

Abstract
Promoter methylation analysis of genes frequently silenced in breast cancer is a promising indicator of breast cancer risk, as these methylation events are thought to occur long before presentation of disease. The numerous exfoliated epithelial cells present in breast milk may provide the breast epithelial DNA needed for detailed methylation analysis and assessment of breast cancer risk. Fresh breast milk samples and health, lifestyle and reproductive history questionnaires were collected from 111 women. Pyrosequencing analysis was conducted on DNA isolated from the exfoliated epithelial cells immunomagnetically separated from the total cell population in the breast milk of 102 women. A total of 65 CpG sites were examined in six tumor suppressor genes: PYCARD (also known as ASC or TMS1), CDH1, GSTP1, RBP1 (also known as CRBP1), SFRP1 and RASSF1. A sufficient quantity of DNA was obtained for meaningful analysis of promoter methylation; women donated an average of 86 ml of milk with a mean yield of 32,700 epithelial cells per ml. Methylation scores were in general low as expected of benign tissue, but analysis of outlier methylation scores revealed a significant relationship between breast cancer risk, as indicated by previous biopsy and methylation score, for several CpG sites in CDH1, GSTP1, SFRP1 and RBP1. Methylation of RASS F1 was positively correlated with women's age irrespective of her reproductive history. Promoter methylation patterns in DNA from breast milk epithelial cells can likely be used to assess breast cancer risk. Additional studies of women at high breast cancer risk are warranted.
Key words: biomarker, pyrosequencing, promoter methylation, breast epithelial cells, breast milk, breast cancer risk, parity, age-related promoter methylation, pregnancy-associated protection from breast cancer
Introduction
Breast cancer is the most common malignant neoplasm of women living in developed nations with an estimated 182,460 new cases and 40,480 deaths in 2008.1 While the widespread use of mammography has led to a decline in the incidence of late stage breast cancer, young women (under 40) have seen the smallest decline in incidence and stand to benefit the most from improved methods for assessing individual breast cancer risk.2 Analysis of epithelial cells in breast fluids (nipple aspirate and ducal lavage) and breast tissue (fine needle aspirate and core biopsy) has been proposed as a valuable aid in assessing individual breast cancer risk.3–6
Deregulation of the epigenome, heritable non-sequence changes in DNA, is widely recognized as a major mechanism in the development and progression of cancer.7–9 One type of epigenetic event frequently detected in breast cancer tissue and in fluid from diseased breasts is the addition of methyl groups on cytosines in CpG rich areas within the promoter regions of various tumor-related genes.10–15 Promoter hypermethylation of tumor suppressor genes, proto-oncogenes and vital cell cycle genes results in changes to the chromatin structure that often lead to transcriptional silencing and disruption of normal cell function.16 Because methylation is thought to occur early in disease and is potentially reversible, it is considered one of the most promising tools for accurate detection, prognosis and treatment.17 Several recent studies have shown that methylation of benign breast epithelium is increased in women at high risk of developing breast cancer.18,19
The promoter methylation of several tumor suppressor genes has been shown to increase with age20,21 and the methylation of Rass association domain family1 protein (RASSF1), in particular, increases with both age and breast-cancer-risk in benign breast epithelium.19 Since the most significant risk factor for breast cancer is age,1 and pregnancy is associated with a transient (3–5 years) increased risk of breast cancer,22,23 as women wait longer to have their first child their risk of developing pregnancy-associated breast cancer increases. In contrast, early pregnancy decreases a woman's lifetime risk of developing breast cancer.24,25 It has been proposed that long lasting changes in gene expression, such as those resulting from methylation or demethylation could be partly responsible for the breast-cancer-protection associated with an early pregnancy. An early pregnancy could plausibly reset a “methylation clock” resulting in a “younger” promoter methylation profile.
To examine whether epithelial cells from milk are a good source of material to study effects such as age-induced methylation, biomarkers associated with risk or parity-induced alterations in methylation status, six genes were chosen (selected regions for each are shown in Figure 1). Promoter methylation of the six genes analyzed in the present study has been detected previously in breast cancer and may increase a woman's risk of developing this disease. PYCARD [PY and CARD domain containing; also known as TMS or TMS1 (target of methylation silencing) and as ASC] is involved in the regulation of apoptosis, activation of inflammatory caspases and the regulation of NFκB activity.26 PYCARD expression suppresses the growth of breast cancer cells and methylation in the promoter region downregulates protein expression.10 Promoter hypermethylation of PYCARD was detected in 40 and 23% of primary breast tumors.10,27 CDH1 (E-Cadherin) functions in maintaining cell-cell adhesions in epithelial tissues. Promoter hypermethylation of CDH1 is frequently detected in breast cancer.28–30 In breast cancer, loss of CDH1 expression correlates with loss of differentiation, increased invasiveness and tumor grade, metastasis and poor prognosis.31 GSTP1 (Glutathione-s-transferase pi) plays a role in protecting cells from cytotoxic and carcinogenic agents.32 Expression of GSTP1 varies from tissue to tissue, however loss of GSTP1 expression has been attributed to DNA hypermethylation.33 Methylation of GSTP1 has been detected in breast cancer tissue14,29,34–36 and breast fluids from cancer patients12,37 and has been correlated with age in benign prostate.38 RBP1 (cellular retinol-binding protein 1, also known as CRBP1) functions in retinol (vitamin A) storage which is required for maintaining the differentiated state of adult epithelium.39 RBP1 is uniformly expressed in normal breast epithelium but downregulated in approximately 24% of human breast cancers40 and the downregulation has been linked to DNA hypermethylation.35,41 SFRP1 (Secreted frizzle related protein 1) is a tumor suppressor gene encoding a WNT signaling antagonist abundantly expressed in normal breast tissue. SFRP1 has been reported to be hypermethylated in >70% of breast cancers.42 RASSF1 (Rass association domain family1 protein), a tumor suppressor gene implicated in the development of breast cancer43 promotes apoptosis, cell cycle arrest and reduces the tumorgenicity of cancer cell lines.44 Aberrant promoter methylation of RASSF1 is frequently detected in the breast cancer tissue13,34,45 and has also been detected in serum of breast cancer patients46 and in fine needle aspirate from benign epithelium of woman at high risk for breast cancer.19
Figure 1.

CpG sites examined in the six genes. The vertical lines represent individual CpG sites within the CpG island of the promoter region. The boxed areas enclose the sites analyzed in each assay. The transcriptional start site is marked with an arrow and the numbers in parentheses define the boxed region relative to the transcriptional start site.
