to protection of the surrounding uninfected cells. Accordingly, the compound efficiently prevented virus spread through a cell population during a 48 h multi-cycle infection initiated at a very low m.o.i. Consistent with the hypothesis, the compound had no detectable influence on a 6 h single-cycle infection initiated at a high m.o.i. The effect of JJ3297 on virus replication was not caused by inhibition of NS1 expression or its mislocalization in the cell. JJ3297 facilitated the induction of an IFN-like antiviral state, resulting in increased resistance to subsequent challenge with vesicular stomatitis virus. The activity of JJ3297 absolutely required the function of cellular RNase L, indicating that an intact IFN system is required for function of the compound. These results support a model in which inhibition of NS1 function results in restoration of the IFN-induced antiviral state and inhibition of virus replication and spread. This represents a new direction for anti-influenza virus drug development that exploits the IFN pathway to challenge virus replication.
INTRODUCTION
Influenza continues to be a significant global public health problem, with 3–5 million severe cases annually, including 250 000–500 000 deaths worldwide (WHO, 2009). The seasonal vaccination programme remains vulnerable to antigenic drift. In addition, newly emergent strains periodically cause pandemics of unpredictable consequence, including the recent swine H1N1 pandemic (Garten et al., 2009; Jain et al., 2009; Pekosz & Glass, 2008; Reid et al., 2001; Taubenberger et al., 2001).
High mutation rates among circulating strains cause problems not only with vaccine efficacy but also with antiviral therapeutics. Currently, there is only one class of drug used for treatment of influenza in the USA, the neuraminidase inhibitors such as oseltamivir. However, 98 % of seasonal A/H1N1 strains in North America in 2009 were resistant to oseltamivir (Layne et al., 2009). Therefore, considering the uncertainty of seasonal vaccine efficacy, the potential for drug resistance to neuraminidase inhibitors and the continuing threat of severe pandemics, the development of novel antivirals is imperative.
We have identified non-structural protein 1 (NS1) as a candidate target for drug development (Basu et al., 2009). NS1 satisfies an important requirement for a viable drug target by being involved in several processes that are crucial for successful virus replication and propagation (Hale et al., 2008). Genetic analyses of NS1 have shown that virus replication, spread and pathogenesis are highly dependent on the function of this protein (Basler & Aguilar, 2008; Basler et al., 2001; Bürger et al., 1985; Donelan et al., 2003; Egorov et al., 1998; Falcón et al., 2005; Garaigorta et al., 2005; García-Sastre et al., 1998; Hatada et al., 1990; Jackson et al., 2008; Kochs et al., 2007b; Koennecke et al., 1981; Ludwig et al., 1995; Shimizu et al., 1983; Talon et al., 2000; Wolstenholme et al., 1980). Among the key features of NS1 is its capacity for RNA binding, including to dsRNA (Chien et al., 2004; Donelan et al., 2003; Hatada & Fukuda, 1992; Hatada et al., 1992, 1997; Lu et al., 1995; Qian et al., 1995; Qiu & Krug, 1994; Qiu et al., 1995; Wang et al., 1999). One of the consequences of NS1 binding to dsRNA is to bind and sequester viral dsRNAs from detection by 2′-5′ oligo(A) synthetase (OAS), a cellular sensor of viral infection (Min & Krug, 2006). Activated OAS synthesizes 2′-5′ oligoadenylates, which in turn stimulate latent RNase L, leading to viral RNA degradation (Silverman, 2007a, b). NS1 also binds and inhibits the activity of protein kinase R (PKR), inhibiting the PKR-dependent phosphorylation of eIF-2α and thereby avoiding shutdown of viral protein synthesis by PKR (Li et al., 2006; Lu et al., 1995; Min et al., 2007). Other important roles of NS1 include binding to the 30 kDa subunit of cleavage and polyadenylation specificity factor (CPSF30), and also to poly(A)-binding protein II (PABPII) (Chen et al., 1999; Kochs et al., 2007a; Krug et al., 2003; Kuo & Krug, 2009; Nemeroff et al., 1998; Noah et al., 2003; Twu et al., 2007). These activities lead to inhibition of 3′-end processing of cellular pre-mRNAs and prevention of their nuclear export, including interferon (IFN) mRNAs synthesized in response to viral infection. Further inhibition of cellular mRNA export is achieved by the interaction of NS1 with components of the nuclear pore complex (Satterly et al., 2007). Finally, NS1 has recently been shown to interact directly with TRIM25, an E3 ubiquitin ligase responsible for activation of retinoic acid-inducible gene I (RIG-I), which normally leads to downstream signalling and stimulation of the IFN response (Guo et al., 2007; Ludwig & Wolff, 2009; Mibayashi et al., 2007; Pichlmair et al., 2006). NS1 binds to TRIM25, inhibits RIG-I ubiquitination and thus prevents the synthesis of cellular IFN (Gack et al., 2009).
We have previously described four inhibitors of influenza replication that act by specific inhibition of NS1 function (Basu et al., 2009). These were identified from a chemical library based on their ability to suppress the slow-growth phenotype of budding yeast expressing NS1 protein. Here, we report a potent novel NS1 inhibitor with some chemical features retained from one of the original four NS1 antagonists. We demonstrate that its biological mechanism of virus inhibition involves prevention of virus spread in cell culture by establishment of an antiviral state that depends on RNase L, a major component of the IFN system.
RESULTS
Inhibition of NS1 function by compound JJ3297
Previously, we described the identification of compound NSC125044, which specifically inhibits NS1 function in yeast and mammalian cells and also specifically inhibits influenza virus replication in cell culture (Basu et al., 2009). Compound JJ3297 was synthesized based on key structural features of NSC125044, along with additional chemical modifications to improve its efficacy. The structure of JJ3297 is shown in Fig. 1. Its design and synthesis will be reported elsewhere (J. J. Jablonski, D. Basu, D. A. Engel and H. M. Geysen, unpublished results). To relate the activity of JJ3297 to NSC125044, each was used to challenge NS1 function in cells infected with influenza virus. In infected cells, one important function of NS1 is to block IFN gene expression (García-Sastre et al., 1998; Hale et al., 2008). Previously, we demonstrated that treatment of infected cells with NSC125044 restored IFN-β mRNA expression, consistent with its ability to inhibit NS1 function. To test the effect of JJ3297 on IFN-β mRNA expression, Madin–Darby canine kidney (MDCK) cells were infected with A/PR/8 at an m.o.i. of 2 in the presence or absence of the compound. As shown in Fig. 2(a) (upper panel), after 6 h of infection and treatment, JJ3297 strongly restored IFN-β mRNA levels, to a degree nearly equal to that seen in uninfected cells treated with poly(I : C). As reported previously for NSC125044, treatment of cells with JJ3297 alone, in the absence of virus infection, had no effect on IFN mRNA levels (Fig. 2a, lower panel), demonstrating that JJ3297 does not act directly to induce IFN production, but rather acts only in the context of infection. These data indicated that JJ3297 reverses the blockade of IFN synthesis that normally occurs in infected cells due to the action of NS1. Previously, we also reported that NS1 expression in Saccharomyces cerevisiae triggered a slow-growth phenotype and that specific inhibition of NS1 function by NSC125044 restored growth of the yeast. As expected, JJ3297 also restored growth of yeast cells expressing NS1 (data not shown). These data demonstrated that JJ3297 and NSC125044 share essential chemical features leading to the inhibition of NS1 function.
