

Abstract
The nuclear factor-kappa B (NF-κB) transcription factor plays a critical role in diverse cellular processes associated with proliferation, cell death, development, as well as innate and adaptive immune responses. NF-κB is normally sequestered in the cytoplasm by a family of inhibitory proteins known as IκBs. The signal pathways leading to the liberation and nuclear accumulation of NF-κB, which can be activated by a wide variety of stimuli, have been extensively studied in the past two decades. After gaining access to the nucleus, NF-κB must be actively regulated to execute its fundamental function as a transcription factor. Recent studies have highlighted the importance of nuclear signaling in the regulation of NF-κB transcriptional activity. A non-Rel subunit of NF-κB, ribosomal protein S3 (RPS3), and numerous other nuclear regulators of NF-κB including Akirin, Nurr1, SIRT6, and others, have recently been identified, unveiling novel and exciting layers of regulatory specificity for NF-κB in the nucleus. Further insights into the nuclear events that govern NF-κB function will deepen our understanding of the elegant control of its transcriptional activity and better inform the potential rational design of therapeutics for NF-κB-associated diseases.
Keywords: RPS3, immunity, DNA binding
Introduction
Nuclear factor-kappa B (NF-κB) was first identified over 20 years ago as a DNA-binding complex governing transcription at the immunoglobulin light chain gene intronic enhancer 1, 2. Soon thereafter, NF-κB emerged as a major regulator not only of inflammatory responses, but also broader cellular activities, including cell proliferation, cell death, and so forth 3-9. This pleiotropic transcription factor can be activated by a diverse spectrum of modulating stimuli, linking NF-κB with an ever-increasing array of genetic targets 3, 10.
The best-known subunits of mammalian NF-κB are five proteins in the Rel domain family: RelA (p65), RelB, c-Rel, p50, and p52, which can homo- and hetero-dimerize through their conserved prototypical Rel homology domain (RHD) 4, 11, 12. NF-κB complexes have long been thought to function dimerically; but functional and biochemical information belied this simple conceptualization. A recent study demonstrated that another essential subunit of the native NF-κB complex, ribosomal protein S3 (RPS3), cooperates with Rel dimers to achieve full binding and transcriptional activity 13. As an integral component, RPS3 plays a critical role in determining the DNA binding affinity and regulatory specificity of NF-κB. This finding revealed new insights into the puzzling but marked differences in the molecular size and DNA binding affinity between native NF-κB complexes derived from nuclear extracts, and those reconstituted from purified Rel proteins 14, 15. Indeed, the molecular machine known as NF-κB consists of both Rel and non-Rel subunits that actually comprise multiple protein complexes with different target gene activation specificities, masquerading as a single NF-κB DNA binding complex in the nucleus 16.
The signal pathways governing the nuclear translocation of NF-κB from the cytoplasmic inhibitory complex initiated by diverse stimuli have been extensively studied in the past two decades 3-5, 17-20. Typically, NF-κB complexes are retained in a latent cytoplasmic form in unstimulated cells through binding to a member of the inhibitor of NF-κB (IκB) protein family, which masks the nuclear localization sequence of associated Rel proteins. Activation of the IκB kinase (IKK) complex induces the phosphorylation and subsequent degradation of IκBs 21. NF-κB is thus liberated by this cytoplasmic “switch” and subsequently translocates into the nucleus, where it acts as a transcription factor by binding to regulatory DNA sequences known as κB sites 22. However, there are significant differences in the precise sequence of different κB sites. Moreover, the presence of Rel family homo- or heterodimers at κB sites appears insufficient for full induction of target genes 13. In particular, it remains unclear as to how a small subset of relevant κB sites can be selectively recognized by NF-κB over the large excess of potential binding sites, given a specific stimulus. Ever-increasing evidence has demonstrated that both the recruitment of NF-κB within the nucleus to target genes and the ensuing transcriptional events are actively regulated 23, 24. Recently, a growing list of novel molecules has been identified to modulate NF-κB transcriptional activity in the nucleus 25, 26. These observations suggest the nuclear “signaling” of NF-κB is more abstruse than initially considered, adding another level of complexity to the complicated but elegantly regulated NF-κB signaling pathway. This review will highlight our current knowledge of the nuclear NF-κB regulation, emphasizing new insights gained through recent studies and focusing on NF-κB from its gain of access to the nucleus through the termination of expression of its target genes.
Non-Rel integral subunit of NF-κB
How NF-κB selectively recognizes certain κB sites to achieve specific gene transcription is an area of intense interest, but remains incompletely understood. It has long been thought that the usage of certain Rel dimers could be dictated by the variability of κB sequences at specific promoters, based on the principle that each κB site variant could preferentially recruit one type of Rel dimer over others 23, 24. However, the κB site sequence in target genes and the specific Rel dimer requirement are not consistently correlated 27. Various κB sequences do impart specific configurations for NF-κB binding, made evident from observations that a single nucleotide change in κB sites affected the formation of productive interactions between Rel subunits and co-activators 28. The presence of other proteins, beyond Rel subunits, in the native NF-κB complex was implied by the largely ignored contradiction in the size of native NF-κB from nuclear extracts (> 200 kD) versus NF-κB reconstituted from purified p50 and p65 proteins (115 kD) 14. Support for this hypothesis also comes from other studies. For example, native NF-κB complexes derived from cells exhibited a 100-fold higher affinity to the canonical Ig κB DNA binding site than the reconstituted p65-p50 heterodimers from recombinant proteins 15. Also, several genome-wide analyses have revealed a large number of NF-κB binding motifs beyond recognizable κB sequences 29-31. These results strongly hint that other integral non-Rel components in the NF-κB complex could not only regulate NF–κB activity, but also participate in the strength and specificity of DNA binding as an essential component.