Unlike other biomarkers or risk factors, promoter methylation is potentially reversible with treatment, making methylation analysis even more desirable as an early screen for detecting increased breast-cancer-risk. This has lead several researches to suggest that nipple aspirate or other non-invasively collected breast fluid could be used to obtain breast epithelial cells for routine screening of breast-cancer-risk using methylation analysis.47,48 Aspiration techniques, however, may be not suitable for widespread screening for breast-cancer-risk as the number of cells obtained can be quite low and the techniques are uncomfortable at best and therefore not likely to be widely accepted as part of a breast-cancer screen. Breast milk, in contrast, is a rich, easily accessible source of epithelial-cell DNA. Importantly, the cells are representative of all the milk-producing lobules in the breast and it is equally easy to obtain samples from both breasts.
The purpose of the present research was threefold: (1) determine if breast milk provides sufficient quantity and quality of breast epithelial cell DNA for epigenetic analyses and breast-cancer-risk assessment, (2) use pyrosequencing to quantitatively examine breast epithelial cell DNA from healthy women for age-related increases in promoter methylation in six tumor suppressor genes and (3) determine if early pregnancy alters promoter methylation patterns.
Our results demonstrate that human milk can serve as an excellent fluid for assessing breast-cancer-risk in lactating women. The large number of epithelial cells and high quality of DNA obtained are sufficient for quantitatively examining methylation of numerous genes, and the frequency and distribution of outlier methylation values indicate that the high CpG-specific methylation scores are not random. Promoter methylation of RASSF1 increased with age and there was some indication that the observed increase differed between women who had an early first birth and those who had their first child after age 25. Additional studies examining promoter methylation in the epithelial cell DNA obtained from the milk of women at high risk of developing breast cancer and diverse reproductive histories are warranted.
Results
Study demographics.
Breast milk, questionnaires and informed consent were obtained from 111 lactating women. Nine of the 111 breast milk samples were excluded from the analyses. Three of the nine excluded samples had a volume less than 10 ml and were not processed and the other six excluded samples yielded too few cells to count and were not processed further. All results are based on the remaining 102 breast milk samples and the women who provided them.
As shown in Figure 2, participants ranged in age from 19–45 years (mean = 32; s.d. = 6) and were nursing children who ranged in age from 30–820 days (mean = 239; s.d. = 177). 63% of the 102 women were nursing their firstborn. The remaining women had between one and four previous births. All of the women considered themselves healthy; none of the women had breast cancer presently or previously; seven women had a prior breast biopsy. The volume of the breast milk samples ranged between 20 and 180 ml (mean = 86; s.d. = 33) and there was no significant correlation between the volume of milk donated and the age of the mother (r = 0.12, F[1,100] = 1.5, p = 0.22), the age of the child (r = −0.01, f[1,99] = 0.54, p = 0.47) or the number of children the mother had previously nursed (r = 0.14, F[1,100] = 2.06, p = 0.16).
Figure 2.

Participant demographics and sample yields from 102 of the 111 women who provided a breast milk sample.
A cell count was made on 90 of the 102 samples prior to the epithelial cell separation revealing an average total-cell population of 203,394 cells per ml (range: 3,917–2,102,857 cells per ml). Among these 90 milk samples there was a marginally significant negative correlation between a mother's age and the number of cells per ml (r = −0.18, F[1,88] = 2.84, p = 0.10). There was no correlation between the child's age and the number of cells in the mother's milk (r = −0.11, F[1,87] = 1.12, p = 0.29). Immunomagnetic cell separation on the total-cell population yielded an average of 32,718 cells per ml in the epithelial-enriched cell fraction (range: 417–241,714 cells per ml) or roughly 20% of the total-cell population. There was no significant relationship between the number of epithelial-selected cells and mother's age (r = −0.14, F[1,99] = 2.17, p = 0.14) or the child's age (r = 0.03, F[1,98] = 0.06, p = 0.80). DNA isolation of the epithelial-enriched cell fractions yielded an average of 2.4 µg per sample and as expected the amount of DNA isolated was directly related to the number of cells in the epithelial-enriched fraction (r = 0.75, F[1,87] = 114.3, p < 0.00).
Pyrosequencing analyses.
Gene-specific amplified products from bisulfite-modified epithelial cell DNA were sent to EpigenDx, Inc., (Worcester, MA, USA) for pyrosequencing analysis in batches of roughly 50 samples per shipment. Methylated and unmethylated controls were included with each batch. Individual “pyrograms” or sequencing readouts and percent of methylated DNA at each CpG site were returned for each of the six genes for all 102 epithelial cell samples. Each pyrogram includes the expected sequence, the nucleotide dispensation order, the sequencing results with at least one sequencing control and one bisulfite modification control. The efficiency of our bisulfite modification is demonstrated in Figure 3 (pyrograms) at position 18 in which a cytosine not followed by a guanine in the genomic DNA (therefore not methylated) was completely converted to uracil after bisulfute treatment and then replaced with thymidine during the PCR amplification as indicated by the complete absence of cytosine in position 18 in either the methylated (top pyrogram) or nonmethyated (bottom pyrogram) DNA. If either the expected sequence was not obtained or the bisulfite conversion was incomplete the pyrosequencing run is flagged with a red code and data are not accepted. A yellow flag indicates that a manual check of the data is needed before accepting or rejecting the data, whereas a blue flag indicates reliable results. The pyrograms from each of the six genes for all 102 samples were examined and after elimination of any unreliable result the sample size for several genes was less than 102.
Figure 3.

Comparison of pyrosequencing results obtained from methylated and non-methylated DNA for nine CpG sites spanning the transcriptional start site in the tumor suppressor gene RASS F1. (A) Pyrograms of methylated (top) and non-methylated (bottom) HMEC DNA show the nucleotide dispensation order on the x-axis, the signal intensity on the y-axis, the sequence being analyzed on the top and the percent methylated (gray columns) for each of the nine CpG sites. The narrower tan-shaded column at dispensation 18 marks a bisulfite-treatment control; as a non-methylated cytosine in the genomic DNA (a cytosine not followed by a guanine) will be completely converted to uracil with bisulfite treatment and then replaced with thymidine during the PCR. The dispensation begins with the addition of enzyme (E) followed by substrate (S) and then the dinucleotides. When no deviation from the predicted sequence is encountered and the bisulfite-conversion is ≥96% complete, the methylation score (%) is framed in a blue square at the top of the gray column and considered a perfect call. Small deviations from the expected sequence and/or bisulfite conversions between 92.5 and 95.5% complete are framed in yellow squares and the sequencing results are manually checked before accepting or rejecting the methylation score. Large deviations and/or bisulfite conversions less than 92.5% complete are framed in red squares (none shown) and the methylation score is rejected. (B) Pyrosequencing results from three separate PCR reactions of bisulfite-modified methylated control DNA (top three lines) and non-methylated control DNA (bottom three lines).