Fig. 1.
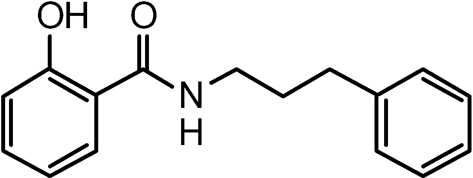
Chemical structure of JJ3297.
Fig. 2.
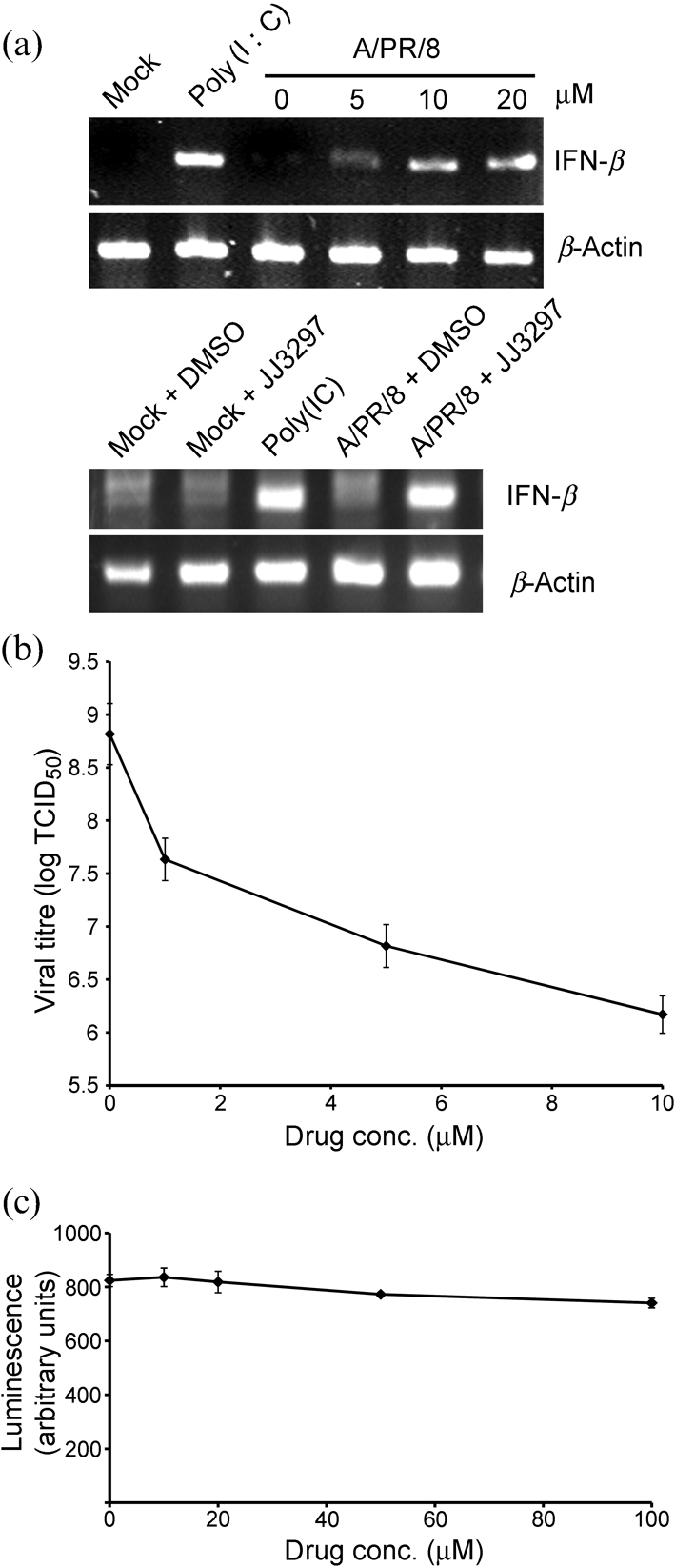
JJ3297-dependent restoration of IFN-β mRNA levels and inhibition of virus replication in MDCK cells. (a) Upper panel: cells were mock infected, treated with poly(I : C) or infected with influenza strain A/PR/8 at an m.o.i. of 2 and treated with increasing concentrations of JJ3297 as indicated or with 1 % DMSO (0 μM). After 6 h, cells were harvested for RT-PCR analysis of IFN-β and β-actin mRNA levels. Lower panel: cells were mock infected, treated with poly(I : C) or infected with influenza A/PR/8 at an m.o.i. of 2 and treated with either 1 % DMSO or 5 μM JJ3297 as indicated. After 6 h, cells were harvested for RT-PCR analysis of IFN-β and β-actin mRNAs. (b) Triplicate cultures were infected with A/PR/8 at an m.o.i. of 0.1 and treated with the indicated concentrations of JJ3297 or 1 % DMSO (0 μM). After 48 h, the supernatants were collected and analysed to determine TCID50. (c) Triplicate cultures were incubated in the presence of JJ3297 at the indicated concentrations for 48 h. Cells were disrupted with CellTiter Glo reagent according to manufacturer's protocol and the luminescent signal was measured, indicating the extent of cell viability.
Inhibition of virus replication
To determine the effect of JJ3297 on virus replication, cells infected at an m.o.i. of 0.1 were treated with increasing concentrations of the compound for 48 h followed by analysis of the culture supernatants by TCID50 assay. As shown in Fig. 2(b), virus replication was inhibited by approximately three orders of magnitude over a 10 μM range of concentration. The 50 % effective concentration (EC50) value for JJ3297 was 0.8 μM (n=3 experiments, one of which is shown in Fig. 2b). This represented a more than tenfold improvement in EC50 compared with NSC125044, as reported previously. Fig. 2(c) shows that JJ3297 was not toxic to MDCK cells up to at least 100 μM over a 48 h incubation. The calculated selective index for JJ3297 was at least 125.
Inhibition of multi-cycle but not single-cycle replication
The ability of JJ3297 to reverse the inhibition of IFN mRNA induction by NS1 suggested a straightforward biological mechanism for inhibition of virus replication. We explored the hypothesis that treatment of infected cells with the compound limits virus replication through restoration of IFN expression, leading to protection of surrounding cells. One prediction of this hypothesis is that JJ3297 would act efficiently in the context of a low-multiplicity (multi-cycle) infection, in which newly released IFN could protect surrounding uninfected cells but would not act in a single-cycle (high m.o.i.) infection in which 100 % of cells are infected prior to the restoration of IFN synthesis caused by JJ3297.
Accordingly, MDCK cells were infected at an m.o.i. of either 0.01 (low) or 3 (high) with A/PR/8 in a multi-cycle infection and incubated in the presence of 5 μM JJ3297 or 1 % DMSO. Treatment with the compound was initiated at the time of infection and samples were collected after 4, 8, 12, 24 and 48 h for determination of TCID50. Fig. 3 illustrates that treatment with JJ3297 had a profound effect on virus replication in the low-multiplicity infection, decreasing the amount of virus in the cell supernatant by approximately 3.5 orders of magnitude. However, the compound had no effect in cells infected at a high m.o.i. These data strongly suggested that inhibition of NS1 by JJ3297 resulted in inhibition of replication and spread through an IFN-dependent mechanism.
