

Abstract
Fibrillin-rich microfibrils are extracellular assemblies that impart structural properties to the connective tissue. To elucidate the contribution of fibrillin-rich microfibrils to organogenesis, we have examined the vascular phenotype of a newly created strain of mice that completely lacks fibrillin-1 and the consequences of combined deficiency of fibrillins 1 and 2 on tissue formation. The results demonstrated that fibrillins 1 and 2 perform partially overlapping functions during aortic development. Fbn1−/− mice died soon after birth from ruptured aortic aneurysm, impaired pulmonary function, and/or diaphragmatic collapse. Analysis of the neonatal Fbn1−/− aorta documented a disorganized and poorly developed medial layer but normal levels of elastin cross-links. Transcriptional profiling revealed that aneurysm progression in Fbn1 null mice is accompanied by unproductive up-regulation of gene products normally involved in tissue repair and vascular integrity, such as plasminogen activator inhibitor-1, activin A, and cysteine-rich angiogenic protein 61. In contrast to Fbn1−/− mice, Fbn2 null mice had a well developed and morphologically normal aortic wall. However, virtually all Fbn1−/−;Fbn2−/− embryos and about half of the Fbn1+/−;Fbn2−/− embryos died in utero and displayed a significantly more severe vascular phenotype than Fbn1−/− mice. Consistent with a specialized function of fibrillin-2, electron microscopy visualized ultrastructurally different microfibrils in Fbn1 null compared with control cell cultures. Collectively, these data demonstrate that involvement of fibrillin-2 in the initial assembly of the aortic matrix overlaps in part with fibrillin-1 and that continued fibrillin-1 deposition is absolutely required for the maturation and function of the vessel during neonatal life.
Elastogenesis is a complex biological process that involves the organized deposition and self-assembly of several macromolecules into microfibrils and elastic fibers (1). Elastic fibers are made of an amorphous core of cross-linked elastin and other molecules and microfibrils; the latter are heterogeneous in composition and can also form macro-aggregates devoid of elastin (2, 3). Fibrillins 1 and 2 are the main structural components of extracellular microfibrils and the defective gene products in Marfan syndrome (MFS)3 and congenital contractural arachnodactyly (CCA), respectively (4, 5). MFS is a pleiotropic disorder of the connective tissue with wide variation in clinical severity (6). Cardiovascular manifestations in the form of aortic dilatation, dissection, and rupture contribute significantly to morbidity and mortality in affected individuals. CCA, on the other hand, is a rare condition akin to MFS but with major manifestations confined to the musculoskeletal system (6). Fibrillins are large cysteine-rich glycoproteins (~350 kDa) composed primarily of multiple repeated domains homologous to the calcium binding epidermal growth factor module and of distinct 8-cys-teine modules (2, 3, 5). Fibrillins polymerize into a characteristic beads-on-a-string microfibril structure, which gives rise to the microfibril lattice by lateral association of the individual microfibril polymers and probable association of other structural components. Fibrillins can form homo- or heteropolymeric microfibrils and can interact with integrins, growth factors, several other matrix components, and latent transforming growth factor β-binding proteins.
Differential expression of elastic fiber components, dynamic cell-matrix interactions, and microfibril-mediated modulation of signaling molecules account for the diverse architectures and functions of elastic networks during the development and growth of individual organ systems (5). The process of aortic media formation is an illustrative example of the interplay between resident cells and elastic fiber components that ultimately results in the highly organized and functionally competent tissue (7, 8). At about mid-gestation, vascular smooth muscle cells (VSMC) deposit fibrillins and tropoelastin molecules into the surrounding matrix and begin to organize them into elastic fibers. The latter process extends into early neonatal life accompanied by the gradual growth of elastic fibers into mature elastic lamellae that separate parallel layers of quiescent VSMC. The resulting organization of the tunica media into a multilayered structure of alternating VSMC and elastic lamellae (also known as the lamellar unit) is the main determinant of arterial function. Genetic studies in mice have demonstrated that different molecules participate in the various steps responsible for the maturation and function of the arterial wall. Fibulin-5 has been reported to regulate initial assembly of the elastic fiber by providing the molecular bridge between tropoelastin in the pericellular space and integrins on the surface of VSMC, whereas lysyl oxidase-like protein-1 is believed to spatially restrict elastin polymerization by interacting with fibulin-5 (9–11). Elastin, on the other hand, is thought to promote the quiescent phenotype of VSMC as well as to influence vascular morphogenesis in response to hemodynamic stress (12, 13). Recently, a similar role in vascular remodeling and VSMC proliferation has also been assigned to fibulin-5 (14). Finally, the enzyme lysyl oxidase has been shown to stabilize the aortic wall by mediating the extracellular conversion of tropoelastin molecules into the insoluble elastic meshwork (15, 16).