The reproducibility of the PCR amplification and pyrosequencing analysis is demonstrated in Figure 3 (bottom), in which bisulfite-treated methylated and nonmethylated DNA from three separate PCR amplifications for RASSF1 was sequenced in three separate batches. As can be seen, the variability among the three batches for both the methylated and nonmethyated DNA is extremely low and site-specific methylation patterns are apparent.
Mean overall methylation for each gene.
Pyrosequncing promoter methylation values below 5% for each of the genes examined is considered typical of normal healthy tissue or background noise.49 We use this standard only to assist in the interpretation of methylation levels and all values of methylation levels are included as real data in all statistical analyses. As expected of healthy women without breast cancer, the methylation scores in the DNA isolated from epithelial cells in breast milk were low. The bottom row of Table 1 presents mean overall methylation, the interquartile range and the number of outliers for each of the six genes. Mean overall methylation was below 5% for PYCARD, CDH1, GSTP1 and RASSF1 and only slightly above 5% for RBP1 (6.49) and SFRP1 (5.8). However, for each of the genes there were several epithelial cell samples with substantially higher levels of overall methylation as indicated by the number of outliers per gene; PYCARD, CDH1 and GSTP1 each had three outliers, while RBP1, SFRP1 and RASSF1 had 4, 15 and 7 outliers, respectively. The relationship of these outliers to the distribution of overall methylation is given in Figure 4. Four of the genes, PYCARD, RBP1, SFRP1 and RASSF1, had epithelial cell samples for women with overall methylation scores above 10%. Of these genes, SFRP and RASSF1 had the greatest number of highly methylated samples.
Table 1.
Mean percent methylated DNA for each of the six genes examined
| PYCARD (n = 100) | CDH1 (n = 93) | GSTP1 (n = 99) | RBP1 (n = 97) | SFRP1 (n = 101) | RASSF1 (n = 102) | |||||||||||||
| CpG | %M | IQR | # OL | %M | IQR | # OL | %M | IQR | # OL | %M | IQR | # OL | %M | IQR | # OL | %M | IQR | # OL |
| 1 | 1.68 | 1.86 | 4 | 3.03 | 2.42 | 4 | 3.17 | 1.74 | 6 | 5.49 | 3.57 | 6 | 5.55 | 5.42 | 6 | 3.86 | 1.78 | 12 |
| 2 | 2.57 | 1.90 | 4 | 1.71 | 1.66 | 3 | 3.58 | 1.85 | 3 | 3.6 | 2.82 | 6 | 6.91 | 5.86 | 8 | 3.68 | 2.49 | 4 |
| 3 | 2.27 | 1.74 | 6 | 3.73 | 2.62 | 5 | 2.16 | 1.27 | 7 | 8.03 | 4.63 | 2 | 6.25 | 4.93 | 9 | 3.68 | 2.74 | 5 |
| 4 | 4.33 | 2.52 | 5 | 3.73 | 3.89 | 1 | 4.42 | 2.54 | 5 | 4.78 | 2.94 | 8 | 4.38 | 3.68 | 8 | 2.17 | 2.00 | 7 |
| 5 | 4.75 | 3.98 | 9 | 1.99 | 1.36 | 2 | 3.06 | 1.59 | 7 | 5.39 | 4.27 | 3 | 5.58 | 3.44 | 14 | 3.73 | 1.73 | 6 |
| 6 | 5.68 | 3.79 | 7 | 4.55 | 3.91 | 3 | 3.1 | 1.74 | 3 | 10.19 | 6.2 | 2 | 7.28 | 4.71 | 6 | 7.43 | 4.40 | 2 |
| 7 | 3.38 | 1.93 | 7 | 5.12 | 4.08 | 2 | 3.09 | 1.65 | 3 | 3.29 | 2.41 | 8 | 3.97 | 2.52 | 9 | 5.02 | 3.58 | 3 |
| 8 | 2.94 | 1.96 | 6 | 6.52 | 4.73 | 1 | 5.09 | 2.66 | 3 | 8.23 | 5.93 | 3 | 6.45 | 4.44 | 6 | 6.16 | 4.64 | 4 |
| 9 | 2.92 | 2.40 | 3 | 1.65 | 1.08 | 4 | 8.23 | 3.95 | 6 | 4.84 | 3.25 | 7 | 6.76 | 3.97 | 9 | |||
| 10 | 3.03 | 2.18 | 7 | 2.4 | 1.43 | 3 | 11.17 | 6.71 | 2 | |||||||||
| 11 | 3.1 | 2.15 | 4 | 2.72 | 1.34 | 5 | 3.18 | 1.92 | 10 | |||||||||
| 12 | 7.38 | 4.91 | 3 | 4.24 | 1.99 | 6 | 7.21 | 5.47 | 3 | |||||||||
| 13 | 2.07 | 1.26 | 1 | 7.27 | 4.49 | 6 | ||||||||||||
| 14 | 8.25 | 6.03 | 8 | |||||||||||||||
| Combined | 3.67 | 1.83 | 3 | 3.56 | 2.19 | 3 | 3.64 | 1.36 | 3 | 6.49 | 3.41 | 4 | 5.8 | 2.39 | 15 | 4.72 | 2.37 | 7 |
%M, percent DNA methylated; IQR, interquartile range; # OL, number of outliers >(75th percentile + 1.5 * IQR). Overall values also shown in Figure 4. BOLD, CpG sites for which the mean methylation score was ≥5%.
Figure 4.
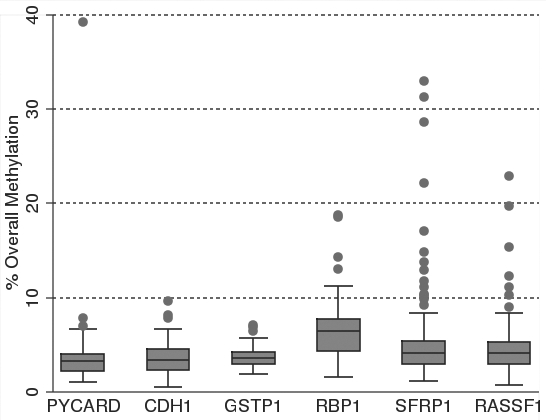
Box and whisker plots of percent overall mean methylation for each of the six genes analyzed in epithelial cell DNA isolated from breast milk. The filled circles represent outliers as defined in the methods. The top whisker represents the highest non-outlier score and the bottom whisker represents the lowest non-outlier score. The top line of the box represents the third quartile, the bottom line of the box the first quartile and the middle line the median. Samples sizes range between 93 and 102 as shown in Table 1.
