

Summary
There is a growing appreciation that patients with seizures are also affected by a number of co-morbid conditions, including an increase in prevalence of depression (Kanner, 2009), sleep apnea (Chihorek et al, 2007), and sudden death (Ryvlin et al, 2006; Tomson et al, 2008). The mechanisms responsible for these associations are unclear. Here we discuss the possibility that underlying pathology in the serotonin (5-HT) system of epilepsy patients lowers the threshold for seizures, while also increasing the risk of depression and sudden death. We propose that post-ictal dysfunction of 5-HT neurons causes depression of breathing and arousal in some epilepsy patients, and this can lead to sudden unexpected death in epilepsy (SUDEP). We further draw parallels between SUDEP and sudden infant death syndrome (SIDS), which may share pathophysiological mechanisms, and which have both been linked to defects in the 5-HT system.
Keywords: Breathing, epilepsy, sudden death, raphé, respiratory control
The 5-HT system and epilepsy
5-HT is a particularly important neurotransmitter, because of the many neurological and psychiatric diseases in which it is involved, and because of the many neuropsychiatric drugs that target it. All of the 5-HT within the brain is produced by a relatively small number of neurons located primarily within the midline raphé nuclei of the midbrain, pons and medulla (Dahlström & Fuxe, 1964) (Fig. 1A). It is not widely appreciated just how extensively 5-HT fibers innervate the brain, but there is a high density of 5-HT terminals and fibers present throughout the CNS, including the hippocampus and neocortex (Fig. 1B).
Figure 1.

Serotonin neurons from the raphé nuclei send diffuse projections throughout the central nervous system. A) Schematic representation of projections from raphe nuclei. DRN, dorsal raphe nucleus; H, hypothalamus; HF, hippocampal formation; MRN, median raphe nucleus; RM, raphe magnus; RO, raphe obscurus; RPa, raphe pallidus; RPo, raphe pontis; Th, thalamus. Modified from Cooper et al (1996). B) Photomicrograph depicting 5-HT receptor distribution within the hippocampus and neocortex. Reproduced with permission from Donovan (2002).
There is a large body of literature on 5-HT and seizures (Bagdy et al, 2007). In general, an increase in extracellular 5-HT levels inhibits many types of seizures and a decrease does the opposite (Kilian & Frey, 1973; Buterbaugh, 1978; Browning et al, 1978; Prendiville & Gale, 1993; Yan et al, 1994; Statnick et al, 1996). Several anti-epileptic drugs including phenytoin, carbamazepine, valproic acid, lamotrigine and zonisamide all cause an increase in extracellular 5-HT, and the elevated 5-HT is thought to contribute to their mechanism of action (Bonnycastle et al, 1957; Okada et al, 1992; Dailey et al, 1997; Ahmad et al, 2005; Bagdy et al, 2007).
There is no evidence for mutations in genes directly involved in 5-HT signaling in human epilepsy syndromes, but genetic deletion of 5-HT2C receptors in mice cause audiogenic seizures (Brennan et al, 1997), and knockout of 5-HT1A receptors reduces the threshold for kainate-induced seizures (Sarnyai et al, 2000). The genetically epilepsy prone rat (GEPR) has an increased frequency of seizures, and this is thought to be due in part to a decrease in levels of 5-HT and norepinephrine{Dailey, 1992 DAILEY1992A /id;Dailey, 1989 DAILEY1989 /id}. A number of other animal models of seizures also appear to involve dysfunction of the 5-HT system (Bagdy et al, 2007).
There are also data from humans indicating a role for 5-HT in epilepsy. For example, there is a decrease in 5-HT1A receptor binding in the brains of patients with epilepsy (Toczek et al, 2003; Savic et al, 2004; Merlet et al, 2004; Giovacchini et al, 2005). In fact, PET scanning using a 5-HT1A receptor ligand may be a more sensitive way to localize epileptic foci in temporal lobe epilepsy patients than MRI or fluorodeoxyglucose PET (Liew et al, 2009). Seizures are a common sequelae of recreational use of the drug 3,4-methylenedioxymethamphetamine (MDMA; Ecstasy) (Ben-Abraham et al, 2003). Seizures early after MDMA use are thought to be due to hyponatremia and/or hyperthermia. However, MDMA has specific effects on the 5-HT system, first causing massive release of 5-HT and then damage to serotonergic synaptic terminals. There is evidence that these changes in 5-HT release mechanisms are involved in the pathophysiology of seizures (Giorgi et al, 2005; 2006).
Co-morbid depression in epilepsy
There is a high incidence of depression in patients with epilepsy, with a lifetime prevalence estimated to be between 17–43% (Wiegartz et al, 1999; Tellez-Zenteno et al, 2007). Compared to the general population, epilepsy patients are about twice as likely to have depression, and are 3.6–5 fold more likely to commit suicide (Nilsson et al, 1997; Rafnsson et al, 2001; Kanner, 2009). Depression could be explained in part by a reaction to chronic illness. However, depression often precedes the diagnosis of epilepsy. For example, a history of attempted suicide is five times more common in newly-diagnosed epilepsy patients (Hesdorffer et al, 2006), and a history of depression is 1.7 to 7 times more common in newly-diagnosed epilepsy patients (Forsgren & Nystrom, 1990; Hesdorffer et al, 2000; Hesdorffer et al, 2006). Patients with major depression are six-fold more likely to have an unprovoked seizure than the general population (Hesdorffer et al, 2000). In both depression and epilepsy there is atrophy of hippocampi and amygdala (in temporal lobe epilepsy) and frontal lobes (in frontal lobe epilepsy), evidence of an overactive hypothalamic-pituitary-adrenal axis, and elevated serum corticosterone levels (Kanner, 2006; Kanner, 2009). These data suggest that there may be shared underlying pathology that predisposes patients to both seizures and depression (a “seizure/depression phenotype”). Dysfunction of the 5-HT system is an obvious explanation for this shared pathology, since defects in the 5-HT system are linked to both conditions{Kanner, 2006 KANNER2006 /id;Jobe, 2003 JOBE2003 /id;Jobe, 1999 JOBE1999 /id}. There is a decrease in 5-HT1A receptor binding on PET scanning in patients who have epilepsy (see above) and depression (Sargent et al, 2000), and this decrease is greater in those patients with both conditions compared to temporal lobe epilepsy alone (Hasler et al, 2007). In patients with comorbid epilepsy and depression there is decreased binding in the epileptic focus, as well as extension into extra-lesional limbic structures (Theodore et al, 2006). In both conditions it has been suggested that there is deficient serotonergic (and noradrenergic) neurotransmission (Kanner, 2009).
If there is shared underlying pathology that can lead to both seizures and depression, then it would be expected that treatments for one condition would improve the symptoms of the other. This appears to be the case, since some treatments for depression are effective against seizures, and some anticonvulsants are effective in management of mood/affective disorders{Jobe, 1999 JOBE1999 /id;Jobe, 2003 JOBE2003 /id}. For example, this is true for both vagus nerve stimulation (VNS) and selective serotonin reuptake inhibitors (SSRIs). There have been numerous studies of VNS for refractory depression (Elger et al, 2000; Rush & Siefert, 2009). It was found in one trial that VNS reduces depressive symptoms by 42% at 24 months (Nahas et al, 2005), an amount that is equivalent to the effect of VNS on seizures. This is interesting, because one of the leading hypotheses about the mechanism of action of VNS is that it causes an increase in firing of norepinephrine neurons in the locus coeruleus acutely and of 5-HT neurons within the midbrain raphé nuclei in the longer term (Dorr & Debonnel, 2006; Manta et al, 2009a; 2009b).