One such non-Rel subunit has been identified as RPS3 in certain NF-κB DNA binding complexes, where it is essential for the recruitment of NF-κB p65 to selected κB sites 13. RPS3 was revealed to interact with p65 in a proteomic screen, and demonstrated to be instrumental for NF-κB transactivation. The featured heterogeneous nuclear protein K (hnRNP K) homology (KH) domain of RPS3, a structural motif that binds single-stranded RNA and DNA with some sequence specificity 32, is essential for its association with p65 13. Several lines of evidence demonstrate that RPS3 functions as an integral part of NF-κB: it associates with the p65-p50-IκB α cytoplasmic sequestration complex in resting cells; upon stimulation, RPS3 specifically translocates to the nucleus and binds to κB sites in a large number of NF-κB-dependent genes in response to diverse stimuli; RPS3 exists in endogenous NF-κB DNA binding complexes, as indicated by the ability of RPS3-specific antibodies to supershift or diminish p65-containing DNA complexes in electrophoretic mobility shift assays (EMSAs); and a significant correlation between RPS3- and p65-dependence in transcribing a subset of NF-κB genes induced by T cell receptor ligation 13. Therefore, RPS3 is an integral subunit of NF-κB rather than a co-activator, given that RPS3 itself possesses little, if any, intrinsic transcriptional activating ability 13. More strikingly, RPS3 protein exerts a dramatic synergistic effect on the DNA binding activity of both p65-p65 and p50-p65, but not p50-p50 complexes in EMSAs 13. As a DNA-binding component, RPS3 dramatically stabilizes NF-κB association with certain cognate κB sites, which provides an important clue to the unexplained extremely high affinity of native NF-κB complexes for κB DNA 15. By contrast, although over 100 proteins have been reported to interact with p65, no other Rel-interacting protein has been clearly demonstrated to be integrated into endogenous NF-κB complexes, or to substantially augment the DNA binding activity of NF-κB as assessed in vitro by utilizing recombinant proteins in EMSAs 16. Moreover, the essential role of RPS3 in regulating NF-κB transcription has been highlighted in certain key physiological processes, including induction of immunoglobulin κ light chain gene expression in B cells, and cell proliferation and cytokine secretion in T cells 13. More recently, more nuclear RPS3 was observed in pro-B, pre-B and immature B cells from receptor-editing model mice (B1-8low α-HEL-κ) compared with the same population of cells from mice with innocuous B cell receptors (B1-8 α-HEL-κ), suggesting RPS3 contributes to NF-κB regulation during B cell receptor editing 33.
The identification of RPS3 underscores the inherent complexity of NF-κB itself. The DNA binding capability of NF-κB is actually conferred not only by the Rel subunits, but also the integral non-Rel subunits, e.g. RPS3. Since RPS3 is only required for selected κB-regulated genes to be activated under certain conditions and preferentially directs NF-κB complex binding to κB sites with certain sequence specificity, we refer to RPS3 as a “specifier” subunit of NF-κB 16. The complexity of NF-κB-regulated transcriptomes suggests that there are actually multiple molecular complexes containing RPS3-like “specifier” subunits with different gene activation specificities that all masquerade as a single NF-κB complex in the nucleus 16. This suggests a new model of NF-κB regulation in which DNA binding activity could be regulated within NF-κB complexes through synergistic interaction between Rel and non-Rel subunits. Whether the RPS3 subunit is essential for other NF-κB activation-inducing stimuli, and whether its targeting of NF-κB complexes to specific κB sites is cell-context specific, deserves further investigation. Undoubtedly, the identification of additional “specifier” subunits of NF-κB will shed more light on the regulatory specificity of NF-κB in the nucleus, particularly the selective recognition and transcription of NF-κB target genes.
Membrane proteins
It is striking that some traditional signaling molecules that normally work close to the plasma membrane have also been shown to expend novel function in regulating NF-κB transcriptional activity in the nucleus. For example, Akt1, a serine/threonine protein kinase that functions immediately downstream of plasma membrane-tethered phosphoinositide 3-kinase (PI-3K), was also observed to translocate into the nucleus and complex with p65 and K-Ras to enhance NF-κB transcriptional activity 34. More surprisingly, similar nuclear functions have been ascribed to some integral membrane proteins, such as the tumor necrosis factor (TNF) receptors (TNFR). Although most studies of TNFRs have focused on their signaling capacity from the plasma membrane and cytoplasm upon recognition of cognate TNF family ligands 35, recent reports demonstrate several TNFR members translocate into the nucleus to interact with distinct NF-κB Rel subunits and regulate the transcription of NF-κB target genes (Table 1).
Table 1. Recently identified participants of the nuclear signaling of NF-κB regulation.
| Nuclear Participants | Functions in NF-κB transactivation | References |
|---|---|---|
| Integral NF-κB Subunit | ||
| RPS3 | Interacts with p65, facilitates NF-κB binding to DNA and preferentially directs NF-κB complex to certain κB sites; enhances NF-κB transactivation | 13 |
| Membrane Proteins and Signal Molecules | ||
| Akt1 | Interacts with p65, and forms a complex with p65 and Ki-Ras in the nucleus; enhances NF-κB transactivation | 34 |
| CD40 | Interacts with c-Rel and is recruited to the promoters of some NF-κB target genes; enhances NF-κB transactivation | 37 |
| BAFF-R | Interacts with c-Rel and is recruited to the promoters of some NF-κB target genes; bridges and enhances the phosphorylation of histone H3 by IKKβ; enhances NF-κB transactivation | 38 |
| Strictly Nuclear Proteins | ||
| Akirin | A highly conserved nuclear factor required for innate immune responses | 41 |
| AKIP1 | Interacts with p65 and retains p65 in the nucleus; recruits PKAc to phsphorylate p65 and enhances the assembly with co-activator CBP/p300 and the binding of NF-κB to target DNA | 43 |
| CHFR | Interacts with p65 and attenuates p65 transcriptional activity | 44 |
| Co-regulators | ||
| AEG-1 | Interacts with p65 and bridges the binding of NF-κB, CBP and basal transcription machinery; functions as a co-activator | 45 |
| ING4 | Interacts with p65 and is recruited to the κB promoters simultaneously with NF-κB; functions as a co-repressor | 46 |
| Histone Modifiers | ||
| SIRT6 | Deacetylates the histone H3 lysine 9 on the κB promoters to attenuate the p65 promoter occupancy; attenuates NF-κB transactivation | 48 |
| EGCG | A novel histone acetyltransferase inhibitor; attenuates the recruitment of p300 and inhibites the acetylation of p65; attenuates NF-κB transactivation | 67 |
| Transcription Factors | ||