Fig. 3.

Influenza replication in the presence or absence of JJ3297 during multi-cycle infection initiated at a low or high m.o.i. MDCK cells were infected with A/PR/8 at an m.o.i. of 0.01 (low) or 3 (high) and treated with 5 μM JJ3297 (‘treated’) or 1 % DMSO (‘untreated’). At the indicated time points, the virus titre was measured by TCID50 analysis.
To test whether JJ3297 in fact inhibited virus spread, immunofluorescence experiments were performed. MDCK cells were infected at an m.o.i. of 0.01 and incubated in the presence of 5 μM JJ3297 or 1 % DMSO. After 72 h, the cells were fixed and stained for viral nucleoprotein (NP). As can be seen in Fig. 4(a), mock-infected cells showed no NP staining, whereas all cells in the DMSO-treated sample showed evidence of viral protein expression after 72 h of infection. By contrast, only a small fraction of cells expressed NP in the sample treated with JJ3297, demonstrating that the compound efficiently inhibited the spread of the virus. Fig. 4(b) shows that, in stark contrast, during a high-multiplicity infection, 100 % of the cells expressed NP whether or not they were treated with JJ3297. This was further investigated by performing a single-cycle infection, in which cells were infected at a high m.o.i. for a total of 5 h in the absence or presence of JJ3297 (Fig. 4c). JJ3297 had no effect on the expression of NP as judged by immunofluorescence microscopy. Western blot analysis of NP during the first cycle of replication also revealed no effect of JJ3297 on NP levels, indicating that the compound's activity did not affect expression of NP (data not shown). As also shown in Fig. 4(c), expression of NS1 protein itself was not affected in the single-cycle infection by treatment with JJ3297, nor was its localization, indicating that the compound does not act by leading to degradation of NS1 or grossly disrupting its subcellular localization.
Fig. 4.

Inhibition of multi-cycle (low m.o.i.) but not single-cycle (high m.o.i.) infections. (a) MDCK cells were mock infected or infected with influenza A/PR/8 at an m.o.i. of 0.01 and treated with 1 % DMSO or 5 μM JJ3297. After 72 h, the cells were fixed and stained for NP (upper panels) or DAPI (lower panels). Arrows indicate DAPI-stained cells expressing NP. (b) MDCK cells were infected at an m.o.i. of 5 and treated as described in (a). (c) MDCK cells were mock infected or infected with A/PR/8 at an m.o.i. of 5 (NS1 stained) or 10 (NP stained) and treated with 1 % DMSO or 5 μM JJ3297. Allowing for a single cycle of replication, the cells were fixed after 5 h and stained for NS1, NP or DAPI as indicated. Magnification: (a, b) ×20; (c) ×40 (NP) and ×60 (NS1 and mock).
Induction of a protective antiviral state in response to JJ3297
We tested for the induction of an antiviral state in infected cells treated with JJ3297. To do this, cells were first infected with A/PR/8 at an m.o.i. of 0.01 in the presence of 5 μM JJ3297 for 48 h. As already shown in Fig. 4(a), this resulted in the vast majority of cells showing no evidence of infection. To test for the establishment of an antiviral state, the culture was then infected with a GFP-expressing vesicular stomatits virus (VSV) construct. This construct contained the gene for GFP in place of the native VSV glycoprotein, VSV-G, and therefore can replicate within cells but cannot be released from infected cells (Takada et al., 1997). After overnight incubation in the presence of VSV–GFP, cells were analysed by fluorescence microscopy. As shown in Fig. 5(a), JJ3297 treatment of influenza virus-infected cells resulted in a significant reduction in the number of cells expressing GFP. As expected, influenza virus-infected cells treated with DMSO instead of JJ3297 exhibited significant cell loss and overwhelming cytopathic effect resulting from 48 h of unrestrained virus replication (data not shown). To quantify the inhibitory effect of JJ3297 on VSV–GFP replication, the cells were harvested and evaluated for GFP expression using flow cytometry, which revealed a twofold reduction in the number of GFP-positive cells (Fig. 5b). This level of protection was consistent in four independent experiments, as shown in Fig. 5(c).
Fig. 5.


Measurement of the antiviral state and IFN production. (a) MDCK cells were mock infected or infected with A/PR/8 at an m.o.i. of 0.01 and treated with 5 μM JJ3297. After 48 h, the cells were infected with VSV–GFP at an m.o.i. of 5, incubated overnight and visualized live for GFP fluorescence and by phase-contrast microscopy. (b) Example of data from flow cytometry. Arrows indicate GFP-positive cells. Mock-infected cells were not infected with A/PR/8, but were treated with JJ3297 for 48 h, followed by infection overnight with VSV–GFP. Infected cells were infected with A/PR/8 for 48 h in the presence of JJ3297, followed by infection overnight with VSV–GFP. (c) Quantification of data from quadruplicate experiments as illustrated in (b). Bars represent the percentage of cells in the population expressing GFP. (d) Wild-type (WT) MEFs or RNase L−/− MEFs were mock infected or infected with A/PR/8 at an m.o.i. of 0.1 and incubated in the presence of 5 μM JJ3297 or 1 % DMSO. After 24 h, supernatants were harvested and the concentration of IFN-β was determined by ELISA. Increasing concentrations of IFN-β standards were included for comparison (left columns). Black horizontal line, IFN-β level of 15.6 pg ml−1 which is the lower limit of detection for the ELISA assay. (e) MEFs are protected from VSV infection by recombinant IFN-β. Cells were pre-treated with 100 U mouse recombinant IFN-β ml−1 for 6 h (where indicated) and then infected with VSV–GFP at an m.o.i. of 5. After overnight incubation, cells were visualized live for GFP fluorescence and by phase-contrast microscopy. (f) Uninfected MDCK cells were incubated in the presence of 1 % DMSO or 5 μM JJ3297. After 72 h, the cells were infected with VSV–GFP at an m.o.i. of 0.5, incubated overnight and visualized live for GFP fluorescence and by phase-contrast microscopy.
To definitively determine the presence of IFN-β in infected cells treated with JJ3297, a quantitative ELISA was performed. Mouse embryonic fibroblast (MEF) cells were mock infected or infected with A/PR/8 at an m.o.i. of 0.1 and treated with DMSO or 5 μM JJ3297. After 24 h, the medium was collected and assayed for the presence of IFN-β. Fig. 5(d) shows that only cells that were infected and treated with JJ3297 produced significant levels of IFN-β, corresponding to 69 pg ml−1. Fig. 5(e) shows the sensitivity of the VSV–GFP construct to pre-treatment of MEF cells with 100 U recombinant murine IFN-β ml−1 for 6 h prior to VSV challenge, revealing a similar level of inhibition of VSV–GFP replication as was shown in Fig. 5(a). To prove that JJ3297 had no direct effect on VSV–GFP replication, MDCK cells were infected with the VSV–GFP construct in the presence or absence of 5 μM JJ3297 for 72 h. A complete lack of effect on VSV–GFP replication is shown in Fig. 5(f). This also proved that JJ3297 by itself does not induce an antiviral state. Taken together, these data demonstrated the presence of IFN-β and the establishment of an IFN-like antiviral state in influenza virus-infected cells treated with compound JJ3297.