CpG site-specific percent methylation.
Between eight and 15 CpG sites were examined in each of six genes for a total of 65 CpGs. The bulk of Table 1 provides the mean percent methylation, interquartile range and number of outliers for each of the CpG sites within the six genes. While the mean overall methylation for the 102 women were above 5% for only two genes, RBP1 and SFRP1, in each of the six genes there were several individual CpG sites with mean percent methylation above 5%. PYCARD, CDH1 and GSTP1 had two sites each with mean methylation scores above 5%, while RBP1, SFRP1 and RASSF1, had nine, six, four sites respectively (shown in bold in Table 1). Of all the CpGs examined, the three sites with the highest mean methylation scores were all in the promoter region of RBP1 (CpGs 6, 10 and 14, had mean percent methylation of 10.19, 11.17 and 8.25 respectively).
Age and overall methylation.
We next examined whether mother's age contributed to the variability observed in methylation. Only for RASSF1 is there a statistically significant linear relationship between age and overall methylation, with overall methylation increasing with age (r = 0.24, b = 0.143, F[1,100] = 6.06, p = 0.016). Overall methylation for RASSF1 increases by just under one and one half percent for each decade of a woman's life. In RASSF1, not surprisingly, variability in overall methylation also increases with age.
Given the significant relationship between overall methylation for RASSF1 and age, and the interest in understanding how early pregnancy reduces a woman's lifetime risk of developing breast cancer, we further considered whether having a first birth before age 25 conditioned this relationship in any way. The increase in overall methylation and methylation variability with increasing age is visually apparent for women who gave birth to their first child after age 25 and for those who had their first birth before that (Fig. 5). However, because of the absence of counterfactuals (no women in the sample could be under 26 at the time of milk sample collection if they had not had a birth until after that age) the relationship among older women is truncated by age and not statistically significant. Despite the small sample of women available (n = 21) the increase in overall methylation for those who had a first birth before age 26 is statistically significant (r = 0.61, F[1,19] = 11.17, p < 0.00). Importantly, having a first birth at a younger first age did not attenuate the relationship between overall RASSF1 methylation and age.
Figure 5.

Relationship between mother's age, age at first birth and percent overall mean methylation in RASSF1 epithelial cell DNA isolated from the breast milk of 102 women. The percent overall mean methylation for RASSF1 in women who first gave birth after they turned 25 years of age is represented as “O” (older at first birth) and the percent overall mean methylation for RASSF1 in women who first gave birth before they turned 25 years of age is represented as “Y” (younger at first birth).
We also examined the relationship between overall methylation for each of the six genes and both age at menarche and current birth parity. Only RBP1 had a significant relationship between overall methylation and age at menarche [r = 0.23, F[1,94] = 5.09, p = 0.03] and this relationship disappeared when controlling for mother's age. Parity had no relationship to overall methylation for any of the six genes regardless of whether or not age was included in the regression.
Age and CpG site-specific percent methylation.
To determine if site-specific percent methylation in breast epithelial cell DNA increased with age we examined the relationship between mother's age and each of the 65 individual CpG sites. For four of the genes PYCARD, CDH1, GSTP1 and SFRP1, there were no significant relationships between mean methylation score and mother's age for any of the CpG sites and for RBP1, only percent methylation of CpG 4 increased significantly with mother's age (r = 0.20, F[1,95] = 4.11, p = 0.05). However, for RASSF1, the only gene for which overall methylation scores increased significantly with age, there are a number of CpG sites in which methylation also increased significantly with age (Fig. 6). The correlation between age and percent methylation was greatest for CpG sites 1, 5, 7 and 9, and the highest methylation scores occurred in CpG sites 1 and 9. The replicated relationship between age and percent methylation for multiple CpG sites suggests that the relationship between age and RASSF1 overall methylation is not due to a statistical artifact or random variation.
Figure 6.

Relationship between mother's age and percent methylation for each of the nine CpG sites analyzed in RASSF1 epithelial cell DNA isolated from the breast milk of 102 women.
Analysis of percent methylation outliers.
One of our goals in analyzing epithelial cells from breast milk is to determine if these cells can be used to assess breast cancer risk. While none of the 102 women who provided milk samples for the present study had breast cancer, there were several epithelial cell samples with percent methylation and overall methylation more typical of those observed in breast cancer tissue. Only with a prospective or retrospective, research design would be able to assess the future diagnostic utility of these outliers for women in our sample. Some analysis of outliers, however, can suggest whether they might plausibly reflect elevated cancer risk or are simply the random product of an extensive repetition of statistical testing and measurement.
First, we can ask the simple question of whether the number and patterns of association among outliers in the data are beyond what would be expected due to random chance. Given that for each epithelial cell sample, 71 separate analyses of methylation data were conducted (65 individual CpG sites and six overall methylation scores) and 102 samples were examined, if we were to assume a 95% confidence level we might expect a mean of 3.55 outlier methylation scores per woman due to random chance alone. Results were only slightly higher than this expectation due to random chance, with the actual average number of outliers per woman 3.68 and a median of 2. Importantly, however, there was an indication in the clustering of outliers within women that the outliers were likely not due to chance alone. Just thirteen women accounted for 44% of all outliers in the data, suggesting these outliers reflect high methylation across CpG sites within specific women and not simple measurement errors.
Second, seven women in our sample had a previous biopsy that may serve as a proxy for elevated risk. Sample sizes for comparisons are small and should be interpreted cautiously. However, using a binomial test of proportions, there is evidence for a significantly higher proportion of methylation outliers for women with previous biopsies than for those with no previous biopsy in seven of the 67 CpG sites (CDH1-1, p = 0.00; 5, p = 0.03; and 6, p = 0.10; GSTP1-10, p = 0.10; SFRP1-4, p = 0.06 and 5, p = 0.04; RBP1-5, p = 0.10). At each of these sites there were also substantively meaningful outliers (i.e., over 5% methylated) and most of these sites include outliers that were also over ten percent methylated. Using a more conservative statistical test, Fisher's Exact Test, the higher proportion of outliers among biopsied women was significant for CDH1-1 (p = 0.03) and SFRP1-4 (p = 0.09) and 5 (p = 0.05) methylation. These results offer some evidence for non-random occurrence of methylation outliers, clustering of those outliers in a small number of women, clearly higher levels of methylation measures among those for biopsied women and evidence for plausible statistical associations between methylation outliers and biopsied women.