| ARNT | Is recruited by CD30 in the endosomes; complexes with RelB and enhances the binding of RelB to κB sites to compete with p65; attenuates NF-κB transactivation | 39 |
| Nurr1 | Is recruited by Ser 468-phosphorylated p65 to dock at the NF-κB complex on target promoters; subsequently recruits the CoREST corepressor complex, resulting in clearance of p65; attenuates NF-κB transactivation | 51 |
| NRF | Interacts with p65 and binds to the IL-8 promoter; required for NF-κB transactivation | 66 |
| Terminators | ||
| p28GANK | Interacts with p65 and exports p65 to the cytoplasm; attenuates NF-κB transactivation | 55 |
| PDLIM2 | Interacts with p65 and translocates nuclear p65 for polyubiquitination and degradation; attenuates NF-κB transactivation | 58 |
| COMMD1 | Interacts with p65 and accelerates the ubiquitination and degradation of p65; attenuates NF-κB transactivation | 59 |
| GCN5 | Interacts with p65, associates with COMMD1 ubiquitin ligase complex, and facilitates proteasomal elimination of nuclear p65; attenuates NF-κB transactivation | 62 |
| MuHV-4 ORF73 | Interacts with p65, poly-ubiquitinates p65 for its subsequent proteasomal degradation of nuclear p65; attenuates NF-κB transactivation | 64 |
For example, the TNFR family member CD40 (TNFRSF5) has been extensively studied as a cell surface receptor, however, it was recently shown to be present not only in the plasma membrane and cytoplasm, but also in the nucleus of human normal and lymphoma cell lines 36. Nuclear CD40 interacts with c-Rel, but not p65, subunits of NF-κB, and complexes with c-Rel on the promoters of some NF-κB target genes, thereby enhancing c-Rel-mediated transactivation 37. Thus, the cooperation between nuclear CD40 and c-Rel appears to be important in regulating genes involved in lymphoma cell proliferation and survival 37.
Similarly, BAFF-R protein (TNFRSF13C) contains a nuclear localization signal sequence and can be present in the nuclei of both normal and neoplastic B cells 38. Nuclear BAFF-R bridges and enhances the phosphorylation of histone H3 by IKKβ, and also associates with c-Rel, for subsequent recruitment to NF-κB target gene promoters for initiation of transcription. Through its novel nuclear function in remodeling chromatin structure and modulating NF-κB transcriptional activity, BAFF-R promotes the survival and proliferation of both normal B cells and B-cell lymphoma. Interestingly, the c-Rel subunit benefits from both CD40 and BAFF-R association to facilitate the transcription of NF-κB target genes; it is possible that those genes that employ both TNFR partners may encompass the normal physiology of B-cell survival and growth, whereas distinct genes specified by CD40 or BAFF-R could be involved in the pathophysiology of diverse B-cell malignancies and diseases.
Finally, CD30, another TNFR family member (TNFRSF8), regulates NF-κB transcription in the nucleus through a novel mechanism. Although it does not translocate to the nucleus itself, CD30 instead recruits the aryl hydrocarbon receptor nuclear translocator (ARNT) after internalization within endosomes 39. As the CD30 signal progresses, ARNT translocates to the nucleus and potently and specifically associates with RelB to enhance binding of RelB to κB sites. This ARNT-RelB complex represses the transcription of NF-κB target genes by competing with p65 for binding to certain κB sites, since ARNT silencing impaired RelB but potentiated RelA recruitment to κB sites 39. In contrast to this repressive function for RelB in competing with p65 after CD30-stimulation, a previous study reported p65, after phosphorylation of serine 276, can repress the DNA binding capacity and transcriptional activity of RelB. This repression of RelB function subsequently induces selective p65-dependent gene activation following TNFR-mediated NF-κB activation 40. These reciprocal regulatory mechanisms for selective NF-κB target gene transcription in response to two different TNFRs could be cell context-specific, or redirected by ARNT during xenobiotic and hypoxic responses.
Strictly nuclear proteins
The sophisticated regulation of NF-κB signaling is has been reinforced by the identification of proteins that localize exclusively in the nucleus to participate in NF-κB-mediated gene transcription once NF-κB gains access to the nucleus. An excellent example comes from a recent study of the highly conserved Akirin protein family 41. Albeit no functions or recognizable domains were previously reported, the nucleus-restricted Akirins were revealed to play critical roles in regulating the transcription of NF-κB-dependent genes 41. Akirin was identified in an unbiased high-throughput RNA interference screen analyzing the innate immune response of Drosophila S2 cells 41. Drosophila Akirin acts in parallel with the NF-κB transcription factor ortholog Relish and is required for defense against Gram-negative bacteria, whereas both the human and mouse genomes contain two homologs of Akirin 41. Although Akirin1 knockout mice have no obvious phenotype, Akirin2 knockout mice are embryonic lethal. Akirin2 appears to execute an essential function downstream in the nuclear regulation of NF-κB, as evidenced by the observation that Akirin2-deficient mouse embryonic fibroblasts are defective in the up-regulation of certain NF-κB target genes induced by Toll-like receptor ligands, tumor necrosis factor and interleukin (IL)-1β 41. Akirins were proposed to bind a still undetermined intermediate protein which in turn interacts with NF-κB. This affects DNA binding or interacts with chromatin or components of the transcriptional machinery, since Akirins apparently do not bind to DNA or NF-κB by themselves 41, 42. Therefore, it is clearly important for future studies to elucidate the precise mechanism by which Akirins modulate NF-κB transcriptional activity in the nucleus. However, the discovery of such a highly conserved nuclear factor required for innate immune responses demonstrates the complexity of NF-κB nuclear regulation and hints that other missing components surely exist but still need to be identified.
Several other nuclear proteins were also recently identified to be involved in NF-κB signaling regulation in the nucleus, such as the protein kinase A-interacting protein 1 (AKIP1) 43 and the CHFR protein, encoded by checkpoint with forkhead and ring finger domains gene 44 (Table 1). AKIP1 was revealed to retain p65 in the nucleus and recruit the catalytic subunit of PKA (PKAc) to phosphorylate p65. Therefore, AKIP1 augments NF-κB transactivation by dramatically enhancing the assembly of p65 with the CBP/p300 co-activator and subsequent binding of NF-κB to target DNA. In contrast, CHFR was shown to physically interact with p65 and antagonize the transcriptional activity of p65 44. Although the precise mechanism by which CHFR attenuates p65 transcriptional activity still needs further definition, a functional relationship between CHFR-regulated IL-8 expression and inflammation/angiogenesis in human cancer cells was discerned in a xenograft model.