The activity of JJ3297 requires a functional IFN response pathway
To conclusively implicate the IFN system in the action of JJ3297, a genetic experiment was performed to test the requirement for a functional IFN pathway in the activity of JJ3297. RNase L is a major downstream effector of IFN action that has been strongly implicated in the innate immune response to influenza virus infection, as well as NS1 activity (Min & Krug, 2006). RNase L is activated in virus-infected cells by 2′-5′ oligoadenylates synthesized in response to IFN and the presence of dsRNA (Silverman, 2007a, b). Therefore, we wondered whether cells lacking RNase L would be sensitive to compound JJ3297. To test this, RNase L−/− and parental wild-type MEF cells were infected with A/PR/8-MA (mouse-adapted strain) at an m.o.i. of 0.1 in the absence or presence of 5 μM JJ3297 for 48 h. As shown in Fig. 6(a), parental MEF cells were readily infected by A/PR/8-MA, as revealed by immunofluorescent staining for NP, and there was almost complete inhibition of virus replication due to JJ3297 treatment. In contrast, RNase L−/− MEF cells were completely resistant to the action of JJ3297, clearly implicating a functional IFN system in its mechanism of action (Fig. 6b). Consistent with this, TCID50 analysis of wild-type and RNase L−/− cells infected with A/PR/8-MA in the absence or presence of JJ3297 also demonstrated that cells lacking RNase L were entirely resistant to JJ3297 action (Fig. 6c). There was no difference in the overall virus titre between the parental and RNase L−/− cells at either 24 or 48 h after infection in the absence of JJ3297 (Fig. 6c, DMSO-treated samples). This was presumably due to the fact that NS1 efficiently blocked the IFN system in the parental cells, thereby circumventing the role of RNase L. Also consistent with these findings, we observed a complete lack of IFN-β induction in the RNase L−/− cells that were infected with A/PR/8 and treated with JJ3297 (Fig. 5d). These data confirmed that compound JJ3297 inhibits influenza replication in a manner that requires an intact IFN system.
Fig. 6.

JJ3297 activity depends on a functional IFN response pathway. (a, b) Parental wild-type (WT) (a) or RNase L−/− (b) MEFs were infected with A/PR/8-MA at an m.o.i. of 0.1 and treated with 5 μM JJ3297 or 1 % DMSO. After 48 h, the cells were fixed and stained for NP. DAPI was used to visualize cell nuclei. (c) Quantification of replication in WT or RNase L−/− MEFs. Supernatants from cells infected with A/PR/8-MA at a low m.o.i. and treated with 5 μM JJ3297 or 1 % DMSO were harvested at 24 and 48 h time points and the virus titre was assayed for samples from three independent cultures by TCID50 determination.
Lack of effect of JJ3297 on NS1 binding to dsRNA in vitro
To explore a possible mechanism of JJ3297 action, the compound was used to challenge an in vitro binding assay for NS1–dsRNA interactions. Bacterially expressed full-length NS1 from A/PR/8 was bound to agarose beads containing immobilized poly(I : C). To ensure that binding of NS1 to the beads was specific, two control experiments were performed. First, the large C-terminal effector domain of NS1, which lacks the dsRNA-binding domain, was incubated with poly(I : C) beads and found not to associate (Fig. 7, lower panel), whereas full-length NS1 associated efficiently (Fig. 7, upper panel, control). Next, full-length NS1 bound to the beads was incubated with a tenfold excess of free poly(I : C) for 2 h at 4 °C. Under these conditions, NS1 was efficiently released from the beads, whereas incubation in binding buffer alone under the same conditions resulted in no release of bound NS1. Having demonstrated the specificity of binding, we then incubated bound NS1 with JJ3297 up to a concentration of 50 μM, which is 50 times higher than the amount of JJ3297 that reduced virus replication tenfold (see Fig. 2b). No effect of JJ3297 on the release of NS1 from the beads was observed (Fig. 7, top panel), suggesting that the compound does not target the NS1–dsRNA interaction in its mechanism of action.
Fig. 7.

Lack of effect of JJ3297 on NS1 binding to dsRNA in vitro. Upper panel: recombinant NS1 (A/PR/8) was bound to poly(I : C) immobilized on agarose beads and challenged with control buffer (lanes 1 and 2), a tenfold excess of free poly(I : C) (lanes 3 and 4), 0.025 % DMSO (lanes 5 and 6) or 50 μM JJ3297 (lanes 7 and 8). Reactions were incubated for 2 h at 4 °C and the pellet (P) and supernatant (S) fractions were analysed by PAGE and Western blotting for NS1. Lower panel: NS1 C-terminal effector domain was incubated with poly(I : C) immobilized on agarose beads and analysed as described above.
DISCUSSION
We have presented the results of our continuing investigation of novel NS1 antagonists that could potentially be used as therapeutics for treatment of influenza in humans. The precise mechanism of action of JJ3297 remains to be elucidated. NS1 disrupts several steps required for the IFN response, and it is likely that at least one of them is affected by JJ3297, either directly or indirectly. For example, it has been shown that inhibiting the interaction between NS1 and CPSF30 restores IFN synthesis in infected cells, establishing this interaction as a potential antiviral target (Das et al., 2010; Krug & Aramini, 2009; Twu et al., 2006). NS1 also binds directly to TRIM25 and prevents it from activating the cellular RNA sensor RIG-I, an important step in activation of the IFN response (Gack et al., 2009). Therefore, JJ3297 might act by preventing or disrupting the TRIM25–NS1 interaction. Another possible function of the compound that could lead to an increase in IFN production would be restoration of efficient mRNA export from the nucleus, which NS1 inhibits by forming a complex with several members of the export machinery (Satterly et al., 2007).
As shown in Fig. 7, we saw no evidence for a direct effect of JJ3297 on in vitro binding of NS1 to dsRNA. This suggests that the main mechanism of JJ3297 is not to disrupt or prevent dsRNA binding to NS1 in the cell. However, as JJ3297 may act indirectly to perturb NS1 activity, it is possible that this could result in a change in the dsRNA association with NS1 in the cell, leading to downstream effects on both IFN production and activity (e.g. by an effect on RNase L activation, which depends on dsRNA through the action of the OAS). Consistent with all of the above mechanisms, we observed that the effect of JJ3297 on virus replication depended strictly on the presence of RNase L. As a major effector of IFN-dependent anti-influenza activity, the loss of RNase L in the knockout cell line removes a downstream step in the IFN pathway into which all of the above mechanisms could feed. In addition, RNase L is required for maximal IFN expression in response to poly(I : C) or Sendai virus infection, and also mediates amplification of induction of IFN gene transcription through the generation of cleaved viral RNAs that activate the IFN response (Malathi et al., 2007). Our observation that IFN induction was undetectable in RNase L−/− MEF cells infected with A/PR/8 and treated with JJ3297 is in line with this possibility. These effects in combination probably account for our observation that RNase L−/− MEF cells are completely resistant to JJ3297 treatment. Therefore, the observed dependence of JJ3297 activity on RNase L does not necessarily imply a role of RNase L in the direct mechanism of JJ3297; rather, it demonstrates the role of the IFN pathway in this mechanism. For instance, it is likely that knockout cells for other IFN pathway components such as RIG-I would also be insensitive to the action of JJ3297. It will be interesting to determine the exact mode of inhibition of NS1 by JJ3297, and experiments addressing this issue are under way.