It is common for neurologists and psychiatrists to be concerned that there are proconvulsive effects of antidepressants. This concern may stem in part from the increase in the risk of seizures in patients with depression even without treatment (see above), as well as a small number of case reports and small series of patients that had seizures after ingesting large quantities of tricyclic antidepressants or SSRIs (Prasher, 1993; Jobe & Browning, 2005). However, there is a large body of literature in support of the opposite conclusion - that SSRIs are anticonvulsants, and that they are safe for treatment of patients with depression and seizures (Jobe & Browning, 2005; Bagdy et al, 2007; Kanner, 2009; Kondziella & Asztely, 2009). In fact, one SSRI, citalopram, has been found to reduce seizures by >50% in 9 of 11 patients treated, which is as efficacious as many anticonvulsants (Favale et al, 2003).
Respiratory dysfunction and seizures: Insights into SUDEP
SUDEP has been defined as the sudden, unexpected, witnessed or unwitnessed, non-traumatic, and nondrowning death of patients with epilepsy with or without evidence of a seizure, excluding documented status epilepticus, and in whom post-mortem examination does not reveal a structural or toxicological cause for death (Nashef, 1997; Tomson et al, 2008) Estimates of the incidence of SUDEP vary depending upon the population studied. SUDEP is the most common cause of death in patients with uncontrolled epilepsy, with an annual incidence in that population of 1.1–5.9 per 1000 (Tomson et al, 2008). The risk is even higher among epilepsy surgery candidates, estimated as 9.3 per 1000 per year (Dasheiff, 1991). SUDEP is estimated to account for up to 50% of deaths in patients with chronic refractory epilepsy, and up to 17% of deaths in all epilepsy patients (Tomson et al, 2005; Lhatoo & Sander, 2005).
The risk factor for SUDEP that has been most consistently found in multiple studies is a high frequency of seizures, especially tonic-clonic seizures (Tomson et al, 2008). There have been a limited number of witnessed cases of SUDEP, and in 90% of those cases death occurred after a seizure (Tomson et al, 2005). Thus, it is generally believed that SUDEP is due to an event that occurs either during or after a seizure. SUDEP cases are commonly found in bed, and it has been reported that they are in the prone position 70–80% of the time (Kloster & Engelskjon, 1999).
For many reasons, it has been difficult to precisely define the causes of SUDEP. For example, most cases are unwitnessed, and it is typical that there is limited information available about the circumstances of death. By definition the autopsy does not show any clear cause of death. Therefore, theories on the causes of SUDEP are based on a synthesis of what is known from a small number of witnessed cases, information gathered at the scene of unwitnessed cases, risk factors identified in epidemiological studies, knowledge of the physiology of seizure patients gathered during long-term monitoring, and data obtained from relevant animal models. These sources of information are incomplete and in some cases of uncertain relevance, but have led to the conclusion that SUDEP is not a single entity, but instead is due to heterogeneous mechanisms that lead to death from either cardiac or respiratory dysfunction.
It is important to note that it is rarely obvious in any specific case of SUDEP whether the primary cause of death was cardiac or respiratory, because there is often not enough information available. For example, unless an EKG and blood pressure were measured, it can’t be concluded that a patient with apnea had a primary respiratory problem, since apnea can be secondary to brain hypoperfusion due to underlying cardiac dysfunction. Likewise, a change in cardiac rhythm can not be interpreted as the primary problem unless there is also respiratory monitoring, because arrhythmias can be secondary to hypoxia and hypercapnia. Unfortunately, these vagaries have not always been considered in the literature. Even when there is respiratory monitoring it is not always possible to rule out a primary respiratory problem. For example, even when there is not frank apnea there can still be hypoventilation if there is a low tidal volume. It is particularly easy to miss hypoventilation in patients who continue to have respiratory movements, but are in the prone position and have blockage of the airway by bedding.
There is evidence that some SUDEP cases are due to cardiac causes (Schuele et al, 2007; Tomson et al, 2008), such as ictal asystole (Ryvlin et al, 2006) or mutations of long QT syndrome genes (Ackerman, 2005; Johnson et al, 2009). However, we will focus our attention in this review on respiratory causes of SUDEP, because there is a closer link between breathing and 5-HT neuron dysfunction.
Respiratory dysfunction in human epilepsy
It is not widely appreciated how commonly apnea and severe hypoventilation occur during and after seizures, but this has been documented in multiple studies of patients in epilepsy monitoring units (Tomson et al, 2008; Blum, 2009). Hughlings Jackson first noted in 1899 that respiratory arrest can occur in monkeys and humans during seizures originating near the uncus, and this can lead to patients “turning blue” (Jackson, 1899). In a study of 17 patients, 10 had apnea in 20 of 47 seizures, with a mean duration of 24 seconds (Nashef et al, 1996). Most of these seizures (16 of 20) were complex partial, and there was a drop in O2 saturation to less than 85% in 10 seizures. All of these apneas were central, and in some cases were also followed by obstructive apneas. More recently, 56 patients with intractable localization-related epilepsy were studied (Bateman et al, 2008), and it was found that oxygen desaturation below 90% occurred in 33% of seizures (whether they were generalized or partial), lasting for an average of 69 seconds (Figure 2A). Central apneas or hypopneas occurred with 53% of seizures. In a subsequent study (Seyal & Bateman, 2009) it was found that the occurrence of apnea in temporal lobe seizures correlated with seizure spread to the contralateral temporal lobe. End-tidal CO2 was measured in a subset of patients in both studies to confirm that oxygen desaturation was due to hypoventilation, and this revealed a substantial increase in PCO2 by 18.6 mm Hg (Fig 2B).
Figure 2.

Seizure-related respiratory apnea in patients with epilepsy. A) Pronounced oxygen desaturation with a complex partial left temporal onset seizure without secondary generalization. Modified with permission from Bateman et al (2008). B) Oxygen saturation (thick line) and ETCO2 (thin line) tracings in an 18-year-old man with a right temporal onset seizure followed by a secondarily generalized convulsion. The peak ETCO2 rise was 64 mm Hg. Oxygen saturation nadir was 78%. Duration of seizure is demarcated by the arrowheads. Duration of apnea is demarcated by the vertical bars. Modified with permission from Seyal and Bateman (2009).
There have been a small number of seizure patients undergoing monitoring in an epilepsy monitoring unit when they died of SUDEP or had a life-threatening episode and were resuscitated (Tomson et al, 2008; Espinosa et al, 2009; Bateman et al, 2010). It is important to recognize that these few examples are not necessarily representative of all SUDEP, but they are instructive about potential mechanisms. All of these cases occurred in association with a seizure. Six of the cases were recently reviewed (Tomson et al, 2008), and it was concluded that two of them were due to apnea, one to an arrhythmia, and three to “electrocerebral shutdown” defined as terminal flattening of the EEG without evidence of fatal cardiac or respiratory arrest. However, respiration was not quantitatively measured in any of these cases, and was assessed in four of them only by visual inspection. The conclusion that there was not a respiratory problem in the three cases of CNS shutdown was based on the lack of “EEG slowing typically observed during anoxia.” (Tomson et al, 2008). However, there is no evidence that EEG slowing is an invariant finding during anoxia, and may not be seen when EEG activity is already suppressed during the post-ictal period (Bateman et al, 2010). In fact, there have been recent reports of two additional cases of SUDEP (Bateman et al, 2010) and four near-SUDEP cases (Bateman et al, 2009) that occurred during epilepsy monitoring. In some of these patients there was post-ictal respiratory depression that was associated with EEG suppression, but not slowing. These data indicate that electrocerebral shutdown is probably not a primary cause of death, but is likely an event that is secondary to ictal or post-ictal hypoventilation or to hypoperfusion from a cardiac event. There have been additional SUDEP cases that were witnessed without monitoring. The witnesses of these cases commonly report post-ictal breathing problems (Langan et al, 2000).