Three mouse strains harboring distinct mutations in the fibrillin-1 (Fbn1) gene have been created thus far that faithfully replicate the spectrum of clinical severity in MFS (17–19). The mutations include the substitution of an obligatory cysteinyl residue in a calcium binding epidermal growth factor motif (C1039G) and a hypomorphic in-frame deletion (mgΔ), which in homozygosity mimic the neonatal lethal form of MFS, and a hypomorphic mutation (mgR) that produces ~15% of the normal amount of wild-type fibrillin-1 and which in homozygosity recapitulates the adult lethal form of MFS. Ruptured aortic aneurysm in these mutant mice is associated with morphologically normal elastic fibers between focal lesions and in unaffected tissues. Hence, the notion followed that fibrillin-1 microfibrils play a predominant role in maintaining tissue integrity rather than controlling aortic development (17, 18). The fact that fibrillin-2 is generally produced earlier than fibrillin-1 and primarily during fetal development was originally interpreted to suggest a predominant role for this molecule in organizing matrix assembly (20). However, the validity of this hypothesis was subsequently questioned by the finding that ablation of fibrillin-2 production in mice affected BMP signaling and digit formation but had no apparent effect on elastogenesis (21).
Whereas the above studies were instrumental in delineating pathogenic events in MFS, they left unresolved the issue of the role of fibrillin-1 microfibrils during development. The present study was, therefore, designed to elucidate the contribution of fibrillin-1 to organogenesis, specifically to aortic development, by creating a new line of mutant mice that completely lacks fibrillin-1. Availability of these mutant mice also enabled us to assess whether or not fibrillins 1 and 2 have redundant functions in organ formation by examining aortic wall development in the progeny of intercrosses of Fbn1 and Fbn2 heterozygous null mice. The results of these studies are the first to document the critical role of fibrillin-1 in the maturation of the aortic wall and of fibrillin-rich micro-fibrils in tissue development. They are also the first to demonstrate partial redundancy between fibrillins 1 and 2 in promoting matrix formation and to indicate the potential for specific functions in this process.
EXPERIMENTAL PROCEDURES
Generation of Fbn1 Null Mice
Engineering and electroporation of the targeting vector as well as selection and analysis of mouse embryonic stem cells and generation of mutant mice were performed as previously described (17, 18). Targeting of the Fbn1 gene substituted ~700 bp of exon 1, which included the ATG codon and the signal peptide coding sequence, with the phosphoglycerate kinase-neo cassette and the alkaline phosphatase gene containing an internal ribosome entry site (Fig. 1A). Four positively targeted embryonic stem cell clones were injected into blastocysts and yielded mutant mice with identical phenotypes. Gene-targeted animals were generated at the Mount Sinai Mouse Genetics Shared Resource Facility (New York). Characterization of the mutation employed Northern and Western analyses on tissue samples and mouse embryonic fibroblasts, respectively (22). Mice were genotyped by PCR amplification using the following primers and conditions: 5′-GCGGACGATACTTGAAGAGG-3′ (forward primer) and 5′-TGCCTACACGGTCTTAATGG-3′ (reverse primer), 95 °C for 1 min, 51 °C for 1 min, and 72 °C for 1 min (wild-type allele); 5′-GAGGCTATTCGGCTATGACT-3′ (forward primer) and 5′-CTCTTCGTCCAGATCATCCT-3′ (reverse primer), 95 °C for 1 min, 57 °C for 1 min, and 72 °C for 1 min (mutant allele). The frequency of the different genotypes in the progeny from the intercrosses between heterozygous null Fbn1 and Fbn2 mice was evaluated using Fischer’s exact test assuming a p value of ≤ 0.05 as significant.
