

Abstract
Cerebral cavernous malformations (CCMs) are human vascular malformations caused by mutations in three genes of unknown function: CCM1, CCM2, and CCM3. CCM3, also known as PDCD10 (programmed cell death 10), was initially identified by its mRNA induction by apoptotic stimuli in vitro. However, the in vivo function of CCM3 has not been determined. Here, we describe mice with a deletion of the CCM3 gene either ubiquitously or specifically in certain cell types, including the vascular endothelium, smooth muscle cells, and neurons. Mice with global or endothelial cell-specific deletion of CCM3 die at embryonic stage, exhibiting defects in embryonic angiogenesis. CCM3 deletion reduces VEGFR2 signaling in embryos and derived endothelial cells. CCM3 is recruited to and stabilizes VEGFR2 in response to stimulation by VEGF, and the C-terminal domain of CCM3 is required for the stabilization of VEGFR2. Indeed, the CCM3 mutants found in human patients with a deletion of the C-terminal domain were labile, and unable to stabilize and activate VEGFR2. These results demonstrate that CCM3 regulates vascular development by modulating VEGFR2 signaling.
Keywords: Cerebral cavernous malformations, CCM3, VEGFR2, Vascular development
Introduction
Cerebral cavernous malformations (CCMs) are a common form of vascular malformation that affect the central nervous system vasculature with a prevalence of 0.1-0.5% in the human population (1-3). CCMs arise primarily in the brain as thin-walled, dilated blood vessels that can cause seizures, headaches and stroke in midlife and are often associated with focal hemorrhage. These lesions can occur sporadically, or as a familial form attributable to mutations in three different genes: CCM1 [also known as Krev or Rap1 interacting trapped 1 (KRIT1)] (4, 5), CCM2 [also known as malcavernin or osmosensing scaffold for mitogen-activated protein kinase kinase kinase-3-(s)](6, 7) and CCM3 (also known as PDCD10) (8, 9). Inheritance of mutations in the genes CCM1, CCM2, and CCM3 usually result in truncations of the resulting proteins. These mutations are autosomal dominant for CCM (10, 11).
All three CCM genes are broadly distributed during development (12, 13) and in neuronal and endothelial cells (14-16). Therefore, the cell type in which CCMs function related to a neuronal vascular phenotype is controversial. Studies from zebrafish support the roles of CCM1 and CCM2 in the cardiovascular system. The loss of santa and valentine (corresponding to CCM1 and CCM2, respectively) display dilated heart and vasculature phenotypes, suggesting roles in the regulation of endothelial-specific cellular morphogenesis (17, 18). The in vivo functions of CCM1 and CCM2 associated with the vascular development have been reported in genetically deficient mice. Homozygous mutant mice with a deletion of CCM1 die during early embryonic development due to vascular defects in arterial morphogenesis (19). Moreover, two reports using in vivo endothelial-specific deletion of CCM2 and in vitro endothelial cell culture demonstrate that the disruption of CCM2 in mice results in early embryonic vascular defects through an endothelial cell autonomous mechanism (20, 21).
Mutations in the CCM1, CCM2 and CCM3 genes cannot be clinically distinguished, suggesting that they may function in common or related pathways (10, 22-24). In vitro biochemical analyses indicate that the protein products of these three genes interact to form the CCM complex (10, 11) CCM3 was initially identified by its induction by apoptotic stimuli in a premyeloid cell line (8). CCM3 has been implicated in the mitogen-activated protein kinase (MAPK) pathway in vitro, in part, because of its binding to serine/threonine kinase 25 (STK25) and to the phosphatase domain of Fas-associated phosphatase (11, 25). However, these in vitro studies related to pathogenesis is not clear. Moreover, the in vivo function of CCM3 has not been defined.
Results
Mice with a global deletion of the CCM3 gene die at E8.5 and display defects in VEGFR2-dependent signaling, vasculogenesis and hematopoiesis
To explore the function of the CCM3 gene in vivo, we created CCM3-flox (CCM3lox/lox) mice using homologous recombination so that the endogenous CCM3 gene contains two lox sites flanking exons 4 and 5 (see fig. S1A and B). CCM3lox/lox mice were first mated with β-actin-Cre deleter mice to generate CCM3+/- mice with a deletion of both the targeting region (the exons 4 and 5) and the Neo gene. Mating between CCM3+/- males and females to generate CCM3-KO embryos with a global deletion of CCM3 was verified by PCR genotyping (fig. S1e), Western blotting, and immunostaining using an antibody directed against CCM3 (fig. S1D-E). We did not recover any CCM3-KO pups at birth, indicating that a global deletion of the CCM3 gene resulted in embryonic lethality. Genotype analysis of embryos failed to detect a normal Mendelian distribution of CCM3-KO embryos after E8.5, and no CCM3-KO embryos could be detected after E9.5 (fig. S1F). CCM3-KO embryos were easily identified through the uterine wall because of their smaller size, and pale and anemic appearance relative to the wild-type embryos at E8.0 (Fig. 1A), suggesting defects in vasculogenesis and hematopoiesis. The yolk sac is a primary site of embryonic hematopoiesis, during which the differentiation and maturation of vascular endothelial cell (vasculogenesis) as well as remodeling (angiogenesis) occur (26). Macroscopic examination of the yolk sac revealed the presence of a dense capillary plexus in the wild-type yolk sac, but complete loss of visible blood cell-filled vessels in the CCM3-KO yolk sac, as determined by hematoxylin and eosin stained cross sections (Fig. 1B). Histological analysis (H&E staining) also revealed a thinner myocardium and a reduction of the trabecular network within the heart, indicating CCM3-KO mice suffer from general defects in the cardiovascular system (Fig. 1C).
Fig. 1. Mice with global deletion of the CCM3 gene (CCM3-KO) die at E8.5 and display defects in VEGFR2 signaling, vasculogenesis and hematopoiesis.
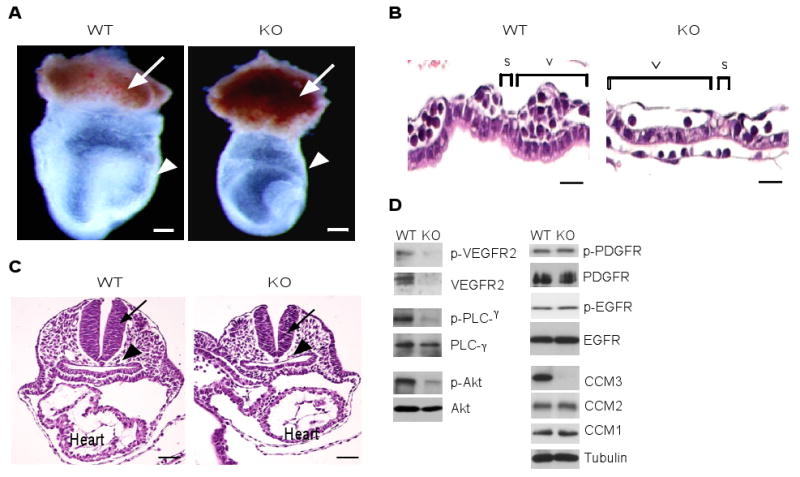
(A) Appearance of wild type and CCM3-KO embryos at E8.0. Embryos were freshly dissected, then photographed. The ectoplacental cone, yolk sac and embryo (inside) are indicated. Scale bar, 200 μm. (B) H&E stained cross-sections of the yolk sac. Yolk sacs from WT and CCM3-KO mice were embedded, followed by H&E staining. The cross-sectional length of the yolk sac vessel, adjacent and parallel to the endoderm layer (v), and the empty space between the vessels (s) are indicated. Scale bar, 20 μm. (C) H&E staining of embryos. Neural tube and dorsal aorta are indicated by an arrow and an arrowhead, respectively. The heart is also labeled. Scale bar, 100 μm. (D) Defects of VEGFR2-dependent signaling in CCM3-KO embryos. Whole embryos of wild type and CCM3-KO were subjected to Western blotting with various antibodies against VEGFR2 signaling molecules, PDGFR, EGFR, and the CCM proteins, as indicated. 5 other embryos of wild type or KO gave similar results. The relative abundance of VEGFR2, p-VEGFR2, the ratios of p-PLC-γ/PLC-γ and p-Akt/Akt are shown, with WT group as 1.0. Data are mean ± SEM (n=6). *, p<0.05.
We then examined mRNA abundance for factors and receptors involved in vasculogenesis and hematopoiesis by quantitative RT-PCR. Although angiogenic factors, including VEGF and angiopoietin-2, were not affected by a deletion of CCM3 (fig. S1G), early vascular and hematopoietic marker genes including vascular endothelial growth factor receptor 2 (VEGFR-2), globin transcription factor1 (GATA-1) and SCP-like extracellular protein 1 (SCL1) [also T-cell acute lymphocytic leukemia 1 (TAL-1)] were reduced. PDGFR or EGFR were not altered, or were only slightly increased (fig. S1G), suggesting a specific expression defect of vasculogenic and hematopoietic genes. The endothelial marker genes Tie-2, PECAM-1 (CD31), Notch1 and Notch-dependent gene Hey-1 were also reduced (fig. S1G), even after normalization with endothelial cell marker PECAM-1 (fig. S1H).