Co-regulators and chromatin modulation
The recognition and binding of NF-κB complexes to target DNA is not sufficient for full gene transcription. A vast array of co-regulators, including co-activators and co-repressors, have been implicated to play a crucial role in nuclear NF-κB signaling, most of which physically associate with Rel subunits. Recent work focused on the astrocyte elevated gene-1 (AEG-1) 45 and the inhibitor of growth 4 (ING4) 46 in malignant glioma has further expanded this extensive list. AEG-1 is not found within the nucleus; however, upon TNFα treatment, it translocates into the nucleus and associates with p65. AEG-1 was also observed to interact with CBP and complex in the promoter of IL-8 gene, thus functioning as a co-activator to bridge NF-κB, CBP and basal transcription machinery 45. In contrast, ING4 was recently identified as a co-repressor of NF-κB transactivation. It is recruited to the κB promoters simultaneously with NF-κB, which correlates with reductions in p65 phosphorylation, p300 expression, and the levels of acetylated histones, but enhances the levels of HDAC-1 46. Thus, ING4 negatively regulates the activity of the NF-κB bound to target gene promoters through its specific interaction with p65 46.
Chromatin structure and epigenetic state are also extremely important for determining proper expression of NF-κB-dependent genes 47. This concept has been reinforced by recent work on SIRT6 of the sirtuin (SIRT) family proteins 48. Besides their critical role in life span regulation, SIRT proteins have also been implicated in modulating NF-κB dependent transcription through multiple mechanisms. SIRT1 employs the strategy of post-translational modulation to suppress the NF-κB p65 transcriptional activity: it physically associates and directly deacetylates p65 at lysine 310 49. In contrast, SIRT6 attenuates NF-κB signaling by functioning at the chromatin level 48. Although it directly binds to p65 like SIRT1, SIRT6 deacetylates the histone H3 lysine 9 (K3K9) on the promoters of NF-κB target genes to decrease promoter occupancy by p65, rather than directly modulating p65 48. The possibility remains that SIRT6 could also be recruited to certain promoters, which are vital for life span regulation, beyond that of NF-κB target genes. However, the contribution of NF-κB signaling to premature and normal aging is underscored by the observation that the haploinsufficiency of p65 rescues the early lethality and degenerative syndrome of Sirt6-deficient mice 48. The interaction of SIRT6 and p65 may provide a therapeutic target for aging-associated diseases; however, the key NF-κB target genes associated with aging remain undefined, and how signal termination by SIRT6 balances the dynamic pattern and target selection of important NF-κB genes at a genome scale will need to be addressed.
Crosstalk between transcription factors
One important strategy to elegantly control and efficiently execute NF-κB transcriptional activity is via crosstalk with various other transcription factors, with these formed complexes resulting in either the synergistic activation or antagonistic inhibition of NF-κB target gene expression. An ever-expanding list of transcription factors has been reported to cooperate with NF-κB for fine-tuning of specific gene transcription. For example, the formation of an NF-κB/activator protein 1 (AP-1) complex leads to activation of both transcription factors, whereas the interaction of NF-κB and the CCAAT/enhancer binding protein β (C/EBPβ) leads to the enhancement of C/EBPβ activity but repression of NF-κB 50. The recently identified association of NF-κB to Nurr1, a transcription factor belonging to the NR4A subfamily of nuclear receptors, is a clear example of co-regulation of NF-κB activity by coupling to other transcription factors 51.
Nurr1 is critical for the expression of the gene encoding tyrosine hydroxylase, a key enzyme in dopamine synthesis, and mutations in Nurr1 are linked to familial Parkinson's disease. However, Nurr1 has also been implicated in an unexpected feedback loop for the expression of proinflammatory neurotoxic mediators in both mouse microglia and astrocytes 51. Nuclear glycogen synthase kinase 3 beta (GSK3β) phosphorylates p65 in the active NF-κB complex at serine 468 which enables the phosphorylated p65 recruits Nurr1 to dock at the NF-κB complex on target inflammatory gene promoters. Subsequently, Nurr1 recruits the CoREST corepressor complex, resulting in clearance of p65 and attenuation of inflammatory gene transcription by NF-κB 51. This study also highlights the so-called “transrepression” mechanism shared by other nuclear receptors in suppressing inflammatory gene expression 52. These nuclear receptors inhibit transcription by preventing the inflammatory signal-specific removal of the co-repressor complexes containing SMRT and NcoR from promoters, rather than by direct binding to the regulatory elements in target promoters. Like other NR4A subfamily members Nur77 and Nor1, Nurr1 itself can also augment the expression of target genes by directly binding to regulatory elements in their promoters. Thus it will be interesting to dissect the relative contributions of transrepression and transactivation in Nurr1-depedent regulation of inflammation, which could be particularly important for validating the Nurr1 nuclear receptor as a potential therapeutic target.
Termination of NF-κB transactivation
Appropriate termination of NF-κB transcriptional activity is required to return NF-κB to latent complexes sequestered in the cytoplasm, thereby maintaining the cell's responsiveness to subsequent stimuli. Thus far, studies have demonstrated that this procedure is elegantly controlled by at least two mechanisms: reshuttling of NF-κB to the cytoplasm by the newly synthesized IκBs, thus creating a negative-feedback loop 53; and termination of NF-κB activity by the ubiquitination-dependent degradation of NF-κB subunits in the nucleus 54.