FIGURE 1. Fbn1 gene targeting strategy.

A, top to bottom, the wild-type genomic region that was targeted with the size of the wild-type EcoRI restriction fragment detected by probe A in the Southern analysis, the targeting vector containing the human alkaline phosphatase (hAP) and the phosphoglycerate kinase -neo (Neo) selectable gene with the arrow indicating the direction of transcription, and the targeted mgN allele with the size of the mutant EcoRI fragment detected by probe A. IRES, internal ribosome entry site. kb, kilo-bases. B, Southern blots of tail DNA from wild-type (+/+), heterozygous (+/−), and homozygous (−/−) mgN mice that was digested with EcoRI and hybridized to probe A; the size of DNA fragments (in kilobases) is indicated on the left side of the autoradiogram. C, Northern blots of RNA from lung tissues of wild-type (+/+) and mgN/mgN (−/−) mice hybridized to Fbn1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) probes. D, Western blots of conditioned media from wild-type (+/+) and mgN/mgN (−/−) mouse embryonic fibroblast cultures using anti-fibrillin-1 or fibrillin-2 antibodies (α); the size of fibrillins (in kDa) is indicated on the left side of the autoradiogram.
Histological and Ultrastructural Analyses
Embryos and newborn mice (4–10 per genotype) were processed as described previously (17, 18). Paraffin-embedded sections were serially sectioned at 7 μm and stained with hematoxylin and eosin (H&E), and adjacent sections were assayed with Weigert stain for elastic fibers and Masson trichrome for collagen. For immunohistochemistry, tissue sections were dewaxed and washed in Tris-buffered saline buffer and then subjected to antigen retrieval, blocking, and primary antibody incubation. Primary antibodies included SMemb (1:100; Covance, Berkeley; CA) and MMP-9 (1:300; Chemicon, Temecula, CA). After removal of the unbound antibodies, sections were incubated with biotin-conjugated goat anti-rabbit, anti-mouse, or anti-rat IgG (1:500; Jackson ImmunoResearch Laboratories, West Grove, PA) for 1 h at room temperature followed by incubation with horseradish peroxidase-conjugated streptavidin (1:500; Zymed Laboratories Inc., San Francisco, CA) for 15 min at room temperature. Sections were finally washed, incubated with amino ethyl carbazole chromogen solution (Zymed Laboratories Inc.) to localize the antibody, and counterstained with H&E. Processed tissue samples were viewed under a light microscope Nikon Eclipse E-800 (Nikon Instruments Inc., Yokohama, Japan), and micrographs were taken with an RT Slider (Diagnostic Instruments Inc, Sterling Heights, MI). For ultrastructural analyses, wild-type and heterozygous and homozygous mutant neonatal fibroblasts were cultured for 1 month. Cell layers scraped up into 50 mM Tris-HCl, pH 7.4, 0.4 M NaCl, and 0.2 g/ml (wet weight) were incubated for 3 h at room temperature in 50 mM Tris-HCl, pH 7.4, 0.4 M NaCl, 5 mM CaC12, 0.1 mg/ml Type 1A collagenase (Sigma), 2 mM phenylmethylsulfonyl fluoride, and 5 mM N-ethylmaleimide. The suspension was centrifuged for 15 min at 7500 × g, the supernatant was applied to Sepharose CL-2B (1.5 × 50 cm, flow rate 0.3 ml/min), equilibrated with 50 mM Tris-HCl, pH 7.4, containing 0.4 M NaCl, and the void volume peak was pooled and concentrated. After dialyzing against water, the microfibrils were visualized by electron microscopy after rotary shadowing or negative staining (23).