An important criterion for an effective viral target protein is that virus replication and/or spread is highly dependent on the function of the protein. This has been demonstrated for NS1 both in cell culture and in animal model systems (Basler & Aguilar, 2008; Basler et al., 2001; Bürger et al., 1985; Donelan et al., 2003; Egorov et al., 1998; Falcón et al., 2005; Garaigorta et al., 2005; García-Sastre et al., 1998; Hatada et al., 1990; Jackson et al., 2008; Kochs et al., 2007b; Koennecke et al., 1981; Ludwig et al., 1995; Shimizu et al., 1983; Talon et al., 2000; Wolstenholme et al., 1980). In particular, ongoing efforts to develop more effective influenza vaccines include the use of live-attenuated strains that contain deleted or truncated forms of NS1 (Palese, 2006; Palese & García-Sastre, 2002; Schickli et al., 2001; Solórzano et al., 2005). Infections of immunocompetent hosts with these viruses result in establishment of protective immunity against future challenge without causing any significant adverse effects (Baskin et al., 2007; Chambers et al., 2009; Hai et al., 2008; Mueller et al., 2010; Richt et al., 2006; Romanova et al., 2009; Talon et al., 2000; Vincent et al., 2007; Wacheck et al., 2010; Wressnigg et al., 2009a, b). The usefulness of these strains as potential vaccine candidates is illustrated by their highly attenuated growth in animals. Therefore, it is likely that a drug that blocks all or part of the multiple functions of NS1 may act efficiently to limit infection in the host. In addition, activation of the IFN system in drug-treated animals or humans might be expected to aid in the establishment of the immune response and clearing the virus from the body (Bracci et al., 2005; Le Bon et al., 2001; Palese, 2006; Proietti et al., 2002; Talon et al., 2000; Tovey et al., 2008). This aspect of JJ3297 activity will be addressed in future animal studies.
Based on the results presented here, we propose the following explanation: (i) when cells are infected with influenza virus at a low m.o.i. in the absence of treatment with JJ3297, NS1 protein effectively inhibits the synthesis of IFN, allowing the replicated virus to spread through the cell population, resulting in a high virus titre in the medium; (ii) treatment with JJ3297 during infection at low multiplicity allows the infected cells to synthesize IFN despite the presence of NS1, leading to induction of an antiviral state in neighbouring uninfected cells and inhibition of virus spread, also resulting in a lowered virus titre in the medium; (iii) treatment with JJ3297 during infection at high multiplicity also leads to the restoration of IFN synthesis in infected cells, but because the compound was found not to interfere with single-cycle infection, and as the virus initially infected the vast majority of cells in the population, no effect on virus spread or medium titre was observed.
METHODS
Cells and viruses.
MDCK cells were grown as monolayers in Iscove's modified Dulbecco's medium with 10 % FBS, 2 mM l-glutamine and antibiotics. Parental and RNase L−/− MEFs, gifts from Robert Silverman (Malathi et al., 2007), were maintained in Dulbecco's modified Eagle's medium with 10 % FBS and antibiotics. Media and sera were from Invitrogen. Viral dilutions and infections were in medium supplemented with 0.3 % BSA, 0.22 % sodium bicarbonate and 0.25 U TPCK-treated trypsin (Invitrogen) ml−1. All compound treatments were initiated at the time of infection. Some cultures were incubated with 100 U recombinant mouse IFN-β (Millipore) ml−1 for 6 h prior to infection. Influenza viruses A/PR/8/34 and its mouse-adapted variant (A/PR/8-MA) were gifts from Thomas Braciale, Department of Pathology, University of Virginia, USA. VSV–GFP was a gift from Judith White (Takada et al., 1997).
Virus replication assays.
Cell monolayers were infected at an m.o.i. of 0.1 for 48 h in the presence or absence of the compound, which was added at the beginning of infection. Virus titres were determined by TCID50 analysis (Reed & Muench, 1938).
Cell viability assay.
Opaque-walled 96-well plates were seeded with 104 MDCK cells per well in 100 μl medium and incubated overnight at 37 °C. The compound was added to triplicate wells for each concentration tested and incubated for 48 h at 37 °C. The plate was equilibrated for 30 min at room temperature and 100 μl CellTiter Glo reagent (Promega) added according to the manufacturer's protocol. The plate was shaken for 2 min and then incubated for 10 min at room temperature. Luminescence was recorded in an LMAX 384 reader (Molecular Devices) with integration time of 1 s.
RT-PCR.
MDCK cells were infected for 6 h with A/PR/8 at an m.o.i. of 2.0, and total RNA was isolated using RNeasy (Qiagen). Total RNA (2 μg) was incubated with random nanomers (New England Biolabs) at a final concentration of 2 μM. Reverse transcription was with 10 U Moloney murine leukemia virus reverse transcriptase μl−1 (New England Biolabs) in the presence of 1 U RNase inhibitor μl−1 (New England Biolabs). Thereafter, 1/20 vol. cDNA was used as the template for PCR amplification (30 cycles of 94 °C for 30 s, 55 °C for 20 s, 72 °C for 30 s). The following primer pairs were used: canine IFN-β (GenBank accession no. NM_001135787), 5′-CCAGTTCCAGAAGGAGGACA-3′ and 5′-CCTGTTGTCCCAGGTGAAGT-3′; and canine β-actin (GenBank accession no. XM_536230), 5′-GGCATCCTGACCCTGAAGTA-3′ and 5′-GGGGTGTTGAAAGTCTCGAA-3′.
Immunofluorescence.
MDCK cells were grown on coverslips (Fisher) for 24 h prior to infection. Infected cells were washed with PBS and fixed by incubation in fresh 4 % paraformaldehyde in PBS for 20 min at room temperature. The fixative was removed and the cells were permeabilized by incubating with pre-chilled 0.5 % Triton X-100 in PBS for 10 min at 4 °C. All subsequent steps were performed at room temperature. The cells were then washed in PBS for three 15 min washes. Staining was performed according to a protocol described previously (Ornelles & Shenk, 1991). First, cells were incubated in blocking buffer [25 mM Tris/HCl (pH 8.0), 137 mM NaCl, 3 mM KCl, 1.5 mM MgCl2, 2.5 % BSA, 13 mM glycine, 0.05 % Tween 20 and 20 % goat serum] by placing the coverslips face down on a 100 μl drop of buffer in a humidified chamber for 1 h. Next, cells were placed on a 100 μl drop of anti-NP mouse mAb (Centers for Disease Control and Prevention) diluted 1 : 1000 in blocking buffer and incubated for 100 min. After six 10 min washes in PBS supplemented with 0.1 % Tween 20, the coverslips were placed face down on a 100 μl drop of a 1 : 2000 dilution (in blocking buffer) of goat anti-mouse secondary antibody conjugated to Alexa 488 (Invitrogen) for 45 min. Following this incubation, the cells were washed six times for 10 min each in PBS supplemented with 0.1 % Tween 20 and twice for 5 min each in unsupplemented PBS. Finally, the cells were mounted on glass slides in Vectashield mounting media with DAPI (Vector Laboratories). NS1 visualization was performed identically, except that the antibodies used were anti-NS1 H1N1 rabbit polyclonal antibody (diluted 1 : 400 in blocking buffer; a gift from Peter Palese, Department of Microbiology, Mount Sinai School of Medicine) and goat anti-rabbit secondary antibody conjugated to Alexa 594 (diluted 1 : 2000 in blocking buffer; Invitrogen). Slides were sealed with nail polish and examined using a Nikon Eclipse E800 fluorescence microscope and a Princeton Instruments charged-coupled-device camera. Expression of GFP in live cells was examined using a Nikon Eclipse TE2000-E fluorescence inverted microscope and a Hamamatsu ORCA-ER digital camera (Improvision Open Lab software).