Discussion
In the present study we used pyrosequencing to examine promoter methylation of six tumor suppressor genes in DNA isolated from epithelial cells present in the breast milk obtained from 102 nursing mothers. Mean overall methylation scores for PYCARD, CDH1, GSTP1, RBP1, SFRP1 and RASSF1 were low, as would be expected of cells from healthy epithelium. However, analysis of outliers in the 62 individual CpG sites revealed a significantly higher level of promoter methylation in specific CpGs of CDH1, GSTP1, SFRP1 and RBP1 among women who had a previous biopsy. In agreement with previous reports from benign breast tissue,19 we found promoter methylation of RASSF1 to increase with age. Taken together these results support the viewpoint that promoter methylation of specific genes in healthy breast cells can be used to assess breast-cancer-risk and suggest that breast milk is an appropriate source of epithelial cells for risk assessment.
If breast tissue is to be used to detect occult breast cancer or to assess breast-cancer-risk, then the tissue obtained must be representative of the total breast epithelium. The three methods currently used for sampling breast tissue in asymptomatic women are nipple aspiration, random periareolar fine needle aspiration and ductal lavage.4 While each of these methods has been used in at least one study revealing a significant breast cancer-associated promoter methylation pattern in women at high risk,3,5,6,11,12,19 the number of cells collected using nipple aspiration and ductal lavage is typically very low (between 10 and a few thousand 1,000) and usually only 1–2 of the 15 duct systems in a breast is surveyed4,48 possibly leading to the inconsistencies observed among studies. In contrast, the large number of cells obtained from milk, 66% of samples in the present study contained one million or more epithelial cells, suggest that the cells originated throughout the breast and provide appropriate DNA for risk assessment. The perception of discomfort associated with fine needle aspiration and ductal lavage as well as the cost of the procedures potentially limit the widespread use of these techniques, either as part of women's routine breast cancer screening or as a collection method in large epidemiological studies. In contrast, collection of breast milk requires no trained personnel, milk from both breasts can be easily obtained, and we found lactating women eager to participate in research and donate a milk sample.
The use of breast milk as a screen for increased breast-cancer-risk is obviously limited to lactating women. However it may provide an early and valuable assay to establish which women should be followed more closely as they age. The potential subpopulation of pregnant women with hypermethylation would be expected to be much higher than the roughly 1 in 3,000 women diagnosed with breast cancer during pregnancy or within the year following birth.50 Furthermore, the population of women at risk for pregnancy-associated breast cancer will likely rise due to changing childbearing trends. A screen based upon lactation may be of particular value to the growing number of women, at higher risk, having their first pregnancy at older ages and among all women of any parity having children at older ages.
In our study we, in fact, hypothesized that we would see differences in age-related methylation patterns between women who had an early first birth (before 25) and those with later first births. In our sample, however, we did not see evidence that age at first birth affected age-related increases in methylation. The testing of this hypothesis is limited by the sample size, the structural absence of counterfactuals across age and cohort trends with younger women in the sample drawn from more recent birth cohorts. Although our data did not reveal differences in age related increases in methylation between women with early and late first births, it does not rule out such a relationship.
A major concern with trying to use cells in milk to assess breast-cancer-risk is that the methylation pattern of exfoliated epithelial cells in milk may simply reflect the state of the lactating breast and reveal little if anything about the long-term health of the breast. Several results from the present study help reduce these concerns; we detected the same age-related increases in RASSF1 promoter methylation that have been previously reported, and the outlier values appear to be nonrandom and associated with increased breast-cancer-risk.
Our initial results from this sample of 102 women at average risk of breast cancer warrant additional research. A more comprehensive assessment of the use of breast milk in screening for hypermethylation, particularly for the comparison across population subgroups such as those with and without early pregnancy, would benefit from a larger sample size with a greater dispersion across ages, cohorts and age at first birth. Future studies could also benefit from sampling strategies designed to increase the sample density of women at a high risk of breast cancer to increase the representation of methylation outliers in statistical analyses such as those presented. Lastly, the procedures developed for milk collection and analysis in this study are clearly suitable for pairing with concurrent and subsequent diagnostic information, such as biopsy data, in a prospective study which attempts to link methylation to breast cancer outcomes. Our current research efforts are directed at obtaining such expanded sample data for future analysis.
Materials and Methods
Study participants and collection of milk sample.
This study was approved by the Internal Review Board of the Human Subjects Protection Office at the University of Massachusetts, Amherst. From 2006 through 2008 lactating women living or visiting within 100 miles of Amherst, Massachusetts were recruited through local advertisements and fliers. One hundred and eleven nursing mothers provided a milk sample and completed a health and lifestyle questionnaire. Each participant was asked to provide up to 100 mL of breast milk by pumping or hand expressing all the milk from one breast and additional milk from the second breast, as needed. The fresh breast milk was transferred to a sterile glass bottle. The majority of the participants lived in western Massachusetts and in most cases a researcher collected the milk and questionnaire from the participant at her home the morning the breast milk was pumped or expressed. Milk samples were maintained at ambient temperature and taken directly to the laboratory. Less than 10% of the milk samples were collected in the evening and these milk samples were kept cool (placed in a cooler with a small amount of ice) and taken to the laboratory the following morning.
Processing of milk samples.
Fresh milk samples were processed immediately upon arrival at the laboratory. 5 ml of the milk was placed in a glass vial and archived at −20°C for future studies. The remaining milk was diluted 1:1 with sterile PBS and centrifuged at 1,000 G for 10 min in glass 50 mL centrifuge tubes. The supernatant including all of the milk fat was transferred to a 250 mL acid washed amber bottle and archived at −20°C for future studies. The cell pellet was resuspended in ∼40 mL PBS and washed 3–5 times until the supernatant was clear. The washed cell pellet was resuspended in 1 mL degassed PBS (with 2 mM EDTA/0.5% BSA) and total cell count determined using a hemocytometer. For cell counts ≤1 × 107, the cells were resuspended in 60 µl of degassed PBS (with 2 mM EDTA/0.5% BSA) with 20 µl of epithelial-specific MACS HEA-125 micro-beads (Miltenyi Biotec, Germany) and 20 µl of Fc blocking reagent (Miltenyi Biotec, Germany). Volumes were appropriately scaled for larger numbers of cells. The cell-microbead suspension was incubated for 15 min at 4°C, brought to a total volume of 1 mL with degassed PBS (with 2 mM EDTA/0.5% BSA) and centrifuged at 210 G for 10 min. This final pellet was resuspended in 0.5 mL of degassed PBS and epithelial cells separated using the MACS paramagnetic column and stand (Miltenyi Biotec, Germany) according to the manufacturer's instructions. Both the epithelial-enriched (positive) cell fraction and epithelial-depleted (negative) cell fractions were frozen until needed.