Tissue Desmosine Assay
Wild-type and mutant thoracic aortas were collected from E16.5 embryos and P10 mice. Tissue samples containing between 0.02 and 0.5 mg of protein were processed as previously described. Desmosine was quantified by radioimmunoassay in 10–40 ml of sample according to Starcher and Conrad (24). Samples from 3–4 animals were tested for each developmental stage and genotype, and 3–7 samples were independently tested for each aorta. Values were expressed as percentage of desmosine cross-links relative to total protein content, and their statistical value was assessed using Student’s t test, assuming a p value ≤0.05 as significant.
Immunoblots
Western blots were performed on protein extracts from aortas of wild-type and mutant mice sacrificed at P14 using antibodies against phosphorylated Smad2 (Cell Signaling, Beverly, CA) or from the post-mortem tissue of mutant mice using anti-MMP-9 antiserum; in both cases, anti-β-tubulin antibody (Upstate Biotechnology Inc., Charlottesville, VA) was used as a control. Protein extracts (25 μg/lane) were electrophoresed in a 10% (w/v) polyacrylamide gel and electroblotted onto an Immobilon-P membrane (Millipore, Billerica, MA). The membrane was then subjected to two sequential incubations of 12 h at 4 °C and 1 h at room temperature in rabbit anti-phosphorylated Smad2 (1:1000) antisera or in mouse anti-MMP-9 antiserum (1:200) and in a peroxidase-labeled anti-rabbit or anti-mouse IgG antibody (1:50,000; Jackson ImmunoResearch Laboratories, West Grove, PA) in the presence of 5% (w/v) skimmed milk in Tris-buffered saline, pH 7.4, and 0.05% (v/v) Tween 20 for 1 h at room temperature. Immunoreactive products were visualized by chemiluminescence using the ECL Plus kit (Amersham Biosciences), and their relative intensity was evaluated with Photoshop software (Adobe Systems Inc., San Jose, CA). Statistical significance of the results was evaluated using the Student’s t test assuming a p value ≤ 0.05 as significant.
RNA Analyses
Total RNA was extracted from the thoracic aortas of wild-type and mutant mice sacrificed at P14 using the RNeasy fibrous tissues kit (Qiagen Inc., Valencia, CA). Real-time PCR was performed on RNA purified from the aortas of wild-type (n = 3) and Fbn1 null (n = 3) mice using the following oligonucleotides: 5′-CTGCTCGCCGGCT-TGTTGGTTC-3′(forward primer) and 5′-CGGCGGGGCAGGTGG-AGAG-3′ (reverse primer) (Cry61/Cnn1; accession number NM_010516); 5′-GTGCAACCCTGGCCGACTTCA-3′ (forward primer) and 5′-GACGCCACTGTGCCGCTCTC-3′ (reverse primer) (Pai-1; accession number NM_008871); 5′-GCAGCTTGGACACGG-GGGATGAG-3′(forward primer) and 5′-GGCGCTGGATGCTGCT-GGACA-3′ (reverse primer) [activin A; accession number NM_008380]; 5′-GAATAACGGCTACAGCAACAGGGTGGTGGA-C-3′ (forward primer) and 5′-AGTGTTGGGGGCCGAGTTGGGAT-AGG-3′ (reverse primer) (Gapdh; accession number NM_001001303). Samples were transcribed with Sensiscript Kit (Qiagen) and PCR-amplified in a 25-μl reaction of IQ SYBR Supermix kit (Bio-Rad) that included 12.5 μl of PCR master mix, 2.5 pmol of each amplification primer, and 30 ng of original template RNA. Each sample was run in triplicate with an initial 10-min reaction at 95 °C for reverse transcription followed by 40 cycles at 95 °C for 15 s and at 60 °C for 1 min. Amplification data were collected using the iCycler system and analyzed with the iCycler IQ 3.1 system software (Bio-Rad). Relative RNA concentrations were calculated from the threshold cycle (CT) at which fluorescence is first detected using the comparative method 2-ΔΔCT (25). Statistical significance of the results was evaluated with the Student’s t test, assuming a p value ≤ 0.05 as significant.