We conclude that the two main causes of death in SUDEP are probably cardiac and respiratory dysfunction, and that a large percentage of the total may be due to hypoventilation. In some cases this may be due to central and/or obstructive apnea, and in other cases it may be due to a decrease in respiratory effort without frank apnea. In some individuals with post-ictal depression of consciousness, there may be blunting of the normal response to external airway obstruction, which should include turning of the head to relieve the obstruction. There is also evidence in some cases for post-ictal neurogenic pulmonary edema that may contribute to hypoxia (Seyal et al, 2010). Each of these mechanisms of respiratory dysfunction could secondarily lead to cardiac arrhythmias, as has been seen in cases of SUDEP and near-SUDEP during inpatient monitoring (Bateman et al, 2010).
Animal models of SUDEP
There have been a number of animal models that have been used to try to understand the mechanisms of SUDEP. All of these have limitations, but they can inform us about whether respiratory dysfunction can be induced by seizures, and whether it can be severe enough to cause death. They do not reveal whether the death in animals is due to the same mechanisms as in human SUDEP.
In a sheep model of status epilepticus, a subset of animals (38%) die within the first 5 minutes of seizure onset (Johnston et al, 1995) (Figure 3). This subset has severe hypoventilation due to central apnea, leading to hypoxia and hypercapnia. This work has been cited as suggesting that SUDEP in humans is due to respiratory arrest, but it is not known if this model is relevant since status epilepticus is excluded in the definition of SUDEP. However, it clearly shows that seizures can cause respiratory arrest that is severe enough to be fatal.
Figure 3.
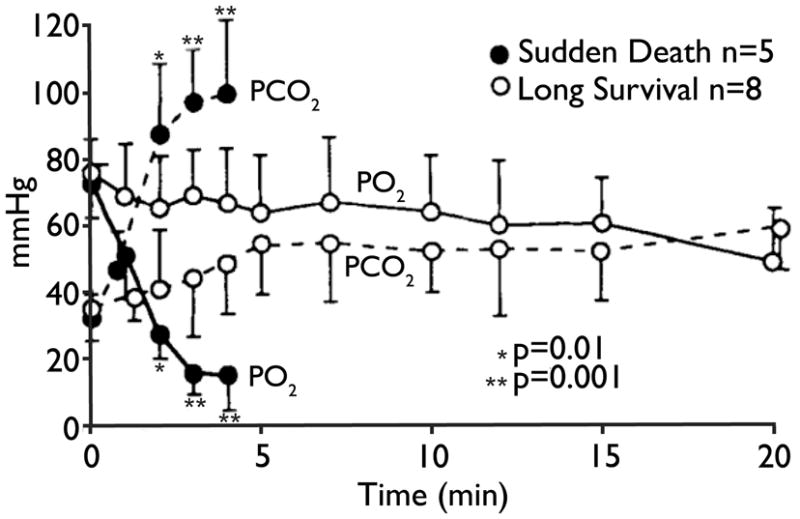
Severe blood gas derangements contribute to seizure related-sudden death. PaO2 and PaCO2 levels during seizures in sudden death (black circles) and long survival (white circles) groups. Dramatic elevations in PaCO2 and declines in PaO2 were demonstrated in sudden death animals. Differences between groups were highly significant (p = 0.001) at 3 and 4 minutes. Reproduced with permission from Johnston et al (1995).
The DBA/2 strain of mouse is susceptible to audiogenic seizures. In those that have tonic extension during their seizures, 88% develop respiratory arrest during the post-ictal period (Tupal & Faingold, 2006). Those with respiratory arrest invariably die without intervention, but essentially all recover if quickly provided respiratory support. This model more closely resembles SUDEP in many respects, but differs in that there is such a high incidence of death after a seizure. In contrast, sudden death is a very low probability event after any given seizure in human epilepsy patients. The high incidence of death after seizures in the DBA/2 mouse suggests that there is a genetic predisposition to respiratory arrest, since this does not occur in all mouse strains. There may be an unusually strong interaction between seizures and the brainstem respiratory centers in DBA/2 mice. If that is the case then there may also be a genetic predisposition to ictal or post-ictal respiratory arrest in some humans.
20% of captive baboons in a colony at the Southwest Foundation for Biomedical Research in San Antonio, Texas have generalized seizures. Those that do also have an increased incidence of sudden unexplained death (Szabo et al, 2009). The mechanisms of death have not yet been defined, but this model may be the closest to human SUDEP due to the relatively low incidence, the spontaneous death, and the fact that it occurs in a primate. This model also offers hope to examine genetic mechanisms leading to predisposition for seizure-associated death.
Links to 5-HT in SUDEP
There are several converging lines of evidence that a genetic or acquired defect in the 5-HT system can predispose an individual to SUDEP. For example, mice with genetic deletion of the 5-HT2C receptor are susceptible to audiogenic seizures, and as with DBA/2 mice they have post-ictal respiratory arrest that invariably leads to death if they are not resuscitated (Brennan et al, 1997). A link to 5-HT has also been established in the DBA/2 mice, because the frequency of respiratory arrest is markedly reduced by pretreatment with an SSRI (Fig. 4A). In the small subset of DBA/2 mice in which seizures do not occur after respiratory arrest, they are induced by the 5-HT receptor antagonist cyproheptadine (Fig 4B) (Tupal & Faingold, 2006). There is also a reduction in expression of certain 5-HT receptor subtypes in these mice (Uteshev et al, 2010). It remains to be determined if these animal models are relevant to human SUDEP, but these observations may be particularly relevant to the use of serotonergic drugs in human epilepsy patients.
Figure 4.

Increasing 5-HT prevents and blocking 5-HT receptors induces seizure-induced respiratory arrest in DBA/2 mice. A) Acute (gray bars) and delayed (black bars) effect of different doses of the serotonin-selective reuptake inhibitor, fluoxetine (i.p.; 30 min prior to first audiogenic seizure trial) in DBA/2 mice that display audiogenic seizure-induced respiratory arrest. Controls (white bars) values are 100% because all mice used for this study were susceptible to seizure-induced respiratory arrest at baseline. *, p < 0.05; #, p < 0.005 (Wilcoxon signed ranks test). B) Acute (gray bars) and delayed (black bars) effect of different doses of the non-selective 5-HT2 receptor antagonist, cyproheptadine (i.p.; 30 min prior to first audiogenic seizure trial) in DBA/2 mice that did not display audiogenic seizure-induced respiratory arrest. Controls (white bars) values are 0% because none of the mice used for this study were susceptible to seizure-induced respiratory arrest at baseline. **, p < 0.01; #, p < 0.005. (Wilcoxon signed ranks test). Modified with permission from Tupal and Faingold (2006).
There are reasons to believe that a defect in 5-HT neurons could lead to SUDEP. 5-HT neurons influence many different brain functions and neural systems, but there are two that may particularly relevant to SUDEP: Control of breathing and arousal.
5-HT neurons have a well-established role in breathing, and appear to act in at least three ways: 1) By stimulating respiratory output (Richerson, 2004; Hodges et al, 2009; Hodges & Richerson, 2010); 2) As chemoreceptors that respond to an increase in systemic CO2 levels by increasing respiratory output (Richerson, 2004; Corcoran et al, 2009), and; 3) To enable plasticity of the respiratory network in response to conditions such as intermittent hypoxia (Feldman et al, 2003). 5-HT neurons project to all of the major respiratory nuclei (Richerson, 2004; Ptak et al, 2009) (Fig. 5A). Lesions of these neurons lead to a decrease in respiratory output, which can be severe under some circumstances (Hodges et al, 2009; Hodges & Richerson, 2010). 5-HT2A and 5-HT4A agonists stimulate respiratory output and reverse the effects of a number of respiratory depressants (Pena & Ramirez, 2002; Richter et al, 2003; Hodges et al, 2009). 5-HT neurons of the medulla also release the neuropeptides substance P and TRH, both of which also have strong stimulatory effects on breathing (Richerson, 2004). The stimulatory effects of 5-HT neurons on breathing are likely to be relevant to human physiology. For example, prior recreational users of MDMA (which causes specific damage to 5-HT neurons; see above) have an increase in the risk of sleep apnea that is greater than the risk due to obesity (McCann et al, 2009). SSRIs have not, however, been found to be particularly effective for treating sleep apnea (Veasey et al, 2006).