We then examined the abundance and activation of VEGFR2, a receptor critical for both vasculogenesis and hematopoiesis (27), and its downstream signaling molecules PLC-γ and Akt by Western blotting. Total protein abundance and phosphorylation of VEGFR2 were both reduced. Although total protein abundance of PLC-γ and Akt was not altered, phosphorylations of these proteins was reduced. In contrast, total and phosphorlation of platelet growth factor receptor (PDGFRβ and epithelial growth factor receptor (EGFR) were not changed by the deletion of CCM3 (Fig. 1D). Mutations in the CCM1, CCM2 and CCM3 genes cannot be clinically distinguished, suggesting that they may function in common or related pathways. The three CCM proteins form a functional complex in which CCM2 bridges CCM1 and CCM3 (11, 25). Therefore, we examined the effect of CCM3 deletion on the abundance of CCM1 and CCM2. However, a deletion of CCM3 had no effect on the abundance of CCM1 or CCM2 during embryogenesis (Fig. 1D). These results indicate that CCM3 may specifically regulate the abundance and activity of VEGFR2 in the embryo.
Mice with a vascular endothelial deletion of the CCM3 gene display angiogenesis defects and reduced VEGFR2 signaling
CCM3 is broadly distributed throughout development (13) and in neuronal and endothelial cells (28). The cell type in which CCMs functions to cause a neuronal vascular phenotype is unknown. To address this question, the CCM3 gene was specifically inactivated in neuronal cells, smooth muscle cells or endothelial cells by mating CCM3lox/lox with the Nestin-Cre deleter (29), SM22α-Cre deleter (30) and Tie2-Cre deleter mice, respectively. We verified the cell-type specificity of the Nestin-Cre and SM22α-Cre deleters by using Cre reporters, PCR genotyping as well as by immunostaining with anti-CCM3 (fig. S2 and S3). Mice with a CCM3 deletion in neuronal cells or smooth muscle cells were viable after birth. A neuronal or smooth muscle cell-specific deletion of CCM3 had no effects on retinal vasculature growth in these mice (fig. S2 and S3). Cre in Tie2-Cre deleter mice is under the control of the Tie2 endothelial-specific promoter (31), and is specifically expressed in vascular endothelial cells, not in hematopoietic lineages (32). The specificity of Tie2-Cre for endothelial cell was further confirmed by a genetic Cre reporter ROSA26YFP line in which Cre-mediated recombination leads to the expression of yellow fluorescence protein (YFP) ROSA26-YFP/Tie2-Cre demonstrated that Tie2-Cre induced YFP expression specifically in the vascular endothelium in embryos (fig. S4A). CCM3 was effectively deleted in vascular endothelium, as determined by genotyping (fig. S4B) and immunostaining of yolk sac with anti-CCM3 (fig. S4C). CCM3lox/+:Tie2-Cre animals were born in the expected Mendelian ratio and exhibited normal life spans compared to CCM3lox/lox. However, we did not recover any pups with CCM3lox/lox:Tie2-Cre at birth, indicating that deletion of CCM3 in vascular endothelial cells also caused embryonic lethality. Genotype analysis of embryos derived from the mating of CCM3lox/lox with Tie2-Cre showed a normal Mendelian distribution of CCM3lox/lox:Tie2-Cre (CCM3-ecKO) embryos after E9.5, and no CCM3-ecKO embryos could be detected after E10.5 (fig. S4D). These data suggest a specific role for CCM3 in endothelium during vascular development.
CCM3-ecKO embryos were easily identified by pale and anemic appearance compared to wild-type embryos, even at E8.5 when wild type and CCM3-ecKO embryos were not different in size (fig. S5A). Indeed, staining with an endothelial cell marker CD31 of E8.5 embryos revealed vascular defects in angiogenesis and major vascular remodeling, as shown by an analysis of the yolk sac (fig. S5B), telencephalic plexus and intersomitic vasculature of embryos (fig. S5C). WT embryo heads contained a highly organized vascular system. Blood vessels in ecKO embryos heads did form, but did not remodel into a finely branched tree, and were often arrested at the uniformly sized primary capillary plexus stage, indicating that angiogenesis was delayed. In the trunk region, the wild type intersomitic vessels were arranged in segments with a highly arborized capillary network. In ecKO embryos, intersomitic vessels lacked a normal segmented pattern, and were poorly organized and less branched (fig. S5C). In H&E stained cross sections of the yolk sac at E8.5, CCM3flox/flox yolk sacs contained blood-filled vessels spaced at regular intervals which were similar to WT. However, CCM3-ecKO yolk sacs had fewer blood vessels. The cross-sectional length of the yolk sac vessel, adjacent and parallel to the endoderm layer (v), the empty space between vessels (s) and the total yolk sac vessel length (t) were measured, and these distances were used to calculate the vessel density (Fig. 2A). Quantitative analyses indicated that the vessel size in ecKO was increased, concomitant with a decrease in vessel density (Fig. 2, B and C). The yolk sac contains two layers of cells, the outside visceral endoderm layer and the inner vessels which surround blood cells. Endothelial cells exhibited positive staining for VEGFR2 whereas blood cells were positively stained for the red blood cell marker Ter-119 (Fig. 2D). CCM3-ecKO yolk sacs exhibited reduced VEGFR2 staining and fewer endothelial and blood cells, suggesting that deletion of CCM3 in endothelial cells causes defects in vasculogenesis and hematopoiesis in the yolk sac. These phenotypic changes are reminiscent of those in mice with a genetic deletion of VEGF or VEGFR2 (26, 33-35). Consistently, proliferation of both endothelial cell and blood cell as measured by phospho-histone 3 was reduced in CCM3-ecKO yolk sac (Fig. 2, E and F). Similarly, CCM3-ecKO embryos exhibited reduced VEGFR2 signaling as well as VEGFR2-dependent gene expression of vascular and hematopoietic markers (fig. S6A and B).
Fig. 2. Mice with a vascular endothelial deletion of the CCM3 gene (CCM3-ecKO) display defects in angiogenesis and hematopoiesis.
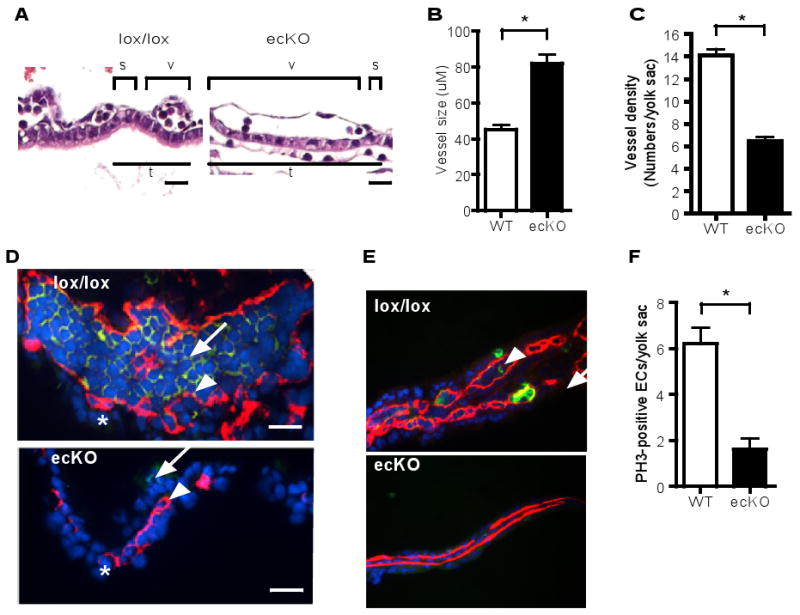
(A to C). H&E stained cross-sections of the yolk sac at E8.5. Yolk sacs from CCM3lox/lox and CCM3-ecKO mice were embedded, followed by H&E staining. The cross-sectional length of the yolk sac vessel, adjacent and parallel to the endoderm layer (v) and the empty space between the vessels (s) as well as total vessel length (t) are measured. The vessel size (μM) and vessel density (number of vessels/yolk sac) are quantified. Scale bar, 20 μm. (D). CCM3-ecKO yolk sacs at E8.5 exhibit reduced VEGFR2 staining. Vessel endothelial cells exhibit positive staining for VEGFR2 (red), whereas blood cells exhibit positive staining for Ter-119 (green). Nuclei were counterstained by DAPI (blue). Scale bar, 10 μm. (E-F) CCM3-ecKO yolk sacs at E8.5 exhibit reduced proliferation. Yolk sacs were stained for phospho-histone-3 (PH3, green) and endothelial cell marker CD31 (red). Blue: DAPI. Scale bar, 100 μm. PH3-positive endothelial cells are indicated by an arrow, with quantification shown in F. PH3-positive blood cell by an arrowhead. Data are mean ± SEM (n=5). *, p<0.01.