RESULTS
Loss of Fibrillin-1 Results in Early Onset Vascular Disease and Neonatal Lethality
A Fbn1 null allele (mgN) was created by targeting part of the exon 1 sequence to investigate the potential role of fibrillin-1 during embryonic development and postnatal growth (Fig. 1A). Germ line transmission of the mgN allele was monitored by Southern hybridization using diagnostic DNA sites (Fig. 1B). Northern analysis of neonatal lung tissue and Western analysis of conditioned media from mouse embryonic fibroblast cultures documented loss of Fbn1 gene activity in homozygous mutant mice (Fig. 1, C and D). The Western data also ruled out the possibility that increased fibrillin-2 production may compensate for loss of fibrillin-1 (Fig. 1D). Reverse transcription-PCR amplification of aortic RNA independently corroborated this last conclusion (data not shown). Heterozygous and homozygous mgN mutant mice were born at the expected Mendelian frequency. Whereas mgN/+ mice were viable and fertile, all mgN/mgN mice died within the first 2 weeks of postnatal life from ruptured aortic aneurysm, impaired pulmonary function, and/or diaphragmatic collapse. Findings at necroscopy included aortic aneurysm with documented hemothorax or hemopericardium, severely affected lungs with abnormal enlargement of the alveolar space, and abdominal organs within the chest cavity (Fig. 2A and data not shown). Additionally, all mgN/mgN mice displayed grossly malformed and elongated ribs, seemingly thinner intercostal muscles, and unusually fragile skin and internal organs upon manipulation of the carcasses (Fig. 2A and data not shown). Interestingly, aneurysm in Fbn1 null mice involved the ascending rather than the root of the aorta, a finding at variance with the phenotype of the other Fbn1 mutant mice and MFS patients (Fig. 2A) (6, 17, 18).
FIGURE 2. Fbn1 null phenotype.

A, left two panels, representative aneurysm of the ascending aorta in a newborn mouse with histological documentation (Weigert staining) of elastic fiber fragmentation and aortic wall rupture (arrow). Right four panels, top, right lungs of wild-type (+/+) and Fbn1 null (−/−) mice showing multiple blebs in the peripheral portion of the latter (arrow); bottom, partial views of the exposed thoraxes of wild-type and mutant mice showing thinner intercostal muscles and abnormal ribs in the latter compared with the former animal. B, cross-sections of aortic tissue around the lesions in mutant mice (−/−) and at corresponding levels in age and sex-matched wild-type littermates stained with H&E, Weigert, trichrome, or anti-MMP-9 antiserum. On the right side of the H&E panels is a magnification of the mutant tissue showing an altered architecture with loss of normal contacts between cells and with the surrounding matrix (yellow arrows). The immunoblot on the right further documents elevated MMP-9 compared with β-tubulin levels in the decaying post-mortem aorta of mutant mice. All the specimens are oriented with the lumen of the vessel at the top; scale bar = 100 μm.
Light microscopic examination of H&E or Weigert-stained cross-sections of aortic lesions from dead mutant mice revealed a significant disorganization of the lamellar units in the medial layer (Fig. 2B). Elastic fibers had an abnormally wavy and thin appearance and were fragmented and discontinuous, whereas VSMC had lost their contacts with neighboring cells and surrounding matrix and were misoriented (Fig. 2B). Similarly, trichrome staining revealed discontinuity of collagen in the aortic lamellae (Fig. 2B). However, additional findings that characterize late lesions of mgR/mgR mice, such as excessive matrix accumulation and advential hyperplasia (18), were virtually absent in the aorta of dead mgN/mgN animals (Fig. 2B and data not shown). Likewise, elevated VSMC expression of MMP-9 was reproducibly seen only in the decaying aortic tissue of dead mgN/mgN mice (Fig. 2B). By contrast, two histopathological findings were unique to mgN/mgN mice. First, the endothelial lining was often detached in both pre- and post-mortem specimens of mutant mice, consistent with the loss of structural connections between the intimal and medial layers (Fig. 3A) (26). Second, the abnormal morphology of the lamellar unit was not restricted to lesions, as in other Fbn1 mutant mice, but involved the whole aorta (Fig. 3B) (17, 18). This last observation raised the possibility that loss of fibrillin-1 microfibrils may interfere with the maturation of the elastic vessel wall in this particular mouse model of ruptured aortic aneurysm.