Figure 5.

Central 5-HT neurons are chemoreceptors. A) Push-pull model of the role of chemosensitive medullary raphe neurons in respiratory chemoreception. It is proposed that the two subtypes of chemosensitive raphe neurons act in opposite ways to influence respiratory output within the medulla and in phrenic motor neurons. The brainstem sites are not specified in this model, but may include the nucleus tractus solitarius, nucleus ambiguus, pre-Botzinger complex and hypoglossal motor nucleus. Reproduced with permission from Richerson et al (2001). B) Left: Membrane potential under control conditions (5% CO2, pH 7.4) and during hypercapnic acidosis. Reproduced from Wang et al, (2002). Right: Ventilation during normoxic hypercapnia conditions (n = 10 WT, 8 Lmx1bf/f/p). Modified from Hodges et al (2008). C) Schematic depicting the proposed model by which serotonergic neurons mediate ventilatory and vigilance state changes in response to changes in CO2/pH (ΔCO2/ΔpH). Increased CO2 (due to rebreathing, airway obstruction or apnea) leads to decreased pH and activation of midbrain serotonergic neurons. These neurons in turn activate thalamocortical circuitry resulting in an elevation of arousal state. Increased CO2 also leads to activation of medullary serotonergic neurons, which stimulate respiratory nuclei to increase ventilation. Both of these pathways serve to restore CO2 homeostasis. 5-HT, serotonin. Reproduced with permission from Buchanan et al (2008).
When studied in brain slices and in cell culture, the majority of 5-HT neurons from mice and rats are strongly stimulated by an increase in PCO2 due to an indirect effect from the resulting decrease in pH (Wang et al, 2001; Richerson, 2004). These neurons are very sensitive, increasing their firing rate 3-fold on average in response to a decrease in pH from 7.4 to 7.2 (Fig. 5B). 5-HT neurons also increase their firing rate in vivo in response to inhalation of CO2 (Veasey et al, 1995). Focal acidosis within the medullary raphé induces an increase in ventilation (Bernard et al, 1996) and genetic lesions of 5-HT neurons reduce the ventilatory response of mice to inhaled CO2 (Hodges et al, 2008). These neurons also appear to be anatomically specialized to detect changes in arterial PCO2, because they have dendrites that wrap around the basilar artery and other large arteries of the medulla (Bradley et al, 2002). For these and other reasons it is believed that 5-HT neurons of the medulla are central respiratory chemoreceptors that regulate the systemic levels of PCO2 (Corcoran et al, 2009).
5-HT neurons of the midbrain are involved in regulation of sleep and arousal. Their specific role has been unclear, but they are considered by some to be part of the ascending arousal system (Saper et al, 2001), causing arousal when they are activated. Since 5-HT neurons in the midbrain, like those in the medulla, are stimulated by acidosis and are near large arteries (Severson et al, 2003), we have proposed that midbrain 5HT neurons are also chemoreceptors, but instead of stimulating breathing in response to hypercapnia they induce arousal when blood CO2 levels rise (Buchanan et al, 2008) (Fig 5C). This arousal response to hypercapnia is very robust in normal individuals (Buchanan & Richerson, 2009b), and can be a life-saving reflex. For example, when a pillow or blanket obstructs the mouth and nose of a person who is asleep, an increase in respiratory effort is unlikely to provide enough air exchange to maintain normal arterial PO2 and PCO2. It is important to also wake up slightly and turn the head to relieve the obstruction. Preliminary data indicate that this reflex is absent after genetic deletion of 5-HT neurons in mice (Buchanan & Richerson, 2009a) consistent with the hypothesis that these neurons are “arousal chemoreceptors.”
These data from animal models lead to an hypothesis for what could cause post-ictal death in some epilepsy patients. It is known that both focal and generalized seizures can invade subcortical structures, and this may in fact underlie the loss of consciousness that occurs in unilateral temporal lobe seizures (Englot & Blumenfeld, 2009). The post-ictal state (i.e. depressed consciousness) may be due in part to stunning of the monoamine neural systems, including 5-HT neurons. Since 5-HT neurons stimulate breathing, a reduction in their firing activity during and after a seizure could lead to ictal and/or post-ictal hypoventilation. There may be genetic determinants that increase the likelihood of this occurring in susceptible individuals. If a patient has a seizure in bed and ends up in the prone position with their face covered by a pillow or blanket, post-ictal depression of 5-HT neurons may prevent that individual from responding appropriately by either turning their head, increasing their breathing, or both. Enhancing the output of the 5-HT system could reduce the likelihood of severe hypoxia, respiratory acidosis, and death in response to this situation. One potential avenue for intervention in humans could be treatment with an SSRI, which is effective in preventing death in the DBA/2 mouse model of SUDEP. It will be important to determine whether SSRIs or 5-HT receptor agonists are effective in preventing seizure-induced respiratory depression, and if there is a decrease in the incidence of SUDEP in patients on these medications.
Taken in combination, the data presented here suggest that some patients could have underlying pathology of the 5-HT system that leads to a “seizure/depression/SUDEP phenotype” rather than just a seizure/depression phenotype. Interictal breathing and arousal may be normal due to compensation, although a defect in the 5-HT system could explain an increased incidence of depression (see above) and sleep apnea (Chihorek et al, 2007) in epilepsy patients. However, a seizure might lead to decompensation, with life-threatening depression of breathing and arousal.
SIDS & SUDEP: Shared common final pathway?
The link between 5-HT neurons and SUDEP is made more plausible by similarities to SIDS (Table 1), which has also recently been linked to defects in the 5-HT system. SIDS is defined as the sudden death of an infant under one year of age that is typically associated with sleep and that remains unexplained after a complete autopsy and death scene investigation (Kinney et al, 2009). This definition has similarities to the definition of SUDEP. Like SUDEP, SIDS is a diagnosis of exclusion. The incidence of SIDS is 0.6 per 1000 live births in the U.S. (Kinney et al, 2009), so as with SUDEP it is relatively low in the relevant population. However, SIDS is still the leading cause of post-neonatal infant death in developed countries, and SUDEP is the leading cause of death in epilepsy patients.
Table 1.
Shared mechanisms in sudden infant death syndrome (SIDS) and sudden unexpected death in epilepsy (SUDEP).
| SIDS | SUDEP | |
|---|---|---|
| Method of diagnosis | Exclusion | Exclusion |
| Routine Autopsy | Normal | Normal |
| Cause of death | Respiratory | Respiratory |
| Cardiac | Cardiac | |
| Arousal | Arousal | |
| Thermoregulatory | ||
| Baseline health | Normal | Often normal except seizures |
| Circumstances of death | Usually found prone | Usually found prone |
| Link to 5-HT | Yes | Yes |
| Incidence | 0.6 per 1000 live births | 0.1-10 per 1000 epilepsy patients |
The causes of SIDS remain unknown, but like SUDEP the leading theories are focused on cardiac and respiratory mechanisms (Hunt & Brouillette, 1987; Kinney et al, 2009). As with SUDEP, there is limited information available for most cases, and it is often difficult to precisely define the cause of death. As with SUDEP, it is likely to be a heterogeneous disorder, and a small percentage of cases have mutations of long QT syndrome genes (Schwartz et al, 1998). However, it is widely believed that the majority of SIDS cases are due to defects in arousal and/or the ventilatory response to hypercapnia and/or hypoxia (Kinney et al, 2009).