A vascular endothelial deletion of the CCM3 gene disrupts vascular integrity in mice
Deletion of the other CCM components CCM1 and CCM2 in mice affects cell-cell junctions and vascular integrity (19-21). To determine if CCM3 deletion has similar effects on vascular integrity, we harvested stage-match wild type and ecKO embryos at E9.0. CCM3-ecKO exhibited vascular defects as indicated by lack of large vitelline arteries in yolk sac (Fig. 3A). However, CCM3-ecKO had no developmental defects in neural tube closure (Fig. 3B). CCM3-ecKO embryos had small heads with enlarged pericardial sac, possibly due to vascular defects in brain and heart. Histological analysis (Fig. 3B) and immunostaining for CD31 (Fig. 3C) revealed that the dorsal aorta and cardinal vein were dilated. Both dorsal aorta and cardinal vein were disorganized and appeared to be discontinuous, indicating the disrupted integrity of the vessels. Mutant embryos also had thinner myocardia and a less developed trabecular network within the heart. We also found that the endocardium (stained by anti-CD31) was dissociated from the myocardium stained with a smooth muscle cell marker smooth muscle α-actin (SMA) in hearts of CCM3-ecKO mice (Fig. 3D), suggesting that a disrupted interactions between endothelial cells and smooth muscle cells in heart. Similarly, the yolk sacs of CCM3-ecKO embryos showed reduced or disrupted cell-cell junctions as measured by coimmunostaining of anti-CD31 with anti-ZO-1 (a tight junctional protein) or with anti-VE-cadherin (an adherent junctional protein) (fig. S7). These phenotypes are similar to the mouse phenotype with the genetic deletions of CCM1 and CCM2 (19-21).
Fig. 3. A vascular endothelial deletion of the CCM3 gene disrupts vascular integrity in mice.
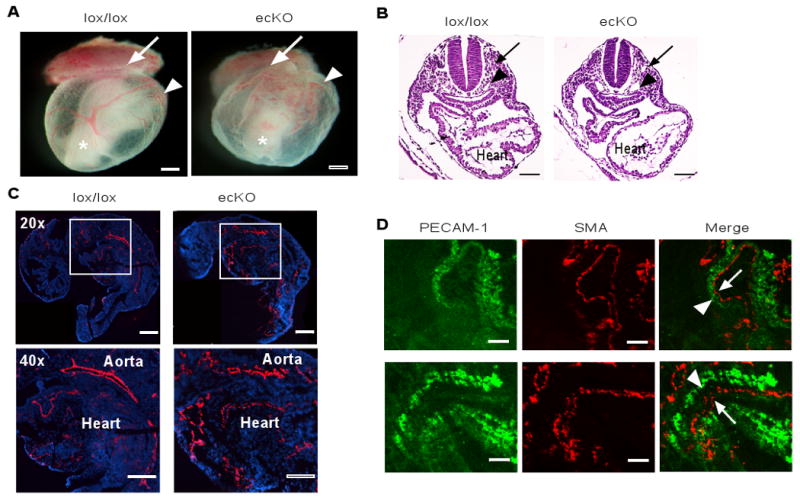
(A) Appearance of WT and CCM3-ecKO embryos at E9.0. Embryos were freshly dissected and photographed. The ectoplacental cone, yolk sac and embryo (inside) are indicated. Scale bar, 200 μm. (B) Histological analysis (H&E staining) of embryos at E9.0. The dorsal aorta, cardinal vein and heart are indicated. Scale bar, 100 μm. (C) Immunostaining (CD31, red) of embryos at E9.0. Blue: DAPI. The aorta and heart are indicated. Scale bar, 200 μm. (D) Co-staining of embryos with antibody against CD31 and SMA. Dissociation of the endocardium (CD31-positive) from the myocardium (SMA-positive) in the hearts of CCM3-ecKO mice at E9.0 is indicated by an arrow. 5 other embryos of wild type or KO gave similar results. Scale bar, 50 μm.
CCM3 deletion in endothelial cell specifically blocks VEGFR2-dependent signaling and angiogenesis
To define the mechanism by which CCM3 regulates VEGFR2 signaling in endothelial cells, we isolated primary brain and lung endothelial cells from CCM3lox/lox mice, and endothelial cells were confirmed by immunostaining with antibody against vWF antibody and by Ac-LDL uptake (36). CCM3lox/lox endothelial cells had no obvious growth phenotype compared to normal mouse lung and brain endothelial cells. The CCM3 gene was effectively deleted by infection with adenovirus expressing Cre, but not by adenovirus expressing LacZ (Fig. 4A). Quantitative RT-PCR revealed that deletion of CCM3 in cultured endothelial cells had no effect on the mRNA abundance of VEGFR2 (fig. S8A). We then checked the effects of CCM3 deletion on protein abundance and VEGF response. Similar to the observation from embryos, CCM3 deletion specifically reduced the abundance of VEGFR2 protein (Fig. 4A). Therefore, loss of CCM3 in endothelial cells seems to primarily affect VEGFR-2 protein stability. VEGF-induced activation of VEGFR2, PLC-γ, Akt and ERK1/2 were determined by Western blotting with phospho-specific antibodies. Phosphorylation of VEGFR2, p-PLC-γ, p-Akt and ERK1/2 were reduced by CCM3 deletion (Fig. 4A). In contrast, CCM3 deletion had no effect on the basic fibroblast growth factors (bFGF) induced phosphorylation of PLC-γ, Akt and ERK1/2, suggesting a specific role of CCM3 in VEGF signaling (fig. S8B). Similar results were obtained from primary human endothelial cell (HUVEC) with CCM3 knockdown by siRNA (Fig. 4B and fig. S8C). These results suggest that the effect of CCM3 on VEGFR2 in embryos is endothelial cell autonomous.
Fig. 4. CCM3 deletion in endothelial cells specifically blocks VEGF-induced VEGFR2 signaling and angiogenesis.
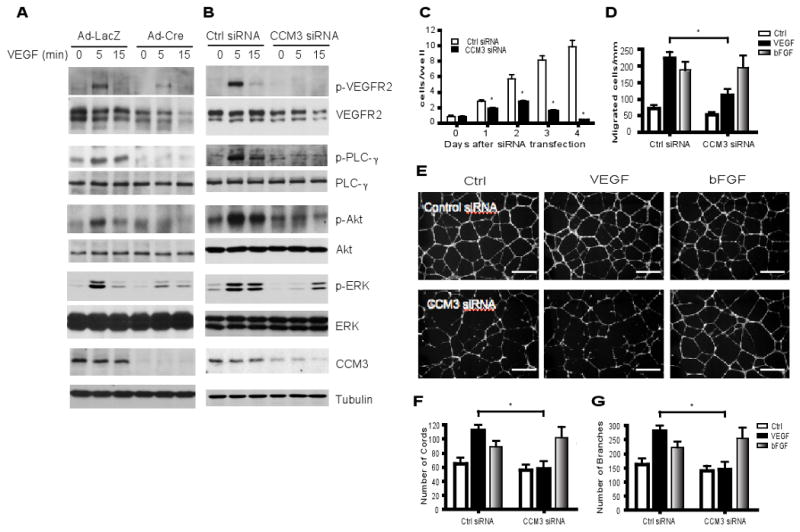
(A) Primary mouse brain endothelial cell (MBEC) were isolated from CCM3lox/lox mice and infected with adenovirus expressing LacZ or Cre recombinase. Cells were cultured overnight in 0.5% FBS followed by VEGF treatment (10 ng/ml) for the indicated times. Cell lysates were subjected to Western blotting with antibodies against VEGFR2 signaling molecules, as indicated. The relative abundance of VEGFR2, the ratios of p-PLC-γ/PLC-γ, p-Akt/Akt and p-ERK/ERK are shown, with untreated control group set as 1.0. Data are mean ± SEM from three independent blots. *, p<0.05. (B to G) Primary human endothelial cells (HUVEC) were transfected with a control siRNA or CCM3 siRNA for 48 hours. B. Cells were serum-starved for 24 hour and treated with VEGF as indicated. Activation of VEGFR2-dependent signaling was determined by Western blot and quantified as in A. Data are mean ± SEM from three independent blots. *, p<0.05. C. Cells were cultured in the presence of VEGF for the indicated times and cell numbers were counted. D. A transwell assay was performed in response to VEGF or bFGF (10 ng/ml each), and migratory cells were quantified. Data are mean ± SEM from three independent experiments. *, p<0.01. (E to G) Cells were subjected to a Matrigel tube formation assay in the presence of VEGF or bFGF (10 ng/ml). Scale bar, 50 μm. The number of tubes and number of branches per field were quantified. Data are mean ± SEM from four independent experiments. *, p<0.05.