FIGURE 3. Histopathology of Fbn1 null aortas.

A, immunostaining with anti-Von Willebrand factor antibody of cross-sections of aortas from a randomly sacrificed (P14) and a dead (pm) Fbn1 null (−/−) mouse showing detachment of the endothelial lining (arrows). Control samples were from sex- and age-matched wild-type (+/+) littermates. The signal was pseudocolored using Adobe Photoshop software to best appreciate the immunostaining. B, Weigert staining of aortic tissues in segments other than the focal lesions of Fbn1 null mice (−/−) and in comparable regions of the wild-type (+/+) aorta. Arrows indicate discontinuities in elastic fibers. Scale bar, 100 μm in both panels.
Loss of Fibrillin-1 Perturbs Maturation of the Aortic Wall
To test the above hypothesis, we compared the development of mutant and wild-type aortas from embryonic day 16.5 (E16.5), when elastic fibers begin to form in the extracellular space (7, 8), to postnatal day 14 (P14), when the majority of mgN/mgN mice die. The analyses documented the appearance of thinner and disorganized elastic lamellae in the mutant mice that developed between P10 and P14 (Fig. 4A). Antibodies against an embryonic VSMC marker (non-muscular myosin heavy chain-B; SMemb) revealed relatively stronger immunostaining in the mutant than in the wild-type tissue during fetal development as well as persistence of SMemb expression postnatally when the gene is normally turned off (Fig. 4B) (27). VSMC proliferation and apoptosis and matrix production were instead largely the same in mutant and wild-type littermates (data not shown). We concluded from these results that loss of fibrillin-1 microfibrils interferes with the normal process of aortic wall maturation, especially during neonatal life when both cellular and architectural effects were seen.
FIGURE 4. Aortic development in Fbn1 null mice.
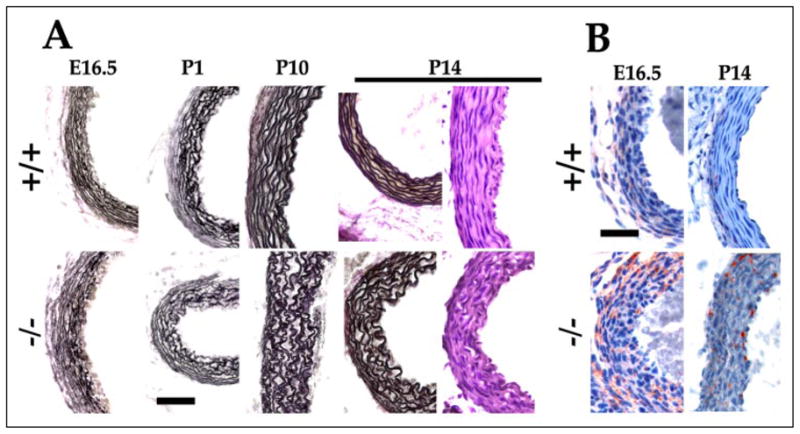
A, cross-sections of aortas from wild-type (+/+) and mgN/mgN (−/−) embryos (E16.5), and randomly sacrificed newborn mice (P1, P10, and P14) were stained with H&E or Weigert for elastic fibers. B, aortas from E16.5 and P14 wild-type (+/+) and mgN/mgN mice immunostained for SMemb, an embryonic VSMC marker. Scale bar, 50 μm in both panels.