Flow cytometry.
Infected cells were trypsinized, collected into 1.5 ml tubes, washed with PBS and fixed by resuspending in fresh 2 % paraformaldehyde in PBS for 10 min. Fixed cells were pelleted (2 min, 300 g), washed with PBS and analysed using a Becton Dickinson FACSCalibur Benchtop Analyser. After isolating the cell population from debris and aggregates using forward and side scatter, the fraction of GFP-positive cells was determined. The threshold for GFP detection was established using a control cell population that was not infected with VSV–GFP.
dsRNA-binding assay.
Recombinant His-tagged NS1 (A/PR/8) or the NS1 C-terminal ‘effector’ domain were purified from Eshcerichia coli using Ni2+ beads. Poly(I : C)-coated agarose beads were prepared from poly(C)-coated beads (Sigma) by incubating them with 2 vols poly(I) (2 mg ml−1; Sigma) in 50 mM Tris/HCl (pH 7.0) and 150 mM NaCl overnight at 4 °C. After washing in the same buffer, 20 μl of a 50 % slurry of bead-bound poly(I : C) was incubated with 50 ng NS1 in binding buffer [50 mM Tris/HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA and 1 % NP-40] in a total volume of 40 μl for 2 h at 4 °C. The beads were washed three times with binding buffer, mixed with reagents identified in the legend to Fig. 7 and incubated for 2 h at 4 °C. The bead fraction and supernatants were separated by PAGE and analysed by Western blotting using polyclonal anti-NS1 antibody (1 : 2000).
Acknowledgments
We thank Tom Braciale, Peter Palese, Robert Silverman and Judith White for gifts of cells, viruses and antibodies. This work was supported by Public Health Service grant R01AI071341 to D. A. E.
References
- Baskin, C. R., Bielefeldt-Ohmann, H., García-Sastre, A., Tumpey, T. M., Van Hoeven, N., Carter, V. S., Thomas, M. J., Proll, S., Solorzano, A. & other authors (2007). Functional genomic and serological analysis of the protective immune response resulting from vaccination of macaques with an NS1-truncated influenza virus. J Virol 81, 11817–11827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler, C. F. & Aguilar, P. V. (2008). Progress in identifying virulence determinants of the 1918 H1N1 and the Southeast Asian H5N1 influenza A viruses. Antiviral Res 79, 166–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basler, C. F., Reid, A. H., Dybing, J. K., Janczewski, T. A., Fanning, T. G., Zheng, H., Salvatore, M., Perdue, M. L., Swayne, D. E. & other authors (2001). Sequence of the 1918 pandemic influenza virus nonstructural gene (NS) segment and characterization of recombinant viruses bearing the 1918 NS genes. Proc Natl Acad Sci U S A 98, 2746–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu, D., Walkiewicz, M. P., Frieman, M., Baric, R. S., Auble, D. T. & Engel, D. A. (2009). Novel influenza virus NS1 antagonists block replication and restore innate immune function. J Virol 83, 1881–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracci, L., Canini, I., Puzelli, S., Sestili, P., Venditti, M., Spada, M., Donatelli, I., Belardelli, F. & Proietti, E. (2005). Type I IFN is a powerful mucosal adjuvant for a selective intranasal vaccination against influenza virus in mice and affects antigen capture at mucosal level. Vaccine 23, 2994–3004. [DOI] [PubMed] [Google Scholar]
- Bürger, H., Steuler, H. & Scholtissek, C. (1985). A mutant of fowl plague virus (influenza A) with an enhanced electrophoretic mobility of RNA segment 8. J Gen Virol 66, 1679–1686. [DOI] [PubMed] [Google Scholar]
- Chambers, T. M., Quinlivan, M., Sturgill, T., Cullinane, A., Horohov, D. W., Zamarin, D., Arkins, S., García-Sastre, A. & Palese, P. (2009). Influenza A viruses with truncated NS1 as modified live virus vaccines: pilot studies of safety and efficacy in horses. Equine Vet J 41, 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z., Li, Y. & Krug, R. M. (1999). Influenza A virus NS1 protein targets poly(A)-binding protein II of the cellular 3′-end processing machinery. EMBO J 18, 2273–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien, C. Y., Xu, Y., Xiao, R., Aramini, J. M., Sahasrabudhe, P. V., Krug, R. M. & Montelione, G. T. (2004). Biophysical characterization of the complex between double-stranded RNA and the N-terminal domain of the NS1 protein from influenza A virus: evidence for a novel RNA-binding mode. Biochemistry 43, 1950–1962. [DOI] [PubMed] [Google Scholar]
- Das, K., Aramini, J. M., Ma, L. C., Krug, R. M. & Arnold, E. (2010). Structures of influenza A proteins and insights into antiviral drug targets. Nat Struct Mol Biol 17, 530–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donelan, N. R., Basler, C. F. & García-Sastre, A. (2003). A recombinant influenza A virus expressing an RNA-binding-defective NS1 protein induces high levels of beta interferon and is attenuated in mice. J Virol 77, 13257–13266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorov, A., Brandt, S., Sereinig, S., Romanova, J., Ferko, B., Katinger, D., Grassauer, A., Alexandrova, G., Katinger, H. & other authors (1998). Transfectant influenza A viruses with long deletions in the NS1 protein grow efficiently in Vero cells. J Virol 72, 6437–6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcón, A. M., Fernandez-Sesma, A., Nakaya, Y., Moran, T. M., Ortín, J. & García-Sastre, A. (2005). Attenuation and immunogenicity in mice of temperature-sensitive influenza viruses expressing truncated NS1 proteins. J Gen Virol 86, 2817–2821. [DOI] [PubMed] [Google Scholar]
- Gack, M. U., Albrecht, R. A., Urano, T., Inn, K. S., Huang, I. C., Carnero, E., Farzan, M., Inoue, S., Jung, J. U. & other authors (2009). Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 5, 439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaigorta, U., Falcón, A. M. & Ortín, J. (2005). Genetic analysis of influenza virus NS1 gene: a temperature-sensitive mutant shows defective formation of virus particles. J Virol 79, 15246–15257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Sastre, A., Egorov, A., Matassov, D., Brandt, S., Levy, D. E., Durbin, J. E., Palese, P. & Muster, T. (1998). Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 252, 324–330. [DOI] [PubMed] [Google Scholar]
- Garten, R. J., Davis, C. T., Russell, C. A., Shu, B., Lindstrom, S., Balish, A., Sessions, W. M., Xu, X., Skepner, E. & other authors (2009). Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 325, 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Z., Chen, L. M., Zeng, H., Gomez, J. A., Plowden, J., Fujita, T., Katz, J. M., Donis, R. O. & Sambhara, S. (2007). NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am J Respir Cell Mol Biol 36, 263–269. [DOI] [PubMed] [Google Scholar]