Next, we examined the effect of CCM3 knockdown on VEGF-dependent in vitro angiogenesis which involves cell proliferation, migration, and morphology remodeling processes. CCM3 knockdown in HUVEC significantly reduced endothelial cell proliferation (Fig. 4C). Cell numbers declined on day 3 post-transfection in CCM3 knockdown group, suggesting that cells may undergo apoptosis. This was confirmed by greater numbers of condensed nuclei in CCM3-knockdown endothelial cells compared to non-transfected cells as measured by DAPI staining (fig. S9, A and B). Endothelial cell apoptosis as induced by CCM3 knockdown was rescued by coexpression of VEGFR2 (fig. S9C). CCM3 knockdown reduced cell migration in response to VEGF as measured by a transwell assay without effects on bFGF-induced chemotaxis (Fig. 4D). Cord formation of HUVEC on a Matrigel is a commonly used in vitro model for angiogenesis (37). CCM3 knockdown significantly inhibited VEGF-induced endothelial cell cord formation, as assessed by the decreased number of cord and branch points (Fig. 4E-G). Consistent with the role of CCM3 in bFGF signaling, CCM3 knockdown had no effect on bFGF-induced endothelial cell cord formation. Finally, we examined the effects of CCM3 knockdown on cell-cell junctions which are important for vascular integrity (38). To this end, CCM3 was knocked down in human dermal microvessel endothelial cells (HDMEC), which form both tight junction and adherent junction in culture. Knockdown of CCM3 disrupted both tight and adherent junctions as measured by staining with ZO-1 and VE-cadherin, respectively (fig. S10). CCM3 knockdown induced internalization of VE-cadherin (fig. S10C and S12).
CCM3 is recruited to and stabilizes VEGFR2 in response to VEGF
To define the mechanism by which CCM3 regulates VEGFR2 signaling, we first determined the association of CCM3 and VEGFR2 in endothelial cells in response to VEGF by a coimmunoprecipitation assay with anti-VEGFR2, followed by Western blotting with anti-CCM3. Association of CCM3 with VEGFR2 could be weakly detected in unstimulated endothelial cells, but was enhanced by VEGF treatment (Fig. 5A), suggesting that CCM3 and VEGFR2 form a complex in endothelial cells in response to VEGF.
Fig. 5. CCM3 is recruited to and stabilizes VEGFR2 in response to VEGF.
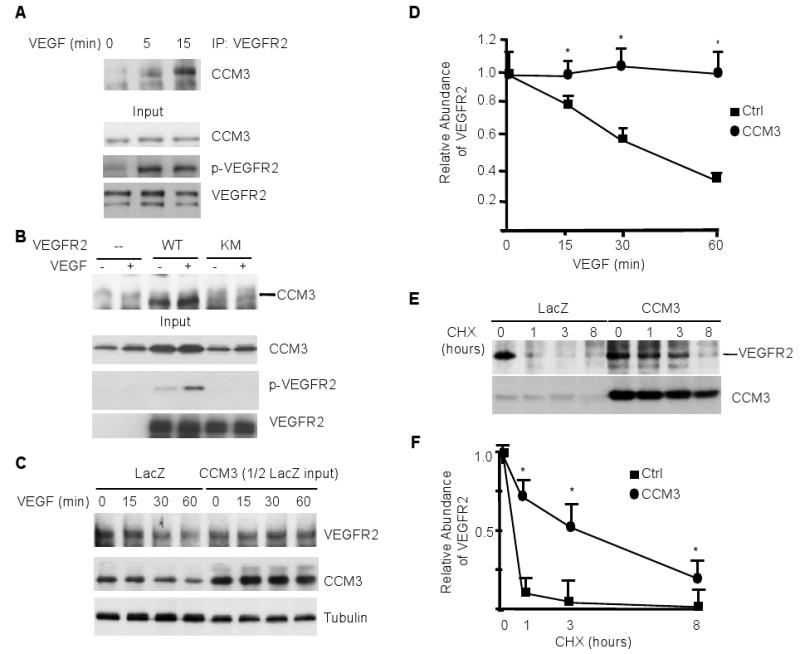
(A) CCM3 is recruited to VEGFR2 in response to VEGF. HUVEC were serum-starved for 12 hours, followed by treatment with VEGF for the indicated times. p-VEGFR2, total VEGFR2 and CCM3 in the input were determined by Western blotting with the respective antibodies. Cells were treated with VEGF (10 ng/ml for 0, 5 and 15 min) and the association of endogenous CCM3 with VEGFR2 was determined by immunoprecipitation with anti-VEGFR2 followed by Western blotting with anti-CCM3. (B) CCM3 binds to an active form of VEGFR2. Flag-tagged CCM3 was coexpressed with VEGFR2-WT or KM (a kinase-inactive mutant) in HEK 293T cells. The association of CCM3 with VEGFR2 was determined by immunoprecipitation with anti-VEGFR2 followed by Western blotting with anti-Flag. (C) CCM3 prevents VEGF-induced VEGFR2 downregulation. HUVEC were infected with adenovirus expressing LacZ or CCM3. 24 hours post-infection, cells were cultured and treated with VEGF for the indicated times. The VEGFR2 and CCM3 proteins were detected as in A. Note that the total protein loaded in the CCM3-expressing cell lysate was 50% less than the LacZ group. The relative levels of the VEGFR2 protein was quantified by setting the untreated group as 1.0. Data are mean ± SEM from three independent blots. *, p<0.05. D-E. CCM3 stabilizes VEGFR2. HUVEC were infected as in C followed by treatment with cycloheximide (10 μg/ml) for the indicated times. Total VEGFR2 and CCM3 protein were determined as in (D). The relative abundance of VEGFR2 protein were quantified by taking the untreated group as 1.0, and the half-life of VEGFR2 was determined in E. Data are mean ± SEM from three independent experiments. *, p<0.05.
To further define the interactions between CCM3 and VEGFR2, we used an overexpression approach in human embryonic kidney (HEK) 293T cells, which lack endogenous VEGFR2. We first determined if CCM3 associates with the active form of VEGFR2. Flag-tagged CCM3 was coexpressed with wild type VEGFR2 or a kinase-inactive form (KM). Association of CCM3 with VEGFR2 was determined by coimmunoprecipitation with anti-VEGFR2, followed by Western blotting anti-Flag. As we have shown previously (39), overexpression of VEGFR2-WT, but not the kinase-inactive form, induced autophosphorylation of VEGFR2 (Fig. 5B). Consistent with the VEGF-induced association of CCM3 with VEGFR2, CCM3 specifically associated with the active form of VEGFR2, which was further enhanced by VEGF treatment (Fig. 5B). In the same assay, CCM3 did not interact with platelet-derived growth factor β (PDGFβ) receptor (PDGFRβ), receptor tyrosine kinase that is closely related to VEGFR2 (fig. S11A). We further confirmed the binding of CCM3 with VEGFR2 in an in vitro pulldown assay using GST-CCM3. GST-CCM3, but not GST, pulled down VEGFR2 from VEGFR2-expressing HEK 293T cell lysates (fig. S11B). These data suggest that CCM3 specifically associates with VEGFR2.
The results from both the embryos and isolated endothelial cells showed that the basal abundance of VEGFR2 was reduced by the deletion of CCM3. CCM3 deletion in endothelial cells also facilitated VEGF-induced decrease in the abundance of VEGFR2. These data support the notion that CCM3 enhances the stability of the VEGFR2 protein. To further test this model, we examined whether CCM3 overexpression would prolong the half-life of the VEGFR2 protein. We first determined if CCM3 delayed the VEGF-induced downregulation of VEGFR2. Mouse brain capillary endothelial cells (MBEC) were infected with adenovirus expressing LacZ or CCM3 and cells were treated with VEGF for various time periods (0 to 60 min). In LacZ-expressing endothelial cells, VEGF reduced total VEGFR2 abundance by 60% at 60 min, as compared with the results observed with the untreated cells. CCM3 expression, in contrast to that observed in CCM3 deletion endothelial cells, led to an increase in the basal abundance of VEGFR2. Therefore, only half amount of CCM3-expressing cell lysate was loaded as input. The results showed that the expression of CCM3 in endothelial cells prevented VEGF-induced downregulation of VEGFR2 (Fig. 5C). To determine whether CCM3 stabilizes VEGFR2 by increasing the half-life of VEGFR2, cells were treated with the protein synthesis inhibitor cycloheximide (CHX) for various time periods (0 to 8 hours). VEGFR2 was labile, whereas CCM3 was very stable, with a half-life close to 24 hours in uninfected endothelial cells (fig. S11C). We then compared the half-life of VEGFR2 in LacZ and CCM3 adenovirus-infected cells. The half-life of VEGFR2 was prolonged from 0.5 hour in LacZ-expressing cells to 4 hours in CCM3-expressing cells (Fig. 5, D and E).