Despite the absence of histological evidence for a fibroproliferative response, an ongoing transcriptional profiling of fibrillin mutant mice nonetheless revealed that genes involved in tissue repair processes are up-regulated in the aorta of mgN/mgN mice.4 Among others, genes found to be abnormally expressed in mgN/mgN mice randomly sacrificed at P14 included those coding for plasminogen activator inhibitor-1 (PAI-1; 4.4-fold increase) and activin A (2.5-fold increase) (28, 29). The DNA microarray experiments also identified a 5-fold increase in the expression of the gene encoding cysteine-rich angiogenic protein 61 (CYR61/CCN1), a matricellular protein that has been implicated in the maintenance of vascular integrity (30). Stimulation of PAI-1, activin A and CYR61/CCN1 production in the fibrillin-1 null aorta was independently validated by real-time PCR and/or immunohistochemistry (Fig. 5). Consistent with Smad-induced stimulation of Pai-1 gene expression (31), Western blots detected nearly 3-fold more phosphorylated Smad2 in the mutant than in the wild-type aorta (Fig. 5). We interpreted these results to indicate that a fibroproliferative response in the fibrillin-1 null aorta fails to manifest histologically due to the very rapid collapse of a structurally impaired and developmentally immature tissue.
FIGURE 5. Altered gene expression in Fbn1 null aorta.

A, left, bar graphs indicate the mean -fold levels ± S.E. of Pai-1, Cyr61/Ccn1, and activin A transcripts in the aortas of P14 wild-type (open bars) and mutant (filled bars) mice (n = 3 per genotype) as evaluated by real-time PCR. The asterisk indicates statistically significant differences between samples. Right, illustrative immunostaining of PAI-1 and CYR61/CCN1 in the aortas of P14 wild-type (+/+) and mutant (−/−) mice; scale bar, 100 μm. B, left, Western analysis of phosphorylated Smad2 and β-tubulin protein levels in the aortas of P14 wild-type (+/+) and mutant (−/−) mice. Right, bar graphs summarizing the data upon normalization of the Western blot experiments. Bars show the means ± S.E., and the asterisk indicates statistically significant differences between control (open bars) and mutant (filled bars) sample (n = 3 per genotype). β-tub, β-tubulin.
Characterization of mice lacking lysyl oxidase activity has recently demonstrated that formation of stable cross-links is required for elastic tissue maturation and integrity (15, 16). Like mgN/mgN mice, those without functional lysyl oxidase die postnatally of ruptured arterial aneurysm and display fragmented elastic fiber architecture and disorganized VSMC morphology. We, therefore, tested whether loss of fibrillin-1 may have also affected the degree of elastin cross-linking in the aortic wall. A desmosine radioimmunoassay performed on aortas collected from E16.5 embryos and P10 mice, however, showed comparable amounts of desmosine in mutant and wild-type samples (Table 1). This result, therefore, excluded a significant contribution of fibrillin-1 deposition to elastin cross-linking, thus uncoupling elastin maturation from the assembly of fibrillin-1 microfibrils. More generally, the data differentiated the pathogenic mechanism of ruptured aortic aneurysm in mice without fibrillin-1 from that of mice without lysyl oxidase.
TABLE 1.
Elastin-derived cross-links
| +/+ | −/− | |
|---|---|---|
| E16.5 | 431.00 ± 150.75 n = 9 | 435.00 ± 20.66 n 3 |
| P10 | 2005.00 ± 100.04 n = 4 | 1932.67 ± 146.46 n = 3 |
The numbers indicate pmol of desmosine per mg of protein reported ± S.D.; the number of animals analyzed in each genotype (n) is indicated.