The IκB negative-feedback loop constitutes the most prevalent and best understood mode of NF-κB regulation. Activation of NF-κB requires the degradation of the IκBs, allowing free NF-κB complexes to translocate to the nucleus. However, transcription of the NFKBIA gene that encodes IκBα is induced immediately after NF-κB enters the nucleus and binds to its promoter. This generates newly synthesized IκBα that enters the nucleus, dissociates NF-κB from DNA, and escorts it back to the cytoplasm. Such a negative-feedback regulation was also revealed for IκBβ and IκBε, albeit with different kinetics of degradation and resynthesis 53. An oncoprotein known as p28GANK was recently discovered to suppress NF-κB by retaining it in the cytoplasm via nuclear export of p65. This process was dependent upon complete ankyrin repeats in p28GANK that are prototypical features among IκB family proteins 55. Importantly, the cycle of induction and suppression of IκB causes antipodal oscillations of nuclear NF-κB. The oscillatory pattern induced by this negative-feedback loop may play a functional role in the expression pattern of certain NF-κB target genes 56.
Additionally, other nuclear signaling events regulate the termination of NF-κB response even in the absence of IκBα 57. The polyubiquitination and degradation of NF-κB subunits in the nucleus is apparently involved in this procedure, which suggests a critical contribution from specific E3 ubiquitin ligases in controlling NF-κB target gene expression 54. Currently, two ubiquitin ligase complexes have been reported to be responsible for the degradation of the NF-κB p65 subunit in the nucleus. PDLIM2 is a ubiquitin ligase that controls the fate of nuclear p65 58. It transfers nuclear p65 to the proteasome-enriched promyelocytic leukemia nuclear bodies where the polyubiquitinated p65 is rapidly degraded, thus terminating the NF-κB activity 58. The other is the “copper metabolism gene MURR1 domain 1” (COMMD1), a ubiquitously expressed NF-κB inhibitor 59. It plays a nonenzymatic role in the EC2S multi-subunit E3 multimeric ligase complex, in which it increases the association of p65 with the substrate-binding protein, suppressors of cytokine signaling 1 (SOCS-1), thus accelerating the ubiquitination and degradation of p65 60, 61. More recently, Mao and colleagues reported that a histone acetyltransferase GCN5 associates with COMMD1 ligase complex, promotes p65 ubiquitination, and represses κB-dependent transcription 62. Moreover, GCN5 binds more avidly to p65 after phosphorylation on serine 468 by the IKK complex, which suggests that specific phosphorylation of p65 contributes to the efficiency of COMMD-1-dependent ubiquitination and the selective disassembly of NF-κB 62, 63. The physiological importance of proteasomal degradation of NF-κB was underscored by a novel viral mechanism to suppress NF-κB activity 64. The open reading frame 73 (ORF 73) protein encoded by the murid herpesvirus-4 (MuHV-4) inhibits host NF-κB transcriptional activity through poly-ubiquitination and subsequent proteasome-dependent nuclear degradation of p65 64. Thus ORF73 is capable of contravening the NF-κB-dependent host immune responses. Collectively, the specific E3 ubiquitin ligases that target p65 clearly contribute to negative control of gene transcription, especially serving to prevent excessive induction of genes involved in the inflammatory response.
Specific inhibition of NF-κB by targeting nuclear signaling
Since NF-κB is present in almost all nucleated cells and is involved in many different cellular pathways and functions, application of these agents to inhibit NF-κB may involve side effects 26. On the other hand, inhibition of NF-κB signaling by targeting downstream nuclear regulators of NF-κB signaling pathways may specifically abolish certain functions of NF-κB without globally abolishing its activity. This concept was illuminated by a recent study showing selective inhibition of interleukin-8 (IL-8) by peptide-mediated disruption of an essential nuclear regulator of NF-κB 65. A constitutively expressed nuclear transcription factor, NF-κB repressing factor (NRF), was shown to interact with p65 and bind to the IL-8 promoter in IL-1-stimulated cells, which is crucial for the transcriptional activation of IL-8 gene 66. To search for a specific inhibitor of IL-8 expression, Bartels and colleagues conducted tandem affinity purification targeting this downstream NRF-p65 interaction, and identified a synthetic peptide corresponding to a.a. 223-238 of NRF that could interfere with the binding of endogenous p65 to NRF 65. Introduction of this inhibitory peptide into the nucleus specifically disturbs the NRF-p65 interaction, thus significantly decreasing endogenous IL-8 gene transcription in response to multiple stimuli 65. Obviously, the adverse side effects and therapeutic benefits of this inhibitory peptide in combating inflammatory and infectious diseases needs further exploration, especially in animal models; however, it could serve to facilitate drug design based on a novel strategy to specifically inhibit NF-κB transcription.
Other than synthetic peptides, natural compounds have also been screened for specific blockers of NF-κB transcriptional activity by targeting events in the nucleus. For example, recent work by Choi and colleagues 67 focused on the critical role of hyperacetylation of p65 for NF-κB activation. The screening study was conducted with natural compounds to explore those with histone acetyltransferase inhibitor (HATi) activity. Through this method, Epigallocatechin-3-gallate (EGCG), the major polyphenol found in green tea, was identified as a novel HATi with broad specificity for the majority of HAT enzymes, but not epigenetic enzymes 67. EGCG attenuated the recruitment of p300 with an increased binding of the histone deacetylase 3, blocking the acetylation of p65 and NF-κB target gene expression in response to myriad stimuli. Furthermore, EGCG treatment completely blocks Epstein-Barr Virus (EBV) infection-induced cytokine expression and subsequent B lymphocyte transformation, both of which require NF-κB transactivation 67.
Concluding remarks
NF-κB has represented a paradigm for signal transduction and gene regulation for over 20 years because of its extensive involvement in key cellular processes, particularly immune and inflammatory responses. An increasing body of literature illustrates that specific signaling in the nucleus is as equally important as cytoplasmic events for controlling the transcriptional activity of NF-κB. The mechanisms of these effects involve the synergistic interaction of Rel and non-Rel subunits within the NF-κB complex, participation of strictly nuclear proteins, cross-talk between other transcription factors, remodeling of chromatin structures, nuclear turnover of NF-κB, and other effects (Figure 1). Although the nuclear components of NF-κB regulation have been intensely studied, numerous unanswered questions remain to be addressed. These include the precise mechanism of RPS3-mediated specific NF-κB transcription; the characterization of unknown additional “specifier” subunits of NF-κB and cofactors restricted to the nucleus; the precise mechanism of chromatin modeling; and the identification of specific E3 ubiquitin ligases that target NF-κB for nuclear degradation.