There is also a large body of data pointing to a defect in the 5-HT system in infants that die of SIDS. These include decreased binding of ligands of 5-HT1A receptors (likely autoreceptors on 5-HT neurons) in the raphé nuclei (Paterson et al, 2006), an increase in the number of immature appearing 5-HT neurons (Paterson et al, 2006), and a decrease in 5-HT levels in the medulla of infants that died of SIDS (Kinney et al, 2009; Duncan et al, 2010). Like SUDEP, the majority of SIDS infants are found in the prone position, and in fact there has been a dramatic decrease in the incidence of SIDS by 50% during the last 2 decades due to the recommendation to place infants to sleep in the supine position (the “Back to Sleep” campaign). Thus, there are significant similarities with the mechanism of SUDEP proposed above, with dysfunction of 5-HT neurons leading to a defect in the normal arousal and respiratory responses to hypercapnia and hypoxia, especially when in the prone position. In the case of SIDS, the defect is likely due to developmental delay in maturation of 5-HT neurons combined with a decrease in 5-HT neuron firing during sleep, whereas in the case of SUDEP there is temporary dysfunction of 5-HT (and likely other monoamine) neurons due to invasion of the brainstem by seizure activity. In both cases, dysfunction of 5-HT neurons leads to reduced ability to respond appropriately to an external stressor during a state of generalized CNS depression (sleep vs post-ictal state).
Conclusions
Knowledge about the normal function of the 5-HT system, and the effect of seizures on breathing and arousal, may help to better understand the cause of death in SUDEP. This may in turn lead to better ways to identify those patients who are at highest risk and to treat them to prevent SUDEP. Given the many similarities between SIDS and SUDEP, lessons learned from one field may benefit the other. For example, the Back to Sleep campaign reduced the incidence of SIDS by 50%, which suggests that it may help to keep patients in the supine position after a seizure. Although there is no evidence for a primary defect in the 5-HT system in SUDEP patients, individuals at highest risk may be those with the greatest seizure-induced respiratory suppression. If so, it may be possible to screen patients to identify the highest risk group. It has not been widely appreciated how many patients have hypoventilation during and after seizures. It seems prudent for epilepsy specialists to monitor breathing more frequently than they have in the past, and to identify and treat respiratory dysfunction when it occurs.
Acknowledgments
Supported by the NICHD, the VAMC, the Bumpus Foundation and Jazz Pharmaceuticals
Footnotes
Disclosure: The authors have no conflicts of interest to declare.
References
- Ackerman MJ. Cardiac causes of sudden unexpected death in children and their relationship to seizures and syncope: genetic testing for cardiac electropathies. Semin Pediatr Neurol. 2005;12:52–8. doi: 10.1016/j.spen.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Ahmad S, Fowler LJ, Whitton PS. Lamotrigine, carbamazepine and phenytoin differentially alter extracellular levels of 5-hydroxytryptamine, dopamine and amino acids. Epilepsy Res. 2005;63:141–9. doi: 10.1016/j.eplepsyres.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Bagdy G, Kecskemeti V, Riba P, Jakus R. Serotonin and epilepsy. J Neurochem. 2007;100:857–73. doi: 10.1111/j.1471-4159.2006.04277.x. [DOI] [PubMed] [Google Scholar]
- Bateman LM, Li CS, Seyal M. Ictal hypoxemia in localization-related epilepsy: analysis of incidence, severity and risk factors. Brain. 2008;131:3239–45. doi: 10.1093/brain/awn277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman LM, Spitz M, Seyal M. Ictal hypoventilation contributes to cardiac arrhythmia and SUDEP: report on two deaths and four potentially life-threatening events in video-EEG monitored patients. Epilepsia. 2009;50:410. doi: 10.1111/j.1528-1167.2009.02513.x. [DOI] [PubMed] [Google Scholar]
- Bateman LM, Spitz M, Seyal M. Ictal hypoventilation contributes to cardiac arrhythmia and SUDEP: Report on two deaths in video-EEG-monitored patients. Epilepsia. 2010 doi: 10.1111/j.1528-1167.2009.02513.x. [DOI] [PubMed] [Google Scholar]
- Ben-Abraham R, Szold O, Rudick V, Weinbroum AA. 'Ecstasy' intoxication: life-threatening manifestations and resuscitative measures in the intensive care setting. Eur J Emerg Med. 2003;10:309–13. doi: 10.1097/00063110-200312000-00013. [DOI] [PubMed] [Google Scholar]
- Bernard DG, Li AH, Nattie EE. Evidence for central chemoreception in the midline raphe. J Appl Physiol. 1996;80:108–15. doi: 10.1152/jappl.1996.80.1.108. [DOI] [PubMed] [Google Scholar]
- Blum AS. Respiratory physiology of seizures. J Clin Neurophysiol. 2009;26:309–15. doi: 10.1097/WNP.0b013e3181b7f14d. [DOI] [PubMed] [Google Scholar]
- Bonnycastle DD, Giarman NJ, Paasonen MK. Anticonvulsant compounds and 5-hydroxytryptamine in rat brain. Br J Pharmacol Chemother. 1957;12:228–31. doi: 10.1111/j.1476-5381.1957.tb00125.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley SR, Pieribone VA, Wang W, Severson CA, Jacobs RA, Richerson GB. Chemosensitive serotonergic neurons are intimately associated with large arteries of the ventral medulla. Nat Neurosci. 2002;5:401–2. doi: 10.1038/nn848. [DOI] [PubMed] [Google Scholar]
- Brennan TJ, Seeley WW, Kilgard M, Schreiner CE, Tecott LH. Sound-induced seizures in serotonin 5-HT2c receptor mutant mice. Nat Genet. 1997;16:387–90. doi: 10.1038/ng0897-387. [DOI] [PubMed] [Google Scholar]
- Browning RA, Hoffmann WE, Simonton RL. Changes in seizure susceptibility after intracerebral treatment with 5,7-dihydroxytryptamine: role of serotonergic neurons. Ann N Y Acad Sci. 1978;305:437–56. doi: 10.1111/j.1749-6632.1978.tb31540.x. [DOI] [PubMed] [Google Scholar]
- Buchanan GF, Hodges MR, Richerson GB. Contribution of Chemosensitive Serotonergic Neurons to Interactions between the Sleep-Wake Cycle and Respiratory Control. In: Monti JM, Pandi-Perumal SR, Jacobs BL, Nutt DJ, editors. Serotonin and sleep: Molecular, functional and clinical aspects. Switzerland: Birkhauser-Verlag; 2008. pp. 529–54. [Google Scholar]
- Buchanan GF, Richerson GB. Effect of genetic deletion of 5-HT neurons on sleep and arousal. Soc Neurosci Abstr. 2009a:35. [Google Scholar]
- Buchanan GF, Richerson GB. Role of chemoreceptors in mediating dyspnea. Respir Physiol Neurobiol. 2009b;167:9–19. doi: 10.1016/j.resp.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buterbaugh GG. Effect of drugs modifying central serotonergic function on the response of extensor and nonextensor rats to maximal electroshock. Life Sci. 1978;23:2393–404. doi: 10.1016/0024-3205(78)90297-7. [DOI] [PubMed] [Google Scholar]