CCM3 mutations found in human patients destabilize VEGFR2 and inhibit VEGFR2 signaling
Several CCM3 mutations, causing a stop codon or a frameshift that generate a N-terminal truncated form of the CCM3 protein, have been reported to be associated with CCM in humans (8). Three common mutations are listed in fig. S12A, containing various numbers (95, 117 and 195) of N-terminal residues (therefore named as CCM3-95, CCM3-117 and CCM3-195, respectively) (fig. S12A). We reasoned that these mutations affect CCM3 function in VEGFR2 signaling. To test this hypothesis, expression plasmids for the three CCM3 truncation mutations were generated (fig. S12B) and transfected into HEK 293T cells. The abundance of all the truncated CCM3 mutants were low, but were enhanced by the presence of the proteasomal inhibitor MG132 (fig. S12C), suggesting the mutant CCM3 proteins are unstable. We then examined the effects of the CCM3 mutants on VEGFR2 signaling in the absence of MG132. Coexpression of CCM3-WT enhanced both the expression and activity of VEGFR2 as determined by Western blotting for total and phospho-VEGFR2 as well as phospho-PLC-γ. In contrast, cotransfection of VEGFR2 with CCM3 mutants led to a reduction of VEGFR2 abundance and activity (Fig. 6A). CCM2, however, had no effects on VEGFR2 protein level and activity in the same assay (Fig. 6B).
Fig. 6. CCM3 mutants found in human patients destabilize VEGFR2 and inhibit VEGFR2 signaling.
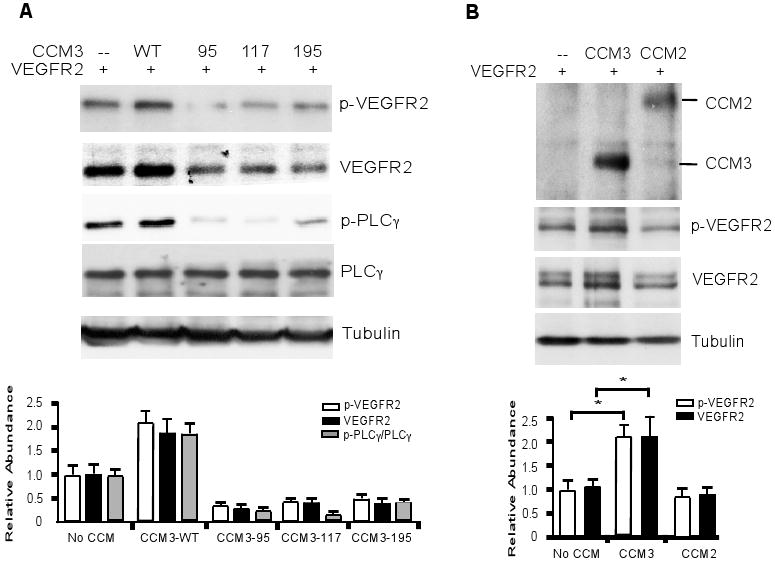
(A) CCM3 mutants do not activate VEGFR2 signaling. VEGFR2 was cotransfected with various CCM3 mutants. Phosphorylation of VEGFR2 and p-PLC-γ were determined. The relative abundance of p-VEGFR2, VEGFR2, as well as the ratio of p-PLC-γ/PLC-γ are shown, with each control group set as 1.0. Data are mean ± SEM from four independent blots. *, p<0.05. (D) CCM3, but not CCM2, stabilizes and activates VEGFR2. VEGFR2 was cotransfected with CCM3 or CCM2. Phosphorylated and total VEGFR2 were determined and quantified as in (C). The relative abundance of p-VEGFR2 and VEGFR2 are shown, with each control group set as 1.0. Data are mean ± SEM from four independent blots. *, p<0.05.
CCM3-WT is colocalized with VEGFR2 on membrane whereas CCM3 N-terminal truncate mutants induce VEGFR2 endocytosis
We next investigated how CCM3 regulates VEGFR2 stability. The observation that CCM3 knockdown induces internalization of VE-cadherin (fig. S10B) led us to examine if CCM3 regulates internalization of VEGFR2. Previous studies suggest a critical role of VE-cadherin as a co-receptor of VEGFR2 that is involved in VEGF signaling (40). VE-cadherin and VEGFR2 together with PECAM-1 comprise a mechanosensory complex in endothelial cells in which PECAM-1 directly transmits mechanical force, VE-cadherin functions as an adaptor, and VEGFR2 activates phosphatidylinositol-3-OH kinase (41). Moreover, VEGFR2 and VE-cadherin both are key mediators of vascular integrity (42). Indeed, VE-cadherin and VEGFR2 showed colocalization on endothelial cell membrane, and knockdown of CCM3 induced internalization of both VE-cadherin and VEGFR2 (fig. S13). We then examined if CCM3 is colocalized with VEGFR2 on endothelial cell membrane, and VEGF induces its internalization, leading to degradation of VEGFR2 (43-47). We found that CCM3 colocalized with VEGFR2 on the membrane in resting endothelial cells, but in intracellular vesicles upon VEGF treatment (fig. S14A), consistent with their association from the immunoprecipitation assay. Quantitative analyses indicated that VEGF treatment significantly induced intracellular colocalization of VEGFR2 with CCM3 (fig. S14, B and C). We next determined if the N-terminal truncated form of a human CCM3 mutant causes VEGFR2 degradation. VEGFR2 was coexpressed with CCM3-WT (aa 1-212), CCM3-C (aa 92-212) or CCM3-N (aa 1-95) in BAEC, and cellular localization of VEGFR2 was determined by indirect immunofluorescence microscopy. VEGFR2 colocalized with CCM3-WT and CCM3-C on the cytoplasmic membrane. However, coexpression of CCM3-N caused VEGFR2 to translocate from the cytoplasmic membrane into intracellular vesicles (Fig. 7A). Quantitative analyses for the colocalization of CCM3-VEGFR2 based on confocal images indicated that CCM3-N expression significantly induced intracellular colocalization of VEGFR2 with CCM3 compared to CCM3-WT (Fig. 7, B and C). Because CCM3-N, which mimicks the human mutant CCM3, is unstable and destabilizes VEGFR2 upon coexpression, we reasoned that CCM3-N induces VEGFR2 translocation into endosomes. Indeed, we found that CCM3-N binds to VEGFR2 in a coimmunoprecipitation assay (Fig. 7D), and coexpression of CCM3-N with VEGFR2 caused a reduction of VEGFR2 on the cell surface with concomitant increase of intracellular colocalization with an endocytic marker GFP-FYVE, in which GFP was fused to a phosphatidylinositol 3-phosphate-binding FYVE domain of the late endosomal protein Hrs (Fig. 7E). These data suggest that CCM3-N facilitates internalization and destabilization of VEGFR2.
Fig. 7. CCM3-WT colocalizes with VEGFR2 on plasma membrane whereas CCM3 N-terminal domain induces VEGFR2 endocytosis.
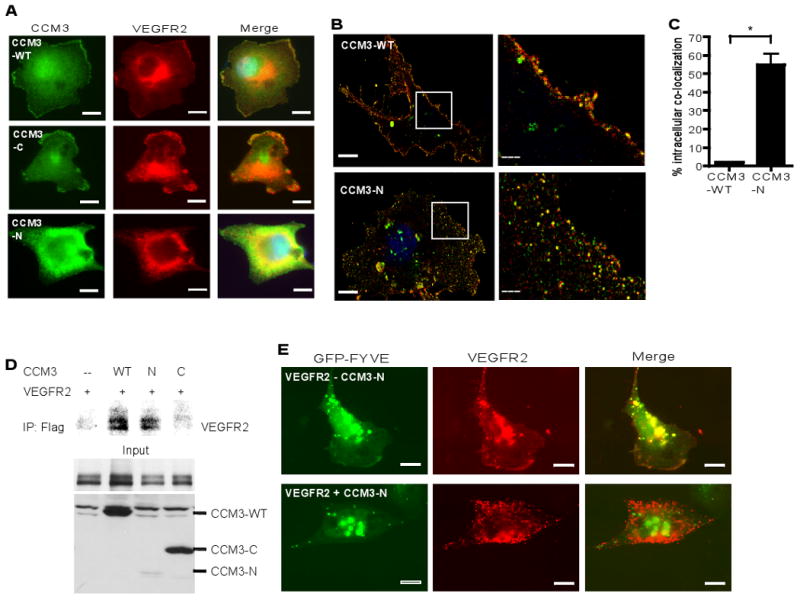
(A) The N-terminal domain of CCM3 induces VEGFR2 endocytosis. VEGFR2 was coexpressed with CCM3-WT (aa 1-212), CCM3-C (aa 92-212) or CCM3-N (aa 1-95) into BAEC, and cellular localization of VEGFR2 was determined by indirect immunofluorescence microscopy with anti-Flag (CCM3, green) and anti-VEGFR2 (red). Scale bar, 20 μm. (B) A higher magnification of confocal images for colocalization of VEGFR2 with CCM3-WT and CCM3-N are shown. An insert from CCM3-WT and CCM3-N is shown on the right. (C) Intracellular colocalization of CCM3-VEGFR2 (yellow dots) was quantified using the Matlab software by a third person in a blind fashion and percentages (yellow dots/total green, red and yellow dots) are shown. Scale bar, 5 μm. A total of 10 cells from each group were analyzed. Data are mean ± SEM (n=10). *, p<0.01. (D) CCM3-N binds to VEGFR2. VEGFR2 was coexpressed with CCM3-WT, CCM3-C or CCM3-N in HEK 293T cells, and association of VEGFR2 with CCM3 proteins was determined by coimmunoprecipitation with anti-Flag, followed by Western blotting with anti-VEGFR2. (E) CCM3-N induces VEGFR2 to endocytic vesicles. VEGFR2 was coexpressed in BAEC with GFP-FYVE, a plasmid that contains two tandem-arranged FYVE domains of the late endosomal protein Hrs, in the absence or presence of CCM3-N. Colocalization of VEGFR2 (red) with GFP-FYVE (green) was analyzed. Representative images from 10 cells in each group are shown. Scale bar, 20 μm.