- Hai, R., Marínez-Sobrido, L., Fraser, K. A., Ayllon, J., García-Sastre, A. & Palese, P. (2008). Influenza B virus NS1-truncated mutants: live-attenuated vaccine approach. J Virol 82, 10580–10590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale, B. G., Randall, R. E., Ortín, J. & Jackson, D. (2008). The multifunctional NS1 protein of influenza A viruses. J Gen Virol 89, 2359–2376. [DOI] [PubMed] [Google Scholar]
- Hatada, E. & Fukuda, R. (1992). Binding of influenza A virus NS1 protein to dsRNA in vitro. J Gen Virol 73, 3325–3329. [DOI] [PubMed] [Google Scholar]
- Hatada, E., Hasegawa, M., Shimizu, K., Hatanaka, M. & Fukuda, R. (1990). Analysis of influenza A virus temperature-sensitive mutants with mutations in RNA segment 8. J Gen Virol 71, 1283–1292. [DOI] [PubMed] [Google Scholar]
- Hatada, E., Takizawa, T. & Fukuda, R. (1992). Specific binding of influenza A virus NS1 protein to the virus minus-sense RNA in vitro. J Gen Virol 73, 17–25. [DOI] [PubMed] [Google Scholar]
- Hatada, E., Saito, S., Okishio, N. & Fukuda, R. (1997). Binding of the influenza virus NS1 protein to model genome RNAs. J Gen Virol 78, 1059–1063. [DOI] [PubMed] [Google Scholar]
- Jackson, D., Hossain, M. J., Hickman, D., Perez, D. R. & Lamb, R. A. (2008). A new influenza virus virulence determinant: the NS1 protein four C-terminal residues modulate pathogenicity. Proc Natl Acad Sci U S A 105, 4381–4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain, S., Kamimoto, L., Bramley, A. M., Schmitz, A. M., Benoit, S. R., Louie, J., Sugerman, D. E., Druckenmiller, J. K., Ritger, K. A. & other authors (2009). Hospitalized patients with 2009 H1N1 influenza in the United States, April–June 2009. N Engl J Med 361, 1935–1944. [DOI] [PubMed] [Google Scholar]
- Kochs, G., García-Sastre, A. & Martínez-Sobrido, L. (2007a). Multiple anti-interferon actions of the influenza A virus NS1 protein. J Virol 81, 7011–7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochs, G., Koerner, I., Thiel, L., Kothlow, S., Kaspers, B., Ruggli, N., Summerfield, A., Pavlovic, J., Stech, J. & other authors (2007b). Properties of H7N7 influenza A virus strain SC35M lacking interferon antagonist NS1 in mice and chickens. J Gen Virol 88, 1403–1409. [DOI] [PubMed] [Google Scholar]
- Koennecke, I., Boschek, C. B. & Scholtissek, C. (1981). Isolation and properties of a temperature-sensitive mutant (ts 412) of an influenza A virus recombinant with a ts lesion in the gene coding for the nonstructural protein. Virology 110, 16–25. [DOI] [PubMed] [Google Scholar]
- Krug, R. M. & Aramini, J. M. (2009). Emerging antiviral targets for influenza A virus. Trends Pharmacol Sci 30, 269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug, R. M., Yuan, W., Noah, D. L. & Latham, A. G. (2003). Intracellular warfare between human influenza viruses and human cells: the roles of the viral NS1 protein. Virology 309, 181–189. [DOI] [PubMed] [Google Scholar]
- Kuo, R. L. & Krug, R. M. (2009). Influenza A virus polymerase is an integral component of the CPSF30–NS1A protein complex in infected cells. J Virol 83, 1611–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layne, S. P., Monto, A. S. & Taubenberger, J. K. (2009). Pandemic influenza: an inconvenient mutation. Science 323, 1560–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bon, A., Schiavoni, G., D'Agostino, G., Gresser, I., Belardelli, F. & Tough, D. F. (2001). Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 14, 461–470. [DOI] [PubMed] [Google Scholar]
- Li, S., Min, J. Y., Krug, R. M. & Sen, G. C. (2006). Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 349, 13–21. [DOI] [PubMed] [Google Scholar]
- Lu, Y., Wambach, M., Katze, M. G. & Krug, R. M. (1995). Binding of the influenza virus NS1 protein to double-stranded RNA inhibits the activation of the protein kinase that phosphorylates the eIF-2 translation initiation factor. Virology 214, 222–228. [DOI] [PubMed] [Google Scholar]
- Ludwig, S. & Wolff, T. (2009). Influenza A virus TRIMs the type I interferon response. Cell Host Microbe 5, 420–421. [DOI] [PubMed] [Google Scholar]
- Ludwig, S., Vogel, U. & Scholtissek, C. (1995). Amino acid replacements leading to temperature-sensitive defects of the NS1 protein of influenza A virus. Arch Virol 140, 945–950. [DOI] [PubMed] [Google Scholar]
- Malathi, K., Dong, B., Gale, M., Jr & Silverman, R. H. (2007). Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448, 816–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mibayashi, M., Martínez-Sobrido, L., Loo, Y. M., Cárdenas, W. B., Gale, M., Jr & García-Sastre, A. (2007). Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol 81, 514–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min, J. Y. & Krug, R. M. (2006). The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: inhibiting the 2′-5′ oligo (A) synthetase/RNase L pathway. Proc Natl Acad Sci U S A 103, 7100–7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min, J. Y., Li, S., Sen, G. C. & Krug, R. M. (2007). A site on the influenza A virus NS1 protein mediates both inhibition of PKR activation and temporal regulation of viral RNA synthesis. Virology 363, 236–243. [DOI] [PubMed] [Google Scholar]
- Mueller, S. N., Langley, W. A., Carnero, E., García-Sastre, A. & Ahmed, R. (2010). Immunization with live attenuated influenza viruses that express altered NS1 proteins results in potent and protective memory CD8+ T-cell responses. J Virol 84, 1847–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeroff, M. E., Barabino, S. M., Li, Y., Keller, W. & Krug, R. M. (1998). Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3′end formation of cellular pre-mRNAs. Mol Cell 1, 991–1000. [DOI] [PubMed] [Google Scholar]
- Noah, D. L., Twu, K. Y. & Krug, R. M. (2003). Cellular antiviral responses against influenza A virus are countered at the posttranscriptional level by the viral NS1A protein via its binding to a cellular protein required for the 3′ end processing of cellular pre-mRNAs. Virology 307, 386–395. [DOI] [PubMed] [Google Scholar]
- Ornelles, D. A. & Shenk, T. (1991). Localization of the adenovirus early region 1B 55-kilodalton protein during lytic infection: association with nuclear viral inclusions requires the early region 4 34-kilodalton protein. J Virol 65, 424–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palese, P. (2006). Making better influenza virus vaccines? Emerg Infect Dis 12, 61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palese, P. & García-Sastre, A. (2002). Influenza vaccines: present and future. J Clin Invest 110, 9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekosz, A. & Glass, G. E. (2008). Emerging viral diseases. Md Med 9, 11–16. [PMC free article] [PubMed] [Google Scholar]
- Pichlmair, A., Schulz, O., Tan, C. P., Naslund, T. I., Liljestrom, P., Weber, F. & Reis e Sousa, C. (2006). RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314, 997–1001. [DOI] [PubMed] [Google Scholar]