DNA isolation, bisulfite modification and PCR amplification for pyrosequencing.
DNA was isolated from both the enriched and depleted cell fractions using the QIAamp DNA isolation kit (Qiagen, Valencia, CA, USA) with the addition of RNAse A and Proteinase K digestion steps as per manufacturer's suggestions. DNA was quantified and quality was assessed using a spectrophotometer (Genequant Amersham Biosciences, Buckinghamshire UK). Bisulfite modification of 1 µg genomic DNA per sample was conducted using the Epitect Bisulfite Kit (Qiagen, Valencia, CA, USA) according to the instructions provided. Bisulfite modified DNA was aliquoted and frozen for use within the next few months.
We refer to each of the six genes by their official symbol and name as listed on the Entrez Gene data base of NCBI as of September 1, 2009. The biotinylated primer sets for CDH1, GSTP1 and RASSF1 were available through Biotages' Pyromark Assay Database (http://techsupport.pyrosequencing.com/). The biotinylated primers for PYCARD, RBP1 and SFRP1 were designed for the CpG island region flanking the transcription start site at the 5′UTR with the assistance of EpigenDx Inc. and their Pyrosequencing design software. Each of the newly designed primer sets were tested for PCR bias in a mixing experiment using in vitro methylated and non-methylated DNA. Locations of the CpG sites examined are shown in Figure 1.
For each of the six genes, 1 µl of bisulfite-modified DNA was amplified in 30 µL of reaction mixture containing 1x buffer, 200 nM primers and 0.2 units of HotStar Taq Polymerase (Qiagen, Valencia, CA, USA) for over 45 cycles at an annealing temperature unique to each primer set (Table S1). Control products were prepared using bisulfite-treated DNA from HMECs treated with m.Sssi methylase (New England Biolabs, Ipswich, MA), non-treated HMECs and non-methylated commercial Fetal DNA (Chemicon, Billerica, MA, USA). Amplified PCR products for the six genes and controls were verified by gel electrophoresis and sent on ice to EpigenDx Inc., in Worcester, MA, USA for pyrosequencing analysis. The PCR product was bound to Streptavidin Sepharose HP (Amersham Biosciences, Uppsala, Sweden) and the Sepharose beads containing the immobilized PCR product were purified, washed and denatured using a 0.2 M NaOH solution and rewashed using the Pyrosequencing Vacuum Prep Tool (Biotage AB) as recommended by the manufacturer. Then 0.2 µM pyrosequencing primer was annealed to the purified single-stranded PCR product. 10 µl of the PCR products were sequenced by Pyrosequencing PSQ96 HS System (Biotage AB) following the manufacturer's instructions (Biotage, Kungsgatan, Sweden). The methylation status of each locus was analyzed individually as a T/C SNP using QCpG software (Biotage, Kungsgatan, Sweden). Resulting pyrograms and percent methylation scores for each CpG site were received from EpigenDX, Inc.
Statistical analyses.
Our analysis is concerned with both evidence of CpG-site-specific methylation within each gene and with overall indications of methylation for each of the six genes. When analyzing CpG-site-specific methylation in a gene, data are presented for each woman, as in some graphs, as the percent methylation at each specific CpG site, or, as in tables, summarized through the mean percent methylation at the CpG site across the sample of women. To examine gene methylation, data are presented, again as in some graphs, for overall methylation (the mean percentage of methylation averaged across the CpG-sites of the gene for each woman in the sample) or summarized across the sample women, as in tables, through the grand mean overall methylation for the gene. Methylation and questionnaire data were merged into a combined data file and statistical analyses were performed using-Stata10: Data Analysis & Statistical Software (Statacorp LP, College Station, TX, USA). In discussing levels of statistical significance exact probability values are provided when specific statistical tests are reported. In some small sample exploratory analyses which seek for any plausible evidence of association we utilize a lower bound of 0.10, corresponding to any evidence of even a “weak” relationship in a one-sided test with given sample sizes.51 In discussing results, abnormally high levels of methylation or sample outliers, are noted. Outliers were identified as values of percent methylation or overall methylation for a woman >[75th percentile + 1.5*(Interquartile Range)]. In any analysis of outliers it is important to bear in mind that any one outlier may reflect genuinely abnormal levels of methylation or simple stochastic measurement error. Hence, association between methylation outliers and other covariates is inferential evidence of an association only if there is an overall association between outliers and a given covariate and only so far as there is no reason to suspect systematic bias in measurement processes and the covariates analyzed.
Supplementary Material
Acknowledgements
The authors thank the Avon Foundation for their generous support.