Discussion
The studies in this report reveal that CCM3 plays a different role from that of CCM1 and CCM2. The first difference is that mice with CCM3 global deletion die at E8.0-E8.5, which is much earlier than CCM1 or CCM2-KO mice. Detailed yolk sac analyses uncovered defects in vasculogenesis and hematopoiesis in CCM3-KO embryos. These data suggest that CCM3 may play a critical role at an earlier stage of embryogenesis than CCM1 and CCM2, which primarily are involved in vascular remodeling and arterial-vein differentiation. Consistent with this vascular phenotype, biochemical analyses support that CCM3 complexes with VEGFR2 and regulates its activity, one of the earliest signaling pathways regulating vascular morphogenesis. In contrast, CCM1 and CCM2 play roles in vascular remodeling and arterial differentiation, most likely by regulating Notch signaling as well as endothelial cell junctions (19-21). Although mutations in the three genes cannot be clinically distinguished, infant carriers of CCM mutations sometimes suffer severe brainstem hemorrhage (48) and childhood hemorrhages correlate with CCM3 mutations (49, 50). Autosomal dominant mutations for CCM genes are seen in 70-80% of familial patients, approximately 40%, 30% and 10% for CCM1, CCM2, and CCM3 respectively (10, 51, 52) with CCM3 being the rarest. This raises the possibility that CCM3 mutations cause early onsets in human lesions, and some of the infants with CCM3 mutations may die in utero.
Our data suggest a following model (Fig. 8): CCM3 contains two functional domains, and the N-terminal domain is for endocytosis and the C-terminal domain for the cytoplasm membrane localization. CCM3 is present in a folded inactive state in resting endothelial cells. In response to VEGF, CCM3 binds to and stabilizes VEGFR2, leading to enhanced activation of the receptor and downstream signaling. The human CCM3 mutants lacking the C-terminal domain fail to retain VEGFR2 on membrane, or facilitate VEGFR2 endocytosis, leading to defects in VEGFR2 signaling and vascular development. Consistently, deletion of CCM3 and VEGFR2 in mice showed a similar phenotype of vascular defects and embryonic lethality (26, 33). The CCM3 mutants detected in human patients were found to be labile and incapable of enhancing VEGFR2 signaling, suggesting a potential mechanism by which CCM3 mutation results in vascular malformation in CCM patients. The reason why CCM is restricted to brain in human adults is not fully understood. This is likely attributed to a unique structure of the neurovascular units in which endothelial cells are surrounded by vascular smooth muscle cells, neurons, astrocytes and macroglia (53). Our findings suggest further investigation on whether the VEGFR2-CCM3 pathway plays a role in the pathogenesis of CCM related disease in humans.
Fig. 8. A model for the role of CCM3 in VEGFR2 signaling.

CCM3 is present in a folded inactive state in resting endothelial cells. In response to VEGF, CCM3 binds to and stabilizes VEGFR2, leading to an enhanced activation of the receptor and downstream signaling. The human CCM3 mutants lacking the C-terminal domain facilitate VEGFR2 endocytosis, leading to defects in VEGFR2 signaling and vascular development.
In vitro studies have indicated that each CCM protein appears to have its own unique function by associating with different signaling proteins, transducing distinct pathways in endothelial cells. Interaction of CCM1 with ICAP1α (integrin cytoplasmic associated protein 1α) has been described (54), providing a link between the CCM complex and integrin β1 signaling. CCM2, which functions as scaffolding protein, regulates the osmotic stress-induced Rac-MEKK3-MKK3-p38MAPK signaling pathway (55). CCM2 also mediates TrkA-specific signaling (56). CCM3 can associate with the serine/threonine protein kinase MST4 to modulate ERK signaling, hereby promoting cell survival (25). In contrast, overexpression of CCM3 also caused endothelial cell apoptosis after serum starvation (57). Our studies of both the CCM3-deletion embryos and derived endothelial cells clearly demonstrate a critical role for CCM3 in VEGFR2-dependent angiogenic signaling in endothelial cells.
Emerging evidence now supports a signaling complex of the three CCM proteins is formed in which CCM2 appears to form the central hub for the complex (10, 11). CCM1, CCM2 and CCM3 seem to share a common function in regulating vascular integrity. CCM1 is an effector of Rap1, a guanosine triphosphatase that maintains the endothelial cell integrity. Specifically, Rap1 increases CCM1 targeting to endothelial junctions where CCM1-CCM2 complex directly associates with the junctional proteins VE-vadherin-β-catenin, suppressing stress fiber formation and stabilizing junctional integrity (58-60). CCM2 can directly associate with and inhibit RhoA activity, decreasing stress fiber formation and endothelial permeability (19). Therefore, a Rap1-CCM1/CCM2-RhoA pathway has been proposed to regulate vascular integrity. Although heart of glass receptor (HEG) in zebrafish has been implicated as an upstream regulator of CCM1/CCM2 signaling by directly binding to CCM2 (21), it is not clear if HEG regulates Rap1-CCM1/CCM2-RhoA pathway. Our data show that CCM3 stabilizes the cell membrane localization of VEGFR2 and VE-cadherin, both of which are key components of vascular integrity (40-42, 45, 61). Therefore, VE-cadherin seems to be a common target of all three CCMs, underlying a potential mechanism by which CCM proteins regulate vascular integrity. Indeed, deletion of CCM3 in endothelial cell disrupts cell-cell junction both in embryos and in cultured endothelial cells. It needs to be determined in the future how CCM3-VEGFR2-VE-cadherin and CCM2 signaling converge on the common pathways.
Supplementary Material
Acknowledgments
This work was supported by NIH grants R01 HL077357, R01 HL085789 and an Established Investigator Award from the American Heart Association (0440172N) to WM; NIH R01 NS046521 to MG; a Scientific Development Grant from the American Heart Association (0835544N) to HC.
Footnotes
This manuscript has been accepted for publication in Science Signaling. This version has not undergone final editing. Please refer to the complete version of record at http://www.sciencesignaling.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.