- Proietti, E., Bracci, L., Puzelli, S., Di Pucchio, T., Sestili, P., De Vincenzi, E., Venditti, M., Capone, I., Seif, I. & other authors (2002). Type I IFN as a natural adjuvant for a protective immune response: lessons from the influenza vaccine model. J Immunol 169, 375–383. [DOI] [PubMed] [Google Scholar]
- Qian, X. Y., Chien, C. Y., Lu, Y., Montelione, G. T. & Krug, R. M. (1995). An amino-terminal polypeptide fragment of the influenza virus NS1 protein possesses specific RNA-binding activity and largely helical backbone structure. RNA 1, 948–956. [PMC free article] [PubMed] [Google Scholar]
- Qiu, Y. & Krug, R. M. (1994). The influenza virus NS1 protein is a poly(A)-binding protein that inhibits nuclear export of mRNAs containing poly(A). J Virol 68, 2425–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, Y., Nemeroff, M. & Krug, R. M. (1995). The influenza virus NS1 protein binds to a specific region in human U6 snRNA and inhibits U6–U2 and U6–U4 snRNA interactions during splicing. RNA 1, 304–316. [PMC free article] [PubMed] [Google Scholar]
- Reed, L. J. & Muench, H. (1938). A simple method of estimating fifty per cent endpoints. Am J Epidemiol 27, 493–497. [Google Scholar]
- Reid, A. H., Taubenberger, J. K. & Fanning, T. G. (2001). The 1918 Spanish influenza: integrating history and biology. Microbes Infect 3, 81–87. [DOI] [PubMed] [Google Scholar]
- Richt, J. A., Lekcharoensuk, P., Lager, K. M., Vincent, A. L., Loiacono, C. M., Janke, B. H., Wu, W. H., Yoon, K. J., Webby, R. J. & other authors (2006). Vaccination of pigs against swine influenza viruses by using an NS1-truncated modified live-virus vaccine. J Virol 80, 11009–11018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanova, J., Krenn, B. M., Wolschek, M., Ferko, B., Romanovskaja-Romanko, E., Morokutti, A., Shurygina, A. P., Nakowitsch, S., Ruthsatz, T. & other authors (2009). Preclinical evaluation of a replication-deficient intranasal ΔNS1 H5N1 influenza vaccine. PLoS ONE 4, e5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterly, N., Tsai, P. L., van Deursen, J., Nussenzveig, D. R., Wang, Y., Faria, P. A., Levay, A., Levy, D. E. & Fontoura, B. M. (2007). Influenza virus targets the mRNA export machinery and the nuclear pore complex. Proc Natl Acad Sci U S A 104, 1853–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schickli, J. H., Flandorfer, A., Nakaya, T., Martíinez-Sobrido, L., García-Sastre, A. & Palese, P. (2001). Plasmid-only rescue of influenza A virus vaccine candidates. Philos Trans R Soc Lond B Biol Sci 356, 1965–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu, K., Mullinix, M. G., Chanock, R. M. & Murphy, B. R. (1983). Temperature-sensitive mutants of influenza A/Udorn/72 (H3N2) virus. III. Genetic analysis of temperature-dependent host range mutants. Virology 124, 35–44. [DOI] [PubMed] [Google Scholar]
- Silverman, R. H. (2007a). Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J Virol 81, 12720–12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman, R. H. (2007b). A scientific journey through the 2–5A/RNase L system. Cytokine Growth Factor Rev 18, 381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solórzano, A., Webby, R. J., Lager, K. M., Janke, B. H., García-Sastre, A. & Richt, J. A. (2005). Mutations in the NS1 protein of swine influenza virus impair anti-interferon activity and confer attenuation in pigs. J Virol 79, 7535–7543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada, A., Robison, C., Goto, H., Sanchez, A., Murti, K. G., Whitt, M. A. & Kawaoka, Y. (1997). A system for functional analysis of Ebola virus glycoprotein. Proc Natl Acad Sci U S A 94, 14764–14769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talon, J., Salvatore, M., O'Neill, R. E., Nakaya, Y., Zheng, H., Muster, T., García-Sastre, A. & Palese, P. (2000). Influenza A and B viruses expressing altered NS1 proteins: a vaccine approach. Proc Natl Acad Sci U S A 97, 4309–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger, J. K., Reid, A. H., Janczewski, T. A. & Fanning, T. G. (2001). Integrating historical, clinical and molecular genetic data in order to explain the origin and virulence of the 1918 Spanish influenza virus. Philos Trans R Soc Lond B Biol Sci 356, 1829–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovey, M. G., Lallemand, C. & Thyphronitis, G. (2008). Adjuvant activity of type I interferons. Biol Chem 389, 541–545. [DOI] [PubMed] [Google Scholar]
- Twu, K. Y., Noah, D. L., Rao, P., Kuo, R. L. & Krug, R. M. (2006). The CPSF30 binding site on the NS1A protein of influenza A virus is a potential antiviral target. J Virol 80, 3957–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twu, K. Y., Kuo, R. L., Marklund, J. & Krug, R. M. (2007). The H5N1 influenza virus NS genes selected after 1998 enhance virus replication in mammalian cells. J Virol 81, 8112–8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent, A. L., Ma, W., Lager, K. M., Janke, B. H., Webby, R. J., García-Sastre, A. & Richt, J. A. (2007). Efficacy of intranasal administration of a truncated NS1 modified live influenza virus vaccine in swine. Vaccine 25, 7999–8009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacheck, V., Egorov, A., Groiss, F., Pfeiffer, A., Fuereder, T., Hoeflmayer, D., Kundi, M., Popow-Kraupp, T., Redlberger-Fritz, M. & other authors (2010). A novel type of influenza vaccine: safety and immunogenicity of replication-deficient influenza virus created by deletion of the interferon antagonist NS1. J Infect Dis 201, 354–362. [DOI] [PubMed] [Google Scholar]
- Wang, W., Riedel, K., Lynch, P., Chien, C. Y., Montelione, G. T. & Krug, R. M. (1999). RNA binding by the novel helical domain of the influenza virus NS1 protein requires its dimer structure and a small number of specific basic amino acids. RNA 5, 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolstenholme, A. J., Barrett, T., Nichol, S. T. & Mahy, B. W. (1980). Influenza virus-specific RNA and protein syntheses in cells infected with temperature-sensitive mutants defective in the genome segment encoding nonstructural proteins. J Virol 35, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2009). Influenza (seasonal). Fact sheet no. 211. World Health Organization.
- Wressnigg, N., Shurygina, A. P., Wolff, T., Redlberger-Fritz, M., Popow-Kraupp, T., Muster, T., Egorov, A. & Kittel, C. (2009a). Influenza B mutant viruses with truncated NS1 proteins grow efficiently in Vero cells and are immunogenic in mice. J Gen Virol 90, 366–374. [DOI] [PubMed] [Google Scholar]
- Wressnigg, N., Voss, D., Wolff, T., Romanova, J., Ruthsatz, T., Mayerhofer, I., Reiter, M., Nakowitsch, S., Humer, J. & other authors (2009b). Development of a live-attenuated influenza B ΔNS1 intranasal vaccine candidate. Vaccine 27, 2851–2857. [DOI] [PubMed] [Google Scholar]