Abbreviations
- CpG
cytosine phosphodiesterbonded to guanine
- PYCARD
PY & CARD domain containing
- CDH1
E-cadherin
- GSTP1
glutathione-S-transferase pi
- RBP-1
retinal binding protein 1
- SFRP1
secreted frizzle related protein 1
- RASSF1
rass association domain family 1
- MACS
magnetic antibody cell separation
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/12961
References
- 1.SEER cancer statistics review 1975–2006. 2008 [Google Scholar]
- 2.Esserman LJ, Shieh Y, Park JW, Ozanne EM. A role for biomarkers in the screening and diagnosis of breast cancer in younger women. Expert Rev Mol Diagn. 2007;7:533–544. doi: 10.1586/14737159.7.5.533. [DOI] [PubMed] [Google Scholar]
- 3.Buehring GC, Letscher A, McGirr KM, Khandar S, Che LH, Hackett AJ. Presence of epithelial cells in nipple aspirate fluid is associated with subsequent breast cancer: A 25-year prospective study. Breast Cancer Res Treat. 2006;98:63–70. doi: 10.1007/s10549-005-9132-5. [DOI] [PubMed] [Google Scholar]
- 4.Fabian CJ, Kimler BF, Mayo MS, Khan SA. Breast-tissue sampling for risk assessment and prevention. Endocr Relat Cancer. 2005;12:185–213. doi: 10.1677/erc.1.01000. [DOI] [PubMed] [Google Scholar]
- 5.Arun B. Ductal lavage and risk assessment of breast cancer. Oncologist. 2004;9:599–605. doi: 10.1634/theoncologist.9-6-599. [DOI] [PubMed] [Google Scholar]
- 6.Tice JA, Miike R, Adduci K, Petrakis NL, King E, Wrensch MR. Nipple aspirate fluid cytology and the gail model for breast cancer risk assessment in a screening population. Cancer Epidemiol Biomarkers Prev. 2005;14:324–328. doi: 10.1158/1055-9965.EPI-04-0289. [DOI] [PubMed] [Google Scholar]
- 7.Gal-Yam EN, Saito Y, Egger G, Jones PA. Cancer epigenetics: modifications, screening and therapy. Annu Rev Med. 2008;59:267–280. doi: 10.1146/annurev.med.59.061606.095816. [DOI] [PubMed] [Google Scholar]
- 8.Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61:3225–3229. [PubMed] [Google Scholar]
- 9.Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4:988–993. doi: 10.1038/nrc1507. [DOI] [PubMed] [Google Scholar]
- 10.Conway KE, McConnell BB, Bowring CE, Donald CD, Warren ST, Vertino PM. TMS1, a novel proapoptotic caspase recruitment domain protein, is a target of methylation-induced gene silencing in human breast cancers. Cancer Res. 2000;60:6236–6242. [PubMed] [Google Scholar]
- 11.Evron E, Dooley WC, Umbricht CB, Rosenthal D, Sacchi N, Gabrielson E, et al. Detection of breast cancer cells in ductal lavage fluid by methylation-specific PCR. Lancet. 2001;357:1335–1336. doi: 10.1016/s0140-6736(00)04501-3. [DOI] [PubMed] [Google Scholar]
- 12.Jeronimo C, Costa I, Martins MC, Monteiro P, Lisboa S, Palmeira C, et al. Detection of gene promoter hyper-methylation in fine needle washings from breast lesions. Clin Cancer Res. 2003;9:3413–3417. [PubMed] [Google Scholar]
- 13.Yan PS, Shi H, Rahmatpanah F, Hsiau TH, Hsiau AH, Leu Y, et al. Differential distribution of DNA methylation within the RASSF1A CpG island in breast cancer. Cancer Res. 2003;63:6178–6186. [PubMed] [Google Scholar]
- 14.Lee JS. GSTP1 promoter hypermethylation is an early event in breast carcinogenesis. Virchows Arch. 2007;450:637–642. doi: 10.1007/s00428-007-0421-8. [DOI] [PubMed] [Google Scholar]
- 15.Fackler MJ, Rivers A, Teo WW, Mangat A, Taylor E, Zhang Z, et al. Hypermethylated genes as biomarkers of cancer in women with pathologic nipple discharge. Clin Cancer Res. 2009;15:3802–3811. doi: 10.1158/1078-0432.CCR-08-1981. [DOI] [PubMed] [Google Scholar]
- 16.Jones P, Baylin S. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dworkin A, Huang TH, Toland A. Epigenetic alterations in the breast: implications for breast cancer detection, prognosis and treatment. Semin Cancer Biol. 2009;19:165–171. doi: 10.1016/j.semcancer.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lewis C, Cler L, Bu D, Zchbauer-Mller S, Milchgrub S, Naftalis E, et al. Promoter hypermethylation in benign breast epithelium in relation to predicted breast cancer risk. Clin Cancer Res. 2005;11:166–172. [PubMed] [Google Scholar]
- 19.Euhus DM, Bu D, Milchgrub S, Xie X, Bian A, Leitch AM, et al. DNA methylation in benign breast epithelium in relation to age and breast cancer risk. Cancer Epidemiol Biomarkers Prev. 2008;17:1051–1059. doi: 10.1158/1055-9965.EPI-07-2582. [DOI] [PubMed] [Google Scholar]
- 20.Issa JJ, Ahuja N, Toyota M, Bronner MP, Brentnall TA. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res. 2001;61:3573–3577. [PubMed] [Google Scholar]
- 21.Fraga MF, Esteller M. Epigenetics and aging: the targets and the marks. Trends in Genetics. 2007;23:413–418. doi: 10.1016/j.tig.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 22.Helewa M, Levesque P, Provencher D, Lea RH, Rosolowich V, Shapiro HM, et al. Breast cancer, pregnancy and breastfeeding. J Obstet Gynaecol Can. 2002;24:164–180. [PubMed] [Google Scholar]
- 23.Schedin P. Pregnancy-associated breast cancer and metastasis. Nat Rev Cancer. 2006;6:281–291. doi: 10.1038/nrc1839. [DOI] [PubMed] [Google Scholar]
- 24.Trichopoulos D, Hsieh CC, MacMahon B, Lin TM, Lowe CR, Mirra AP, et al. Age at any birth and breast cancer risk. Internat J Cancer. 1983;31:701–704. doi: 10.1002/ijc.2910310604. [DOI] [PubMed] [Google Scholar]
- 25.Albrektsen G, Heuch I, Hansen S, Kvle G. Breast cancer risk by age at birth, time since birth and time intervals between births: exploring interaction effects. Brit J Cancer. 2005;92:167–175. doi: 10.1038/sj.bjc.6602302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Srinivasula S, Poyet J, Razmara M, Datta P, Zhang Z, Alnemri E. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. JBC. 2002;277:21119–21122. doi: 10.1074/jbc.C200179200. [DOI] [PubMed] [Google Scholar]