References and Notes
- 1.Otten P, Pizzolato GP, Rilliet B, Berney J. 131 cases of cavernous angioma (cavernomas) of the CNS, discovered by retrospective analysis of 24,535 autopsies. Neurochirurgie. 1989;35:82–83. 128–131. [PubMed] [Google Scholar]
- 2.Rigamonti D, Hadley MN, Drayer BP, Johnson PC, Hoenig-Rigamonti K, Knight JT, Spetzler RF. Cerebral cavernous malformations. Incidence and familial occurrence. N Engl J Med. 1988;319:343–347. doi: 10.1056/NEJM198808113190605. [DOI] [PubMed] [Google Scholar]
- 3.Revencu N, Vikkula M. Cerebral cavernous malformation: new molecular and clinical insights. J Med Genet. 2006;43:716–721. doi: 10.1136/jmg.2006.041079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sahoo T, Johnson EW, Thomas JW, Kuehl PM, Jones TL, Dokken CG, Touchman JW, Gallione CJ, Lee-Lin SQ, Kosofsky B, Kurth JH, Louis DN, Mettler G, Morrison L, Gil-Nagel A, Rich SS, Zabramski JM, Boguski MS, Green ED, Marchuk DA. Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1) Hum Mol Genet. 1999;8:2325–2333. doi: 10.1093/hmg/8.12.2325. [DOI] [PubMed] [Google Scholar]
- 5.Eerola I, Plate KH, Spiegel R, Boon LM, Mulliken JB, Vikkula M. KRIT1 is mutated in hyperkeratotic cutaneous capillary-venous malformation associated with cerebral capillary malformation. Hum Mol Genet. 2000;9:1351–1355. doi: 10.1093/hmg/9.9.1351. [DOI] [PubMed] [Google Scholar]
- 6.Liquori CL, Berg MJ, Siegel AM, Huang E, Zawistowski JS, Stoffer T, Verlaan D, Balogun F, Hughes L, Leedom TP, Plummer NW, Cannella M, Maglione V, Squitieri F, Johnson EW, Rouleau GA, Ptacek L, Marchuk DA. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet. 2003;73:1459–1464. doi: 10.1086/380314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denier C, Goutagny S, Labauge P, Krivosic V, Arnoult M, Cousin A, Benabid AL, Comoy J, Frerebeau P, Gilbert B, Houtteville JP, Jan M, Lapierre F, Loiseau H, Menei P, Mercier P, Moreau JJ, Nivelon-Chevallier A, Parker F, Redondo AM, Scarabin JM, Tremoulet M, Zerah M, Maciazek J, Tournier-Lasserve E. Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am J Hum Genet. 2004;74:326–337. doi: 10.1086/381718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergametti F, Denier C, Labauge P, Arnoult M, Boetto S, Clanet M, Coubes P, Echenne B, Ibrahim R, Irthum B, Jacquet G, Lonjon M, Moreau JJ, Neau JP, Parker F, Tremoulet M, Tournier-Lasserve E. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet. 2005;76:42–51. doi: 10.1086/426952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guclu B, Ozturk AK, Pricola KL, Bilguvar K, Shin D, O'Roak BJ, Gunel M. Mutations in apoptosis-related gene, PDCD10, cause cerebral cavernous malformation 3. Neurosurgery. 2005;57:1008–1013. doi: 10.1227/01.neu.0000180811.56157.e1. [DOI] [PubMed] [Google Scholar]
- 10.Stahl S, Gaetzner S, Voss K, Brackertz B, Schleider E, Surucu O, Kunze E, Netzer C, Korenke C, Finckh U, Habek M, Poljakovic Z, Elbracht M, Rudnik-Schoneborn S, Bertalanffy H, Sure U, Felbor U. Novel CCM1, CCM2, and CCM3 mutations in patients with cerebral cavernous malformations: in-frame deletion in CCM2 prevents formation of a CCM1/CCM2/CCM3 protein complex. Hum Mutat. 2008;29:709–717. doi: 10.1002/humu.20712. [DOI] [PubMed] [Google Scholar]
- 11.Voss K, Stahl S, Schleider E, Ullrich S, Nickel J, Mueller TD, Felbor U. CCM3 interacts with CCM2 indicating common pathogenesis for cerebral cavernous malformations. Neurogenetics. 2007;8:249–256. doi: 10.1007/s10048-007-0098-9. [DOI] [PubMed] [Google Scholar]
- 12.Denier C, Gasc JM, Chapon F, Domenga V, Lescoat C, Joutel A, Tournier-Lasserve E. Krit1/cerebral cavernous malformation 1 mRNA is preferentially expressed in neurons and epithelial cells in embryo and adult. Mech Dev. 2002;117:363–367. doi: 10.1016/s0925-4773(02)00209-5. [DOI] [PubMed] [Google Scholar]
- 13.Petit N, Blecon A, Denier C, Tournier-Lasserve E. Patterns of expression of the three cerebral cavernous malformation (CCM) genes during embryonic and postnatal brain development. Gene Expr Patterns. 2006;6:495–503. doi: 10.1016/j.modgep.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 14.Guzeloglu-Kayisli O, Amankulor NM, Voorhees J, Luleci G, Lifton RP, Gunel M. KRIT1/cerebral cavernous malformation 1 protein localizes to vascular endothelium, astrocytes, and pyramidal cells of the adult human cerebral cortex. Neurosurgery. 2004;54:943–949. doi: 10.1227/01.neu.0000114512.59624.a5. discussion 949. [DOI] [PubMed] [Google Scholar]
- 15.Guzeloglu-Kayisli O, Kayisli UA, Amankulor NM, Voorhees JR, Gokce O, DiLuna ML, Laurans MS, Luleci G, Gunel M. Krev1 interaction trapped-1/cerebral cavernous malformation-1 protein expression during early angiogenesis. J Neurosurg. 2004;100:481–487. doi: 10.3171/ped.2004.100.5.0481. [DOI] [PubMed] [Google Scholar]
- 16.Seker A, Pricola KL, Guclu B, Ozturk AK, Louvi A, Gunel M. CCM2 expression parallels that of CCM1. Stroke. 2006;37:518–523. doi: 10.1161/01.STR.0000198835.49387.25. [DOI] [PubMed] [Google Scholar]
- 17.Hogan BM, Bussmann J, Wolburg H, Schulte-Merker S. ccm1 cell autonomously regulates endothelial cellular morphogenesis and vascular tubulogenesis in zebrafish. Hum Mol Genet. 2008;17:2424–2432. doi: 10.1093/hmg/ddn142. [DOI] [PubMed] [Google Scholar]
- 18.Mably JD, Chuang LP, Serluca FC, Mohideen MA, Chen JN, Fishman MC. santa and valentine pattern concentric growth of cardiac myocardium in the zebrafish. Development. 2006;133:3139–3146. doi: 10.1242/dev.02469. [DOI] [PubMed] [Google Scholar]
- 19.Whitehead KJ, Chan AC, Navankasattusas S, Koh W, London NR, Ling J, Mayo AH, Drakos SG, Marchuk DA, Davis GE, Li DY. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat Med. 2009;15:177–184. doi: 10.1038/nm.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whitehead KJ, Plummer NW, Adams JA, Marchuk DA, Li DY. Ccm1 is required for arterial morphogenesis: implications for the etiology of human cavernous malformations. Development. 2004;131:1437–1448. doi: 10.1242/dev.01036. [DOI] [PubMed] [Google Scholar]
- 21.Kleaveland B, Zheng X, Liu JJ, Blum Y, Tung JJ, Zou Z, Chen M, Guo L, Lu MM, Zhou D, Kitajewski J, Affolter M, Ginsberg MH, Kahn ML. Regulation of cardiovascular development and integrity by the heart of glass-cerebral cavernous malformation protein pathway. Nat Med. 2009;15:169–176. doi: 10.1038/nm.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plummer NW, Zawistowski JS, Marchuk DA. Genetics of cerebral cavernous malformations. Curr Neurol Neurosci Rep. 2005;5:391–396. doi: 10.1007/s11910-005-0063-7. [DOI] [PubMed] [Google Scholar]
- 23.Gore AV, Lampugnani MG, Dye L, Dejana E, Weinstein BM. Combinatorial interaction between CCM pathway genes precipitates hemorrhagic stroke. Dis Model Mech. 2008;1:275–281. doi: 10.1242/dmm.000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pagenstecher A, Stahl S, Sure U, Felbor U. A two-hit mechanism causes cerebral cavernous malformations: complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells. Hum Mol Genet. 2009;18:911–918. doi: 10.1093/hmg/ddn420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma X, Zhao H, Shan J, Long F, Chen Y, Chen Y, Zhang Y, Han X, Ma D. PDCD10 interacts with Ste20-related kinase MST4 to promote cell growth and transformation via modulation of the ERK pathway. Mol Biol Cell. 2007;18:1965–1978. doi: 10.1091/mbc.E06-07-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coultas L, Chawengsaksophak K, Rossant J. Endothelial cells and VEGF in vascular development. Nature. 2005;438:937–945. doi: 10.1038/nature04479. [DOI] [PubMed] [Google Scholar]
- 27.Shibuya M, Claesson-Welsh L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res. 2006;312:549–560. doi: 10.1016/j.yexcr.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 28.Tanriover G, Boylan AJ, Diluna ML, Pricola KL, Louvi A, Gunel M. PDCD10, the gene mutated in cerebral cavernous malformation 3, is expressed in the neurovascular unit. Neurosurgery. 2008;62:930–938. doi: 10.1227/01.neu.0000318179.02912.ca. discussion 938. [DOI] [PubMed] [Google Scholar]
- 29.Sclafani AM, Skidmore JM, Ramaprakash H, Trumpp A, Gage PJ, Martin DM. Nestin-Cre mediated deletion of Pitx2 in the mouse. Genesis. 2006;44:336–344. doi: 10.1002/dvg.20220. [DOI] [PubMed] [Google Scholar]