Figure 1. The nuclear regulation of NF-κB.
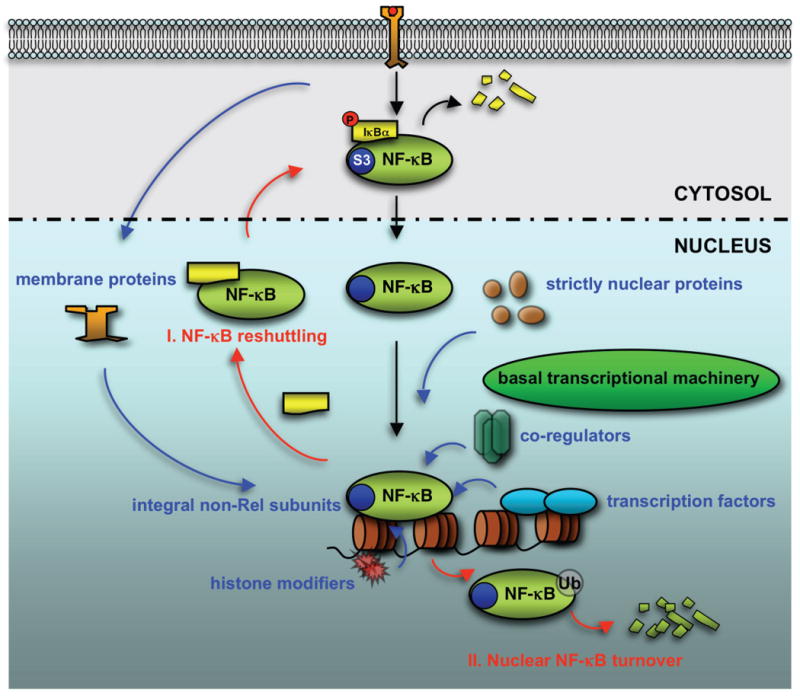
After it gains access to the nucleus, NF-κB transcriptional activity is elegantly controlled by a number of nuclear regulators, as illustrated in blue, including non-Rel subunits such as RPS3 (S3) within the NF-κB complex, proteins restricted to the nucleus or translocated from the plasma membrane, other transcription factors, and chromatin modifiers. Two distinct pathways to terminate the NF-κB transactivation, as illustrated in red, involve (I) IκB-mediated restitution of NF-κB to the cytoplasm and (II) ubiquitination-dependent nuclear degradation.
Besides its extensive role in regulating the immune response and cellular metabolism, a growing number of studies demonstrate that aberrant NF-κB signaling is critical for the development of various diseases, especially inflammatory diseases and tumors. This makes NF-κB signaling pathway even more tantalizing as a focus for research on pharmacological interventions. Thus far, numerous therapeutic agents that inhibit either activation or function of NF-κB have been identified, including a variety of natural and synthetic molecules 68. Targeting specific downstream nuclear regulators of NF-κB signal pathways could represent an innovative approach to block NF-κB activation in specific contexts and avoid unwanted global effects. Therefore, a more complete elucidation of the mechanisms that dictate nuclear regulatory specificity of NF-κB will not only revise or add to our fundamental knowledge of gene regulation, but also reveal better and more specific targets for pharmacological interventions.
Acknowledgments
We are grateful to Andrew Snow, Amanda Weaver, and Michael Bern for critically reading the manuscript. We apologize to those who also made important contributions to the issues discussed and could not be cited due to space limit. The research in the authors' laboratory is supported by the Intramural Research Program of the NIAID, NIH. F.W. is a recipient of an NIH grant 1K99CA137171.
References
- 1.Sen R, Baltimore D. Inducibility of kappa immunoglobulin enhancer-binding protein NF-kappa B by a posttranslational mechanism. Cell. 1986;47:921–928. doi: 10.1016/0092-8674(86)90807-x. [DOI] [PubMed] [Google Scholar]
- 2.Lenardo M, Pierce JW, Baltimore D. Protein-binding sites in Ig gene enhancers determine transcriptional activity and inducibility. Science. 1987;236:1573–1577. doi: 10.1126/science.3109035. [DOI] [PubMed] [Google Scholar]
- 3.Lenardo MJ, Baltimore D. NF-kappa B: a pleiotropic mediator of inducible and tissue-specific gene control. Cell. 1989;58:227–229. doi: 10.1016/0092-8674(89)90833-7. [DOI] [PubMed] [Google Scholar]
- 4.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 5.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 6.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 7.Kang SM, Tran AC, Grilli M, Lenardo MJ. NF-kappa B subunit regulation in nontransformed CD4+ T lymphocytes. Science. 1992;256:1452–1456. doi: 10.1126/science.1604322. [DOI] [PubMed] [Google Scholar]
- 8.Su H, Bidere N, Zheng L, et al. Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science. 2005;307:1465–1468. doi: 10.1126/science.1104765. [DOI] [PubMed] [Google Scholar]
- 9.Bidere N, Ngo VN, Lee J, et al. Casein kinase 1alpha governs antigen-receptor-induced NF-kappaB activation and human lymphoma cell survival. Nature. 2009;458:92–96. doi: 10.1038/nature07613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grilli M, Chiu JJ, Lenardo MJ. NF-kappa B and Rel: participants in a multiform transcriptional regulatory system. Int Rev Cytol. 1993;143:1–62. doi: 10.1016/s0074-7696(08)61873-2. [DOI] [PubMed] [Google Scholar]
- 11.Chen LF, Greene WC. Shaping the nuclear action of NF-kappaB. Nat Rev Mol Cell Biol. 2004;5:392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- 12.Rothwarf DM, Karin M. The NF-kappaB activation pathway: a paradigm in information transfer from membrane to nucleus. Sci STKE. 1999;1999:RE1. doi: 10.1126/stke.1999.5.re1. [DOI] [PubMed] [Google Scholar]
- 13.Wan F, Anderson DE, Barnitz RA, et al. Ribosomal protein S3: a KH domain subunit in NF-kappaB complexes that mediates selective gene regulation. Cell. 2007;131:927–939. doi: 10.1016/j.cell.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 14.Urban MB, Schreck R, Baeuerle PA. NF-kappaB contacts DNA by a heterodimer of the p50 and p65 subunit. EMBO J. 1991;10:1817–1825. doi: 10.1002/j.1460-2075.1991.tb07707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phelps CB, Sengchanthalangsy LL, Malek S, Ghosh G. Mechanism of kappaB DNA binding by Rel/NF-kappaB dimers. J Biol Chem. 2000;275:24392–24399. doi: 10.1074/jbc.M003784200. [DOI] [PubMed] [Google Scholar]
- 16.Wan F, Lenardo MJ. Specification of DNA binding activity of NF-kappaB proteins. Cold Spring Harb Perspect Biol. 2009 doi: 10.1101/cshperspect.a000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lenardo MJ, Fan CM, Maniatis T, Baltimore D. The involvement of NF-kappaB in beta-interferon gene regulation reveals its role as widely inducible mediator of signal transduction. Cell. 1989;57:287–294. doi: 10.1016/0092-8674(89)90966-5. [DOI] [PubMed] [Google Scholar]