- Chihorek AM, Abou-Khalil B, Malow BA. Obstructive sleep apnea is associated with seizure occurrence in older adults with epilepsy. Neurology. 2007;69:1823–7. doi: 10.1212/01.wnl.0000279334.78298.d5. [DOI] [PubMed] [Google Scholar]
- Cooper JR, Bloom FE, Roth HR. The biochemical basis of neuropharmacology. 7. New York: Oxford University Press; 1996. [Google Scholar]
- Corcoran AE, Hodges MR, Wu Y, et al. Medullary serotonin neurons and central CO2 chemoreception. Respir Physiol Neurobiol. 2009;168:49–58. doi: 10.1016/j.resp.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlström A, Fuxe K. Evidence for the existence of monoamine containing neurons in the central nervous system: I. Demonstration of monoamines in cell bodies of brain stem neurons. Acta Physiol Scand. 1964;62:1–55. [PubMed] [Google Scholar]
- Dailey JW, Mishra PK, Ko KH, Penny JE, Jobe PC. Serotonergic abnormalities in the central nervous system of seizure-naive genetically epilepsy-prone rats. Life Sci. 1992;50:319–26. doi: 10.1016/0024-3205(92)90340-u. [DOI] [PubMed] [Google Scholar]
- Dailey JW, Reigel CE, Mishra PK, Jobe PC. Neurobiology of seizure predisposition in the genetically epilepsy-prone rat. [Review] [55 refs] Epilepsy Res. 1989;3:3–17. doi: 10.1016/0920-1211(89)90063-6. [DOI] [PubMed] [Google Scholar]
- Dailey JW, Reith ME, Yan QS, Li MY, Jobe PC. Anticonvulsant doses of carbamazepine increase hippocampal extracellular serotonin in genetically epilepsy-prone rats: dose response relationships. Neuroscience Letters. 1997;227:13–6. doi: 10.1016/s0304-3940(97)00288-7. [DOI] [PubMed] [Google Scholar]
- Dasheiff RM. Sudden unexpected death in epilepsy: a series from an epilepsy surgery program and speculation on the relationship to sudden cardiac death. J Clin Neurophysiol. 1991;8:216–22. [PubMed] [Google Scholar]
- Donovan SL, Mamounas LA, Andrews AM, Blue ME, McCasland JS. GAP-43 is critical for normal development of the serotonergic innervation in forebrain. J Neurosci. 2002;22:3543–52. doi: 10.1523/JNEUROSCI.22-09-03543.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorr AE, Debonnel G. Effect of vagus nerve stimulation on serotonergic and noradrenergic transmission. J Pharmacol Exp Ther. 2006;318:890–8. doi: 10.1124/jpet.106.104166. [DOI] [PubMed] [Google Scholar]
- Duncan JR, Paterson DS, Hoffman JM, et al. Brainstem serotonergic deficiency in sudden infant death syndrome. JAMA. 2010;303:430–7. doi: 10.1001/jama.2010.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elger G, Hoppe C, Falkai P, Rush AJ, Elger CE. Vagus nerve stimulation is associated with mood improvements in epilepsy patients. Epilepsy Res. 2000;42:203–10. doi: 10.1016/s0920-1211(00)00181-9. [DOI] [PubMed] [Google Scholar]
- Englot DJ, Blumenfeld H. Consciousness and epilepsy: why are complex-partial seizures complex? Prog Brain Res. 2009;177:147–70. doi: 10.1016/S0079-6123(09)17711-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa PS, Lee JW, Tedrow UB, Bromfield EB, Dworetzky BA. Sudden unexpected near death in epilepsy: malignant arrhythmia from a partial seizure. Neurology. 2009;72:1702–3. doi: 10.1212/WNL.0b013e3181a55f90. [DOI] [PubMed] [Google Scholar]
- Favale E, Audenino D, Cocito L, Albano C. The anticonvulsant effect of citalopram as an indirect evidence of serotonergic impairment in human epileptogenesis. Seizure. 2003;12:316–8. doi: 10.1016/s1059-1311(02)00315-1. [DOI] [PubMed] [Google Scholar]
- Feldman JL, Mitchell GS, Nattie EE. Breathing: Rhythmicity, plasticity, chemosensitivity. Ann Rev Neurosci. 2003;26:239–66. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsgren L, Nystrom L. An incident case–referent study of epileptic seizures in adults. Epilepsy Res. 1990;6:66–81. doi: 10.1016/0920-1211(90)90010-s. [DOI] [PubMed] [Google Scholar]
- Giorgi FS, Lazzeri G, Natale G, et al. MDMA and seizures: a dangerous liaison? Ann N Y Acad Sci. 2006;1074:357–64. doi: 10.1196/annals.1369.035. [DOI] [PubMed] [Google Scholar]
- Giorgi FS, Pizzanelli C, Ferrucci M, et al. Previous exposure to (+/−) 3,4-methylenedioxymethamphetamine produces long-lasting alteration in limbic brain excitability measured by electroencephalogram spectrum analysis, brain metabolism and seizure susceptibility. Neurosci. 2005;136:43–53. doi: 10.1016/j.neuroscience.2005.07.036. [DOI] [PubMed] [Google Scholar]
- Giovacchini G, Toczek MT, Bonwetsch R, et al. 5-HT 1A receptors are reduced in temporal lobe epilepsy after partial-volume correction. J Nucl Med. 2005;46:1128–35. [PMC free article] [PubMed] [Google Scholar]
- Hasler G, Bonwetsch R, Giovacchini G, et al. 5-HT1A receptor binding in temporal lobe epilepsy patients with and without major depression. Biol Psychiatry. 2007;62:1258–64. doi: 10.1016/j.biopsych.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesdorffer DC, Hauser WA, Annegers JF, Cascino G. Major depression is a risk factor for seizures in older adults. Ann Neurol. 2000;47:246–9. [PubMed] [Google Scholar]
- Hesdorffer DC, Hauser WA, Olafsson E, Ludvigsson P, Kjartansson O. Depression and suicide attempt as risk factors for incident unprovoked seizures. Ann Neurol. 2006;59:35–41. doi: 10.1002/ana.20685. [DOI] [PubMed] [Google Scholar]
- Hodges MR, Richerson GB. The role of medullary serotonin (5-HT) neurons in respiratory control: Contributions to eupneic ventilation, CO2 chemoreception and thermoregulation. J Appl Physiol. 2010 doi: 10.1152/japplphysiol.01270.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges MR, Tattersall GJ, Harris MB, et al. Defects in breathing and thermoregulation in mice with near-complete absence of central serotonin neurons. J Neurosci. 2008;28:2495–505. doi: 10.1523/JNEUROSCI.4729-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges MR, Wehner M, Aungst J, Smith JC, Richerson GB. Transgenic mice lacking serotonin neurons have severe apnea and high mortality during development. J Neurosci. 2009;29:10341–9. doi: 10.1523/JNEUROSCI.1963-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt CE, Brouillette RT. Sudden infant death syndrome: 1987 perspective. J Pediatr. 1987;110:669–78. doi: 10.1016/s0022-3476(87)80001-x. [DOI] [PubMed] [Google Scholar]
- Jackson JH. On asphyxia in slight epileptic paroxysms. Lancet. 1899;153:79–80. [Google Scholar]
- Jobe PC. Common pathogenic mechanisms between depression and epilepsy: an experimental perspective. Epilepsy Behav. 2003;4 (Suppl 3):S14–S24. doi: 10.1016/j.yebeh.2003.08.020. [DOI] [PubMed] [Google Scholar]