- 30.Lepore JJ, Cheng L, Min Lu M, Mericko PA, Morrisey EE, Parmacek MS. High-efficiency somatic mutagenesis in smooth muscle cells and cardiac myocytes in SM22alpha-Cre transgenic mice. Genesis. 2005;41:179–184. doi: 10.1002/gene.20112. [DOI] [PubMed] [Google Scholar]
- 31.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 32.Kano A, Wolfgang MJ, Gao Q, Jacoby J, Chai GX, Hansen W, Iwamoto Y, Pober JS, Flavell RA, Fu XY. Endothelial cells require STAT3 for protection against endotoxin-induced inflammation. J Exp Med. 2003;198:1517–1525. doi: 10.1084/jem.20030077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 34.Yang J, Boerm M, McCarty M, Bucana C, Fidler IJ, Zhuang Y, Su B. Mekk3 is essential for early embryonic cardiovascular development. Nat Genet. 2000;24:309–313. doi: 10.1038/73550. [DOI] [PubMed] [Google Scholar]
- 35.Deng Y, Yang J, McCarty M, Su B. MEKK3 is required for endothelium function but is not essential for tumor growth and angiogenesis. Am J Physiol Cell Physiol. 2007;293:C1404–1411. doi: 10.1152/ajpcell.00058.2007. [DOI] [PubMed] [Google Scholar]
- 36.Zhang H, He Y, Dai S, Xu Z, Luo Y, Wan T, Luo D, Jones D, Tang S, Chen H, Sessa WC, Min W. AIP1 functions as an endogenous inhibitor of VEGFR2-mediated signaling and inflammatory angiogenesis in mice. J Clin Invest. 2008;118:3904–3916. doi: 10.1172/JCI36168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arnaoutova I, George J, Kleinman HK, Benton G. The endothelial cell tube formation assay on basement membrane turns 20: state of the science and the art. Angiogenesis. 2009;12:267–274. doi: 10.1007/s10456-009-9146-4. [DOI] [PubMed] [Google Scholar]
- 38.Dejana E, Tournier-Lasserve E, Weinstein BM. The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell. 2009;16:209–221. doi: 10.1016/j.devcel.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 39.Zhang R, Xu Y, Ekman N, Wu Z, Wu J, Alitalo K, Min W. Etk/Bmx transactivates vascular endothelial growth factor 2 and recruits phosphatidylinositol 3-kinase to mediate the tumor necrosis factor-induced angiogenic pathway. J Biol Chem. 2003;278:51267–51276. doi: 10.1074/jbc.M310678200. [DOI] [PubMed] [Google Scholar]
- 40.Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, Balconi G, Spagnuolo R, Oostuyse B, Dewerchin M, Zanetti A, Angellilo A, Mattot V, Nuyens D, Lutgens E, Clotman F, de Ruiter MC, Gittenberger-de Groot A, Poelmann R, Lupu F, Herbert JM, Collen D, Dejana E. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 1999;98:147–157. doi: 10.1016/s0092-8674(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 41.Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437:426–431. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- 42.Weis S, Shintani S, Weber A, Kirchmair R, Wood M, Cravens A, McSharry H, Iwakura A, Yoon YS, Himes N, Burstein D, Doukas J, Soll R, Losordo D, Cheresh D. Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J Clin Invest. 2004;113:885–894. doi: 10.1172/JCI20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duval M, Bedard-Goulet S, Delisle C, Gratton JP. Vascular endothelial growth factor-dependent down-regulation of Flk-1/KDR involves Cbl-mediated ubiquitination. Consequences on nitric oxide production from endothelial cells. J Biol Chem. 2003;278:20091–20097. doi: 10.1074/jbc.M301410200. [DOI] [PubMed] [Google Scholar]
- 44.Ewan LC, Jopling HM, Jia H, Mittar S, Bagherzadeh A, Howell GJ, Walker JH, Zachary IC, Ponnambalam S. Intrinsic tyrosine kinase activity is required for vascular endothelial growth factor receptor 2 ubiquitination, sorting and degradation in endothelial cells. Traffic. 2006;7:1270–1282. doi: 10.1111/j.1600-0854.2006.00462.x. [DOI] [PubMed] [Google Scholar]
- 45.Lampugnani MG, Orsenigo F, Gagliani MC, Tacchetti C, Dejana E. Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J Cell Biol. 2006;174:593–604. doi: 10.1083/jcb.200602080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Labrecque L, Royal I, Surprenant DS, Patterson C, Gingras D, Beliveau R. Regulation of vascular endothelial growth factor receptor-2 activity by caveolin-1 and plasma membrane cholesterol. Mol Biol Cell. 2003;14:334–347. doi: 10.1091/mbc.E02-07-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jopling HM, Odell AF, Hooper NM, Zachary IC, Walker JH, Ponnambalam S. Rab GTPase regulation of VEGFR2 trafficking and signaling in endothelial cells. Arterioscler Thromb Vasc Biol. 2009;29:1119–1124. doi: 10.1161/ATVBAHA.109.186239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Surucu O, Sure U, Gaetzner S, Stahl S, Benes L, Bertalanffy H, Felbor U. Clinical impact of CCM mutation detection in familial cavernous angioma. Childs Nerv Syst. 2006;22:1461–1464. doi: 10.1007/s00381-006-0202-8. [DOI] [PubMed] [Google Scholar]
- 49.Denier C, Labauge P, Bergametti F, Marchelli F, Riant F, Arnoult M, Maciazek J, Vicaut E, Brunereau L, Tournier-Lasserve E. Genotype-phenotype correlations in cerebral cavernous malformations patients. Ann Neurol. 2006;60:550–556. doi: 10.1002/ana.20947. [DOI] [PubMed] [Google Scholar]
- 50.Ng BH, Mulyadi E, Pereira JK, Ghedia S, Pinner J, Mowat D, Vonau M. Familial cerebral cavernous haemangioma diagnosed in an infant with a rapidly growing cerebral lesion. Australas Radiol. 2006;50:583–590. doi: 10.1111/j.1440-1673.2006.01638.x. [DOI] [PubMed] [Google Scholar]
- 51.Dubovsky J, Zabramski JM, Kurth J, Spetzler RF, Rich SS, Orr HT, Weber JL. A gene responsible for cavernous malformations of the brain maps to chromosome 7q. Hum Mol Genet. 1995;4:453–458. doi: 10.1093/hmg/4.3.453. [DOI] [PubMed] [Google Scholar]
- 52.Craig HD, Gunel M, Cepeda O, Johnson EW, Ptacek L, Steinberg GK, Ogilvy CS, Berg MJ, Crawford SC, Scott RM, Steichen-Gersdorf E, Sabroe R, Kennedy CT, Mettler G, Beis MJ, Fryer A, Awad IA, Lifton RP. Multilocus linkage identifies two new loci for a mendelian form of stroke, cerebral cavernous malformation, at 7p15-13 and 3q25.2-27. Hum Mol Genet. 1998;7:1851–1858. doi: 10.1093/hmg/7.12.1851. [DOI] [PubMed] [Google Scholar]
- 53.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 54.Zawistowski JS, Stalheim L, Uhlik MT, Abell AN, Ancrile BB, Johnson GL, Marchuk DA. CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis. Hum Mol Genet. 2005;14:2521–2531. doi: 10.1093/hmg/ddi256. [DOI] [PubMed] [Google Scholar]
- 55.Uhlik MT, Abell AN, Johnson NL, Sun W, Cuevas BD, Lobel-Rice KE, Horne EA, Dell'Acqua ML, Johnson GL. Rac-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat Cell Biol. 2003;5:1104–1110. doi: 10.1038/ncb1071. [DOI] [PubMed] [Google Scholar]
- 56.Harel L, Costa B, Tcherpakov M, Zapatka M, Oberthuer A, Hansford LM, Vojvodic M, Levy Z, Chen ZY, Lee FS, Avigad S, Yaniv I, Shi L, Eils R, Fischer M, Brors B, Kaplan DR, Fainzilber M. CCM2 mediates death signaling by the TrkA receptor tyrosine kinase. Neuron. 2009;63:585–591. doi: 10.1016/j.neuron.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 57.Chen L, Tanriover G, Yano H, Friedlander R, Louvi A, Gunel M. Apoptotic Functions of PDCD10/CCM3, the Gene Mutated in Cerebral Cavernous Malformation 3. Stroke. 2009 doi: 10.1161/STROKEAHA.108.527135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Glading A, Han J, Stockton RA, Ginsberg MH. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J Cell Biol. 2007;179:247–254. doi: 10.1083/jcb.200705175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Serebriiskii I, Estojak J, Sonoda G, Testa JR, Golemis EA. Association of Krev-1/rap1a with Krit1, a novel ankyrin repeat-containing protein encoded by a gene mapping to 7q21-22. Oncogene. 1997;15:1043–1049. doi: 10.1038/sj.onc.1201268. [DOI] [PubMed] [Google Scholar]
- 60.Knox AL, Brown NH. Rap1 GTPase regulation of adherens junction positioning and cell adhesion. Science. 2002;295:1285–1288. doi: 10.1126/science.1067549. [DOI] [PubMed] [Google Scholar]
- 61.Shay-Salit A, Shushy M, Wolfovitz E, Yahav H, Breviario F, Dejana E, Resnick N. VEGF receptor 2 and the adherens junction as a mechanical transducer in vascular endothelial cells. Proc Natl Acad Sci U S A. 2002;99:9462–9467. doi: 10.1073/pnas.142224299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.