- 18.Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Courtois G, Israel A. NF-kappaB defects in humans: the NEMO/incontinentia pigmenti connection. Sci STKE. 2000;2000:PE1. doi: 10.1126/stke.2000.58.pe1. [DOI] [PubMed] [Google Scholar]
- 20.Scheidereit C. IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene. 2006;25:6685–6705. doi: 10.1038/sj.onc.1209934. [DOI] [PubMed] [Google Scholar]
- 21.Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- 22.Chen FE, Huang DB, Chen YQ, Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA. Nature. 1998;391:410–413. doi: 10.1038/34956. [DOI] [PubMed] [Google Scholar]
- 23.Natoli G, Saccani S, Bosisio D, Marazzi I. Interactions of NF-kappaB with chromatin: the art of being at the right place at the right time. Nat Immunol. 2005;6:439–445. doi: 10.1038/ni1196. [DOI] [PubMed] [Google Scholar]
- 24.Hoffmann A, Natoli G, Ghosh G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene. 2006;25:6706–6716. doi: 10.1038/sj.onc.1209933. [DOI] [PubMed] [Google Scholar]
- 25.Sun SC, Ley SC. New insights into NF-kappaB regulation and function. Trends Immunol. 2008;29:469–478. doi: 10.1016/j.it.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mankan AK, Lawless MW, Gray SG, Kelleher D, McManus R. NF-kappaB regulation: the nuclear response. J Cell Mol Med. 2009;13:631–643. doi: 10.1111/j.1582-4934.2009.00632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffmann A, Leung TH, Baltimore D. Genetic analysis of NF-kappaB/Rel transcription factors defines functional specificities. EMBO J. 2003;22:5530–5539. doi: 10.1093/emboj/cdg534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leung TH, Hoffmann A, Baltimore D. One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell. 2004;118:453–464. doi: 10.1016/j.cell.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 29.Martone R, Euskirchen G, Bertone P, et al. Distribution of NF-kappaB-binding sites across human chromosome 22. P Nat Acad Sci USA. 2003;100:12247–12252. doi: 10.1073/pnas.2135255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schreiber J, Jenner RG, Murray HL, et al. Coordinated binding of NF-kappaB family members in the response of human cells to lipopolysaccharide. P Nat Acad Sci USA. 2006;103:5899–5904. doi: 10.1073/pnas.0510996103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lim CA, Yao F, Wong JJ, et al. Genome-wide mapping of RELA(p65) binding identifies E2F1 as a transcriptional activator recruited by NF-kappaB upon TLR4 activation. Mol Cell. 2007;27:622–635. doi: 10.1016/j.molcel.2007.06.038. [DOI] [PubMed] [Google Scholar]
- 32.Siomi H, Matunis MJ, Michael WM, Dreyfuss G. The pre-mRNA binding K protein contains a novel evolutionarily conserved motif. Nucleic Acids Res. 1993;21:1193–1198. doi: 10.1093/nar/21.5.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cadera EJ, Wan F, Amin RH, et al. NF-kappaB activity marks cells engaged in receptor editing. J Exp Med. 2009;206:1803–1816. doi: 10.1084/jem.20082815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwon O, Kim KA, He L, et al. Complex formation of p65/RelA with nuclear Akt1 for enhanced transcriptional activation of NF-kappaB. Biochem Bioph Res Co. 2008;365:771–776. doi: 10.1016/j.bbrc.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 35.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 36.Lin-Lee YC, Pham LV, Tamayo AT, et al. Nuclear localization in the biology of the CD40 receptor in normal and neoplastic human B lymphocytes. J Biol Chem. 2006;281:18878–18887. doi: 10.1074/jbc.M513315200. [DOI] [PubMed] [Google Scholar]
- 37.Zhou HJ, Pham LV, Tamayo AT, et al. Nuclear CD40 interacts with c-Rel and enhances proliferation in aggressive B-cell lymphoma. Blood. 2007;110:2121–2127. doi: 10.1182/blood-2007-02-073080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fu L, Lin-Lee YC, Pham LV, et al. BAFF-R promotes cell proliferation and survival through interaction with IKKbeta and NF-kappaB/c-Rel in the nucleus of normal and neoplastic B-lymphoid cells. Blood. 2009;113:4627–4636. doi: 10.1182/blood-2008-10-183467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wright CW, Duckett CS. The aryl hydrocarbon nuclear translocator alters CD30-mediated NF-kappaB-dependent transcription. Science. 2009;323:251–255. doi: 10.1126/science.1162818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacque E, Tchenio T, Piton G, Romeo PH, Baud V. RelA repression of RelB activity induces selective gene activation downstream of TNF receptors. P Nat Acad Sci USA. 2005;102:14635–14640. doi: 10.1073/pnas.0507342102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goto A, Matsushita K, Gesellchen V, et al. Akirins are highly conserved nuclear proteins required for NF-kappaB-dependent gene expression in drosophila and mice. Nat Immunol. 2008;9:97–104. doi: 10.1038/ni1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beutler B, Moresco EM. Akirins versus infection. Nat Immunol. 2008;9:7–9. doi: 10.1038/ni0108-7. [DOI] [PubMed] [Google Scholar]
- 43.Gao N, Asamitsu K, Hibi Y, Ueno T, Okamoto T. AKIP1 enhances NF-kappaB-dependent gene expression by promoting the nuclear retention and phosphorylation of p65. J Biol Chem. 2008;283:7834–7843. doi: 10.1074/jbc.M710285200. [DOI] [PubMed] [Google Scholar]