- Jobe PC, Browning RA. The serotonergic and noradrenergic effects of antidepressant drugs are anticonvulsant, not proconvulsant. Epilepsy Behav. 2005;7:602–19. doi: 10.1016/j.yebeh.2005.07.014. [DOI] [PubMed] [Google Scholar]
- Jobe PC, Dailey JW, Wernicke JF. A noradrenergic and serotonergic hypothesis of the linkage between epilepsy and affective disorders. [Review] [508 refs] Critical Reviews in Neurobiology. 1999;13:317–56. doi: 10.1615/critrevneurobiol.v13.i4.10. [DOI] [PubMed] [Google Scholar]
- Johnson JN, Hofman N, Haglund CM, Cascino GD, Wilde AA, Ackerman MJ. Identification of a possible pathogenic link between congenital long QT syndrome and epilepsy. Neurology. 2009;72:224–31. doi: 10.1212/01.wnl.0000335760.02995.ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston SC, Horn JK, Valente J, Simon RP. The role of hypoventilation in a sheep model of epileptic sudden death. Ann Neurol. 1995;37:531–7. doi: 10.1002/ana.410370416. [DOI] [PubMed] [Google Scholar]
- Kanner AM. Epilepsy, suicidal behaviour, and depression: do they share common pathogenic mechanisms? Lancet Neurol. 2006;5:107–8. doi: 10.1016/S1474-4422(06)70331-3. [DOI] [PubMed] [Google Scholar]
- Kanner AM. Depression and epilepsy: a review of multiple facets of their close relation. Neurol Clin. 2009;27:865–80. doi: 10.1016/j.ncl.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Kilian M, Frey HH. Central monoamines and convulsine thresholds in mice and rats. Neuropharmacology. 1973;12:681–92. doi: 10.1016/0028-3908(73)90121-4. [DOI] [PubMed] [Google Scholar]
- Kinney HC, Richerson GB, Dymecki SM, Darnall RA, Nattie EE. The brainstem and serotonin in the sudden infant death syndrome. Annu Rev Pathol. 2009;4:517–50. doi: 10.1146/annurev.pathol.4.110807.092322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloster R, Engelskjon T. Sudden unexpected death in epilepsy (SUDEP): a clinical perspective and a search for risk factors. J Neurol Neurosurg Psychiatry. 1999;67:439–44. doi: 10.1136/jnnp.67.4.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondziella D, Asztely F. Don't be afraid to treat depression in patients with epilepsy! Acta Neurol Scand. 2009;119:75–80. doi: 10.1111/j.1600-0404.2008.01088.x. [DOI] [PubMed] [Google Scholar]
- Langan Y, Nashef L, Sander JW. Sudden unexpected death in epilepsy: a series of witnessed deaths. J Neurol Neurosurg Psychiatry. 2000;68:211–3. doi: 10.1136/jnnp.68.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lhatoo SD, Sander JW. Cause-specific mortality in epilepsy. Epilepsia. 2005;46 (Suppl 11):36–9. doi: 10.1111/j.1528-1167.2005.00406.x. [DOI] [PubMed] [Google Scholar]
- Liew CJ, Lim YM, Bonwetsch R, et al. 18F-FCWAY and 18F-FDG PET in MRI-negative temporal lobe epilepsy. Epilepsia. 2009;50:234–9. doi: 10.1111/j.1528-1167.2008.01789.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manta S, Dong J, Debonnel G, Blier P. Enhancement of the function of rat serotonin and norepinephrine neurons by sustained vagus nerve stimulation. J Psychiatry Neurosci. 2009a;34:272–80. [PMC free article] [PubMed] [Google Scholar]
- Manta S, Dong J, Debonnel G, Blier P. Optimization of vagus nerve stimulation parameters using the firing activity of serotonin neurons in the rat dorsal raphe. Eur Neuropsychopharmacol. 2009b;19:250–5. doi: 10.1016/j.euroneuro.2008.12.001. [DOI] [PubMed] [Google Scholar]
- McCann UD, Sgambati FP, Schwartz AR, Ricaurte GA. Sleep apnea in young abstinent recreational MDMA (“ecstasy”) consumers. Neurology. 2009;73:2011–7. doi: 10.1212/WNL.0b013e3181c51a62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlet I, Ostrowsky K, Costes N, et al. 5-HT1A receptor binding and intracerebral activity in temporal lobe epilepsy: an [18F]MPPF-PET study. Brain. 2004;127:900–13. doi: 10.1093/brain/awh109. [DOI] [PubMed] [Google Scholar]
- Nahas Z, Marangell LB, Husain MM, et al. Two-year outcome of vagus nerve stimulation (VNS) for treatment of major depressive episodes. J Clin Psychiatry. 2005;66:1097–104. doi: 10.4088/jcp.v66n0902. [DOI] [PubMed] [Google Scholar]
- Nashef L. Sudden Unexpected Death in Epilepsy: Terminology and Definitions. Epilepsia. 1997;38:S6–S8. doi: 10.1111/j.1528-1157.1997.tb06130.x. [DOI] [PubMed] [Google Scholar]
- Nashef L, Walker F, Allen P, Sander JW, Shorvon SD, Fish DR. Apnoea and bradycardia during epileptic seizures: relation to sudden death in epilepsy. J Neurol Neurosurg Psychiatry. 1996;60:297–300. doi: 10.1136/jnnp.60.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson L, Tomson T, Farahmand BY, Diwan V, Persson PG. Cause-specific mortality in epilepsy: a cohort study of more than 9,000 patients once hospitalized for epilepsy. Epilepsia. 1997;38:1062–8. doi: 10.1111/j.1528-1157.1997.tb01194.x. [DOI] [PubMed] [Google Scholar]
- Okada M, Kaneko S, Hirano T, et al. Effects of zonisamide on extracellular levels of monoamine and its metabolite, and on Ca2+ dependent dopamine release. Epilepsy Res. 1992;13:113–9. doi: 10.1016/0920-1211(92)90066-3. [DOI] [PubMed] [Google Scholar]
- Paterson DS, Trachtenberg FL, Thompson EG, et al. Multiple serotonergic brainstem abnormalities in sudden infant death syndrome. JAMA. 2006;296:2124–32. doi: 10.1001/jama.296.17.2124. [DOI] [PubMed] [Google Scholar]
- Pena F, Ramirez JM. Endogenous activation of serotonin-2A receptors is required for respiratory rhythm generation in vitro. J Neurosci. 2002;22:11055–64. doi: 10.1523/JNEUROSCI.22-24-11055.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasher VP. Seizures associated with fluoxetine therapy. Seizure. 1993;2:315–7. doi: 10.1016/s1059-1311(05)80148-7. [DOI] [PubMed] [Google Scholar]
- Prendiville S, Gale K. Anticonvulsant effect of fluoxetine on focally evoked limbic motor seizures in rats. Epilepsia. 1993;34:381–4. doi: 10.1111/j.1528-1157.1993.tb02425.x. [DOI] [PubMed] [Google Scholar]
- Ptak K, Yamanishi T, Aungst J, et al. Raphe neurons stimulate respiratory circuit activity by multiple mechanisms via endogenously released serotonin and substance P. J Neurosci. 2009;29:3720–37. doi: 10.1523/JNEUROSCI.5271-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafnsson V, Olafsson E, Hauser WA, Gudmundsson G. Cause-specific mortality in adults with unprovoked seizures. A population-based incidence cohort study. Neuroepidemiology. 2001;20:232–6. doi: 10.1159/000054795. [DOI] [PubMed] [Google Scholar]
- Richerson GB. Serotonin neurons as CO2 sensors that maintain pH homeostasis. Nature Reviews Neuroscience. 2004;5:449–61. doi: 10.1038/nrn1409. [DOI] [PubMed] [Google Scholar]