- 27.Mirza S, Sharma G, Prasad CP, Parshad R, Srivastava A, Gupta SD, et al. Promoter hypermethylation of TMS1, BRCA1, ERα and PRB in serum and tumor DNA of invasive ductal breast carcinoma patients. Life Sci. 2007;81:280–287. doi: 10.1016/j.lfs.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 28.Li S, Rong M, Iacopetta B. DNA hypermethylation in breast cancer and its association with clinicopathological features. Cancer Lett. 2006;237:272–280. doi: 10.1016/j.canlet.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 29.Shinozaki M, Hoon DSB, Giuliano AE, Hansen NM, Wang H, Turner R, et al. Distinct hypermethylation profile of primary breast cancer is associated with sentinel lymph node metastasis. Clin Cancer Res. 2005;11:2156–2162. doi: 10.1158/1078-0432.CCR-04-1810. [DOI] [PubMed] [Google Scholar]
- 30.Caldeira JRF, Prando E, Quevedo F, Neto FAM, Rainho C, Rogatto S. CDH1 promoter hypermethylation and E-cadherin protein expression in infiltrating breast cancer. BMC Cancer. 2006;6:48–57. doi: 10.1186/1471-2407-6-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berx G, Van Roy F. The E-cadherin/catenin complex: an important gatekeeper in breast cancer tumorigenesis and malignant progression. Breast Can Res. 2001;3:289–293. doi: 10.1186/bcr309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Su F, Hu X, Jia W, Gong C, Song E, Hamar P. Glutathione S transferase pi indicates chemotherapy resistance in breast cancer. J Surg Res. 2003;113:102–108. doi: 10.1016/s0022-4804(03)00200-2. [DOI] [PubMed] [Google Scholar]
- 33.Zhang YJ, Chen Y, Ahsan H, Lunn RM, Chen SY, Lee PH, et al. Silencing of glutathione S-transferase P1 by promoter hypermethylation and its relationship to environmental chemical carcinogens in hepatocellular carcinoma. Cancer Lett. 2005;221:135–143. doi: 10.1016/j.canlet.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 34.Agrawal A, Murphy RF, Agrawal DK. DNA methylation in breast and colorectal cancers. Mod Pathol. 2007;20:711–721. doi: 10.1038/modpathol.3800822. [DOI] [PubMed] [Google Scholar]
- 35.Esteller M, Guo M, Moreno V, Peinado M, Capella G, Galm O, et al. Hypermethylation-associated inactivation of the cellular retinol-binding-protein 1 gene in human cancer. Cancer Res. 2002;62:5902–5905. [PubMed] [Google Scholar]
- 36.Radpour R, Kohler C, Haghighi MM, Fan AX, Holzgreve W, Zhong XY. Methylation profiles of 22 candidate genes in breast cancer using high-throughput MALDI-TOF mass array. Oncogene. 2009;28:2969–2978. doi: 10.1038/onc.2009.149. [DOI] [PubMed] [Google Scholar]
- 37.Krassenstein R, Sauter E, Dulaimi E, Battagli C, Ehya H, Klein-Szanto A, et al. Detection of breast cancer in nipple aspirate fluid by CpG island hypermethylation. Clin Cancer Res. 2004;10:28–32. doi: 10.1158/1078-0432.ccr-0410-3. [DOI] [PubMed] [Google Scholar]
- 38.Kwabi-Addo B, Chung W, Shen L, Ittmann M, Wheeler T, Jelinek J, et al. Age-related DNA methylation changes in normal human prostate tissues. Clin Cancer Res. 2007;13:3796–3802. doi: 10.1158/1078-0432.CCR-07-0085. [DOI] [PubMed] [Google Scholar]
- 39.Farias E, Ong D, Ghyselinck N, Nakajo S, Kuppumbatti Y, Mira y Lopez R. Cellular retinol-binding protein I, a regulator of breast epithelial retinoic acid receptor activity, cell differentiation and tumorigenicity. J Natl Cancer Inst. 2005;97:21–29. doi: 10.1093/jnci/dji004. [DOI] [PubMed] [Google Scholar]
- 40.Kuppumbatti YS, Bleiweiss IJ, Mandeli JP, Waxman S, Mira-Y-Lopez R. Cellular retinol-binding protein expression and breast cancer. J Natl Cancer Inst. 2000;92:475–480. doi: 10.1093/jnci/92.6.475. [DOI] [PubMed] [Google Scholar]
- 41.Arapshian A, Bertran S, Kuppumbatti YS, Nakajo S, Mira-y-Lopez R. Epigenetic CRBP downregulation appears to be an evolutionarily conserved (human and mouse) and oncogene-specific phenomenon in breast cancer. Mol Cancer. 2004;3:13–25. doi: 10.1186/1476-4598-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Veeck J, Niederacher D, An H, Klopocki E, Wiesmann F, Betz B, et al. Aberrant methylation of the wnt antagonist SFRP1 in breast cancer is associated with unfavourable prognosis. Oncogene. 2006;25:3479–3488. doi: 10.1038/sj.onc.1209386. [DOI] [PubMed] [Google Scholar]
- 43.Donninger H, Vos M, Clark G. The RASSF1A tumor suppressor. J Cell Sci. 2007;120:3163–3172. doi: 10.1242/jcs.010389. [DOI] [PubMed] [Google Scholar]
- 44.Agathanggelou A, Cooper W, Latif F. Role of the ras-association domain family 1 tumor suppressor gene in human cancers. Cancer Res. 2005;65:3497–3508. doi: 10.1158/0008-5472.CAN-04-4088. [DOI] [PubMed] [Google Scholar]
- 45.Lehmann U, Langer F, Feist H, Glockner S, Hasemeier B, Kreipe H. Quantitative assessment of promoter hypermethylation during breast cancer development. Am J Pathol. 2002;160:605–612. doi: 10.1016/S0002-9440(10)64880-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shukla S, Mirza S, Sharma G, Parshad R, Gupta S, Ralhan R. Detection of RASSF1A and RARbeta hypermethylation in serum DNA from breast cancer patients. Epigenetics. 2006;1:88–93. doi: 10.4161/epi.1.2.2679. [DOI] [PubMed] [Google Scholar]
- 47.Suijkerbuijk KP, van der Wall E, Vooijs M, van Diest PJ. Molecular analysis of nipple fluid for breast cancer screening. Pathobiology. 2008;75:149–152. doi: 10.1159/000123853. [DOI] [PubMed] [Google Scholar]
- 48.Euhus D, Bu D, Ashfaq R, Xie X, Bian A, Leitch AM, et al. Atypia and DNA methylation in nipple duct lavage in relation to predicted breast cancer risk. Cancer Epidemiol Biomarkers Prev. 2007;16:1812–1821. doi: 10.1158/1055-9965.EPI-06-1034. [DOI] [PubMed] [Google Scholar]
- 49.Shaw RJ, Liloglou T, Rogers SN, Brown JS, Vaughan ED, Lowe D, et al. Promoter methylation of P16, RARbeta, E-cadherin, cyclin A1 and cytoglobin in oral cancer: quantitative evaluation using pyrosequencing. Br J Cancer. 2006;94:561–568. doi: 10.1038/sj.bjc.6602972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keinan-Boker L, Lerner-Geva L, Kaufman B, Meirow D. Pregnancy-associated breast cancer. Is Med Assoc J. 2008;10:722–727. [PubMed] [Google Scholar]
- 51.Raftery A. Bayesian model selection in social research. Sociol Methodol. 1995;25:111–163. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.