- 44.Kashima L, Toyota M, Mita H, et al. CHFR, a potential tumor suppressor, downregulates interleukin-8 through the inhibition of NF-kappaB. Oncogene. 2009;28:2643–2653. doi: 10.1038/onc.2009.123. [DOI] [PubMed] [Google Scholar]
- 45.Sarkar D, Park ES, Emdad L, et al. Molecular basis of Nuclear Factor-kappaB activation by astrocyte elevated gene-1. Cancer Res. 2008;68:1478–1484. doi: 10.1158/0008-5472.CAN-07-6164. [DOI] [PubMed] [Google Scholar]
- 46.Nozell S, Laver T, Moseley D, et al. The ING4 tumor suppressor attenuates NF-kappaB activity at the promoters of target genes. Mol Cell Biol. 2008;28:6632–6645. doi: 10.1128/MCB.00697-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vanden Berghe W, Ndlovu MN, Hoya-Arias R, et al. Keeping up NF-kappaB appearances: epigenetic control of immunity or inflammation-triggered epigenetics. Biochem Pharmacol. 2006;72:1114–1131. doi: 10.1016/j.bcp.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 48.Kawahara TL, Michishita E, Adler AS, et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell. 2009;136:62–74. doi: 10.1016/j.cell.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yeung F, Hoberg JE, Ramsey CS, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zwergal A, Quirling M, Saugel B, et al. C/EBP beta blocks p65 phosphorylation and thereby NF-kappaB-mediated transcription in TNF-tolerant cells. J Immunol. 2006;177:665–672. doi: 10.4049/jimmunol.177.1.665. [DOI] [PubMed] [Google Scholar]
- 51.Saijo K, Winner B, Carson CT, et al. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137:47–59. doi: 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bensinger SJ, Tontonoz P. A Nurr1 pathway for neuroprotection. Cell. 2009;137:26–28. doi: 10.1016/j.cell.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 53.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298:1241–1245. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 54.Natoli G, Chiocca S. Nuclear ubiquitin ligases, NF-kappaB degradation, and the control of inflammation. Sci Signal. 2008;1:pe1. doi: 10.1126/stke.11pe1. [DOI] [PubMed] [Google Scholar]
- 55.Chen Y, Li HH, Fu J, et al. Oncoprotein p28 GANK binds to RelA and retains NF-kappaB in the cytoplasm through nuclear export. Cell Res. 2007;17:1020–1029. doi: 10.1038/cr.2007.99. [DOI] [PubMed] [Google Scholar]
- 56.Ashall L, Horton CA, Nelson DE, et al. Pulsatile stimulation determines timing and specificity of NF-kappaB-dependent transcription. Science. 2009;324:242–246. doi: 10.1126/science.1164860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saccani S, Marazzi I, Beg AA, Natoli G. Degradation of promoter-bound p65/RelA is essential for the prompt termination of the uclear factor kappaB response. J Exp Med. 2004;200:107–113. doi: 10.1084/jem.20040196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tanaka T, Grusby MJ, Kaisho T. PDLIM2-mediated termination of transcription factor NF-kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nat Immunol. 2007;8:584–591. doi: 10.1038/ni1464. [DOI] [PubMed] [Google Scholar]
- 59.Ganesh L, Burstein E, Guha-Niyogi A, et al. The gene product Murr1 restricts HIV-1 replication in resting CD4+ lymphocytes. Nature. 2003;426:853–857. doi: 10.1038/nature02171. [DOI] [PubMed] [Google Scholar]
- 60.Ryo A, Suizu F, Yoshida Y, et al. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell. 2003;12:1413–1426. doi: 10.1016/s1097-2765(03)00490-8. [DOI] [PubMed] [Google Scholar]
- 61.Maine GN, Mao X, Komarck CM, Burstein E. COMMD1 promotes the ubiquitination of NF-kappaB subunits through a cullin-containing ubiquitin ligase. EMBO J. 2007;26:436–447. doi: 10.1038/sj.emboj.7601489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mao X, Gluck N, Li D, et al. GCN5 is a required cofactor for a ubiquitin ligase that targets NF-kappaB/RelA. Gene Dev. 2009;23:849–861. doi: 10.1101/gad.1748409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Geng H, Wittwer T, Dittrich-Breiholz O, Kracht M, Schmitz ML. Phosphorylation of NF-kappaB p65 at Ser468 controls its COMMD1-dependent ubiquitination and target gene-specific proteasomal elimination. EMBO Rep. 2009;10:381–386. doi: 10.1038/embor.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rodrigues L, Filipe J, Seldon MP, et al. Termination of NF-kappaB activity through a gammaherpesvirus protein that assembles an EC5S ubiquitin-ligase. EMBO J. 2009;28:1283–1295. doi: 10.1038/emboj.2009.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bartels M, Schweda AT, Dreikhausen U, et al. Peptide-mediated disruption of NFkappaB/NRF interaction inhibits IL-8 gene activation by IL-1 or Helicobacter pylori. J Immunol. 2007;179:7605–7613. doi: 10.4049/jimmunol.179.11.7605. [DOI] [PubMed] [Google Scholar]
- 66.Nourbakhsh M, Kalble S, Dorrie A, et al. The NF-kappa b repressing factor is involved in basal repression and interleukin (IL)-1-induced activation of IL-8 transcription by binding to a conserved NF-kappa b-flanking sequence element. J Biol Chem. 2001;276:4501–4508. doi: 10.1074/jbc.M007532200. [DOI] [PubMed] [Google Scholar]
- 67.Choi KC, Jung MG, Lee YH, et al. Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Res. 2009;69:583–592. doi: 10.1158/0008-5472.CAN-08-2442. [DOI] [PubMed] [Google Scholar]
- 68.Gilmore TD, Herscovitch M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene. 2006;25:6887–6899. doi: 10.1038/sj.onc.1209982. [DOI] [PubMed] [Google Scholar]