- Richerson GB, Wang W, Tiwari JK, Bradley SR. Chemosensitivity of serotonergic neurons in the rostral ventral medulla. Resp Physiol. 2001;129:175–89. doi: 10.1016/s0034-5687(01)00289-4. [DOI] [PubMed] [Google Scholar]
- Richter DW, Manzke T, Wilken B, Ponimaskin E. Serotonin receptors: guardians of stable breathing. Trends in Molecular Medicine. 2003;9:542–8. doi: 10.1016/j.molmed.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Rush AJ, Siefert SE. Clinical issues in considering vagus nerve stimulation for treatment- resistant depression. Exp Neurol. 2009;219:36–43. doi: 10.1016/j.expneurol.2009.04.015. [DOI] [PubMed] [Google Scholar]
- Ryvlin P, Montavont A, Kahane P. Sudden unexpected death in epilepsy: from mechanisms to prevention. Curr Opin Neurol. 2006;19:194–9. doi: 10.1097/01.wco.0000218238.90711.f4. [DOI] [PubMed] [Google Scholar]
- Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. TINS. 2001;24:726–31. doi: 10.1016/s0166-2236(00)02002-6. [DOI] [PubMed] [Google Scholar]
- Sargent PA, Kjaer KH, Bench CJ, et al. Brain serotonin1A receptor binding measured by positron emission tomography with [11C]WAY-100635: effects of depression and antidepressant treatment. Arch Gen Psychiatry. 2000;57:174–80. doi: 10.1001/archpsyc.57.2.174. [DOI] [PubMed] [Google Scholar]
- Sarnyai Z, Sibille EL, Pavlides C, Fenster RJ, McEwen BS, Toth M. Impaired hippocampal-dependent learning and functional abnormalities in the hippocampus in mice lacking serotonin1A receptors. Proc Natl Acad Sci U S A. 2000;97:14731–6. doi: 10.1073/pnas.97.26.14731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savic I, Lindstrom P, Gulyas B, Halldin C, Andree B, Farde L. Limbic reductions of 5-HT1A receptor binding in human temporal lobe epilepsy. Neurology. 2004;62:1343–51. doi: 10.1212/01.wnl.0000123696.98166.af. [DOI] [PubMed] [Google Scholar]
- Schuele SU, Widdess-Walsh P, Bermeo A, Luders HO. Sudden unexplained death in epilepsy: the role of the heart. Cleve Clin J Med. 2007;74 (Suppl 1):S121–S127. doi: 10.3949/ccjm.74.suppl_1.s121. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ, Stramba-Badiale M, Segantini A, et al. Prolongation of the QT interval and the sudden infant death syndrome. N Engl J Med. 1998;338:1709–14. doi: 10.1056/NEJM199806113382401. [DOI] [PubMed] [Google Scholar]
- Severson CA, Wang WG, Pieribone VA, Dohle CI, Richerson GB. Midbrain serotonergic neurons are central pH chemoreceptors. Nat Neurosci. 2003;6:1139–40. doi: 10.1038/nn1130. [DOI] [PubMed] [Google Scholar]
- Seyal M, Bateman LM. Ictal apnea linked to contralateral spread of temporal lobe seizures: Intracranial EEG recordings in refractory temporal lobe epilepsy. Epilepsia. 2009;50:2557–62. doi: 10.1111/j.1528-1167.2009.02245.x. [DOI] [PubMed] [Google Scholar]
- Seyal M, Bateman LM, Albertson TE, Lin TC, Li CS. Respiratory changes with seizures in localization-related epilepsy: Analysis of periictal hypercapnia and airflow patterns. Epilepsia. 2010 doi: 10.1111/j.1528-1167.2009.02518.x. [DOI] [PubMed] [Google Scholar]
- Statnick MA, Maring-Smith ML, Clough RW, et al. Effect of 5,7-dihydroxytryptamine on audiogenic seizures in genetically epilepsy-prone rats. Life Sci. 1996;59:1763–71. doi: 10.1016/0024-3205(96)00519-x. [DOI] [PubMed] [Google Scholar]
- Szabo CA, Knape KD, Leland MM, et al. Mortality in captive baboons with seizures: a new model for SUDEP? Epilepsia. 2009;50:1995–8. doi: 10.1111/j.1528-1167.2009.02073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellez-Zenteno JF, Patten SB, Jette N, Williams J, Wiebe S. Psychiatric comorbidity in epilepsy: a population-based analysis. Epilepsia. 2007;48:2336–44. doi: 10.1111/j.1528-1167.2007.01222.x. [DOI] [PubMed] [Google Scholar]
- Theodore WH, Giovacchini G, Bonwetsch R, et al. The effect of antiepileptic drugs on 5-HT-receptor binding measured by positron emission tomography. Epilepsia. 2006;47:499–503. doi: 10.1111/j.1528-1167.2006.00458.x. [DOI] [PubMed] [Google Scholar]
- Toczek MT, Carson RE, Lang L, et al. PET imaging of 5-HT1A receptor binding in patients with temporal lobe epilepsy. Neurology. 2003;60:749–56. doi: 10.1212/01.wnl.0000049930.93113.20. [DOI] [PubMed] [Google Scholar]
- Tomson T, Nashef L, Ryvlin P. Sudden unexpected death in epilepsy: current knowledge and future directions. Lancet Neurol. 2008;7:1021–31. doi: 10.1016/S1474-4422(08)70202-3. [DOI] [PubMed] [Google Scholar]
- Tomson T, Walczak T, Sillanpaa M, Sander JW. Sudden unexpected death in epilepsy: a review of incidence and risk factors. Epilepsia. 2005;46 (Suppl 11):54–61. doi: 10.1111/j.1528-1167.2005.00411.x. [DOI] [PubMed] [Google Scholar]
- Tupal S, Faingold CL. Evidence supporting a role of serotonin in modulation of sudden death induced by seizures in DBA/2 mice. Epilepsia. 2006;47:21–6. doi: 10.1111/j.1528-1167.2006.00365.x. [DOI] [PubMed] [Google Scholar]
- Uteshev VV, Tupal S, Mhaskar Y, Faingold CL. Abnormal serotonin receptor expression in DBA/2 mice associated with susceptibility to sudden death due to respiratory arrest. Epilepsy Res. 2010;88:183–8. doi: 10.1016/j.eplepsyres.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Veasey SC, Fornal CA, Metzler CW, Jacobs BL. Response of serotonergic caudal raphe neurons in relation to specific motor activities in freely moving cats. J Neurosci. 1995;15:5346–59. doi: 10.1523/JNEUROSCI.15-07-05346.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veasey SC, Guilleminault C, Strohl KP, Sanders MH, Ballard RD, Magalang UJ. Medical therapy for obstructive sleep apnea: a review by the Medical Therapy for Obstructive Sleep Apnea Task Force of the Standards of Practice Committee of the American Academy of Sleep Medicine. Sleep. 2006;29:1036–44. doi: 10.1093/sleep/29.8.1036. [DOI] [PubMed] [Google Scholar]
- Wang W, Bradley SR, Richerson GB. Quantification of the response of rat medullary raphe neurones to independent changes in pHo and PCO2. Journal of Physiology. 2002;540:951–70. doi: 10.1113/jphysiol.2001.013443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Zaykin AV, Tiwari JK, Bradley SR, Richerson GB. Acidosis-stimulated neurons of the medullary raphe are serotonergic. J Neurophysiol. 2001;85:2224–35. doi: 10.1152/jn.2001.85.5.2224. [DOI] [PubMed] [Google Scholar]
- Wiegartz P, Seidenberg M, Woodard A, Gidal B, Hermann B. Co-morbid psychiatric disorder in chronic epilepsy: recognition and etiology of depression. Neurology. 1999;53:S3–S8. [PubMed] [Google Scholar]
- Yan QS, Jobe PC, Cheong JH, Ko KH, Dailey JW. Role of serotonin in the anticonvulsant effect of fluoxetine in genetically epilepsy-prone rats. Naunyn-Schmiedebergs Arch Pharmacol. 1994;350:149–52. doi: 10.1007/BF00241089. [DOI] [PubMed] [Google Scholar]