Graded Deficiencies of Fibrillins Result in Developmental Failure
Previous work has shown that, unlike mice without fibrillin-1, those lacking fibrillin-2 are viable and have a normal life span (21). Consistent with these findings in mice and the absence of vascular manifestations in CCA patients, the morphology of the aortic wall in Fbn2−/− embryos, newborns, and young mice was remarkably similar to that of wild-type littermates, suggesting that fibrillin-2 is either dispensable for tissue development or functionally redundant with fibrillin-1 (Fig. 6). To discriminate between these two possibilities and, thus, clarify the role of fibrillin-rich microfibrils during development, we genotyped immediately after birth (P1) the progeny (n = 104) of Fbn1+/− and Fbn2+/− intercrosses and found that the incidence of Fbn−/−;Fbn2−/− mice was statistically far below the expected value (Table 2). This result provided genetic evidence for a functional overlap between the fibrillin proteins in addition to indicating that complete loss of extracellular microfibrils is incompatible with embryonic viability. The latter conclusion was corroborated by the genotypes of embryos at 16.5 days (n = 39) and 14.5 days (n = 93) of gestation, which narrowed down the time of death of Fbn1−/−;Fbn2 −/− embryos to some time after E14.5. Although grossly indistinguishable from wild-type embryos, histological analyses of E14.5 embryos suggested impaired or delayed elastogenesis in the medial layer of the Fbn1−/−;Fbn2 −/− aorta compared with the wild-type counterpart (Fig. 7). The same histological differences were noted in comparing the aortas of Fbn1−/−;Fbn2 −/− with those of Fbn1−/− or Fbn2−/− embryos (Figs. 4, 6, and 7, and data not shown).
FIGURE 6. Elastic fibers in the aorta of Fbn2 null mice.
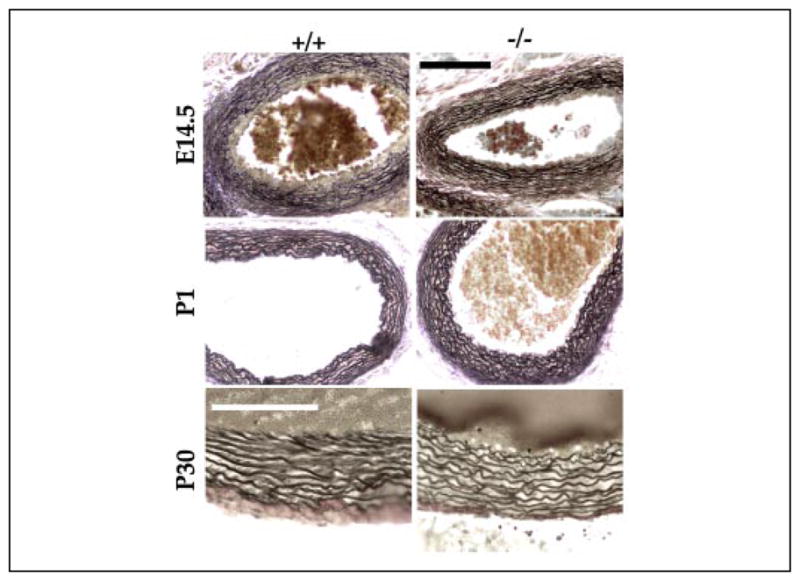
Weigert-stained cross-sections of ascending aortas from wild-type (WT) (+/+) and Fbn2 null (−/−) embryos (E14.5) and randomly sacrificed P1 and P30 mice. Scale bars, 100 μm in the top four panels and 20 μm in the bottom two.
TABLE 2.
Heterozygous Fbn1 and Fbn2 null intercrosses
| Genotype |
Frequency (%) |
||
|---|---|---|---|
| Fbn1 | Fbn2 | Predicted | Observed |
| % | |||
| +/+ | +/+ | 6.25 | 9.62 |
| +/+ | +/− | 12.5 | 13.46 |
| +/+ | −/− | 6.25 | 7.69 |
| +/− | +/+ | 12.5 | 13.46 |
| +/− | +/− | 25 | 26.92 |
| +/− | −/− | 12.5 | 5.77a |
| −/− | +/+ | 6.25 | 5.77 |
| −/− | +/− | 12.5 | 16.35 |
| −/− | −/− | 6.25 | 0.96a |
Statistically relevant differences with p values of <0.04 (Fbn1+/−;Fbn2−/−) and <0.012 (Fbn1−/−;Fbn2−/−).
FIGURE 7. Phenotype of double fibrillin mutant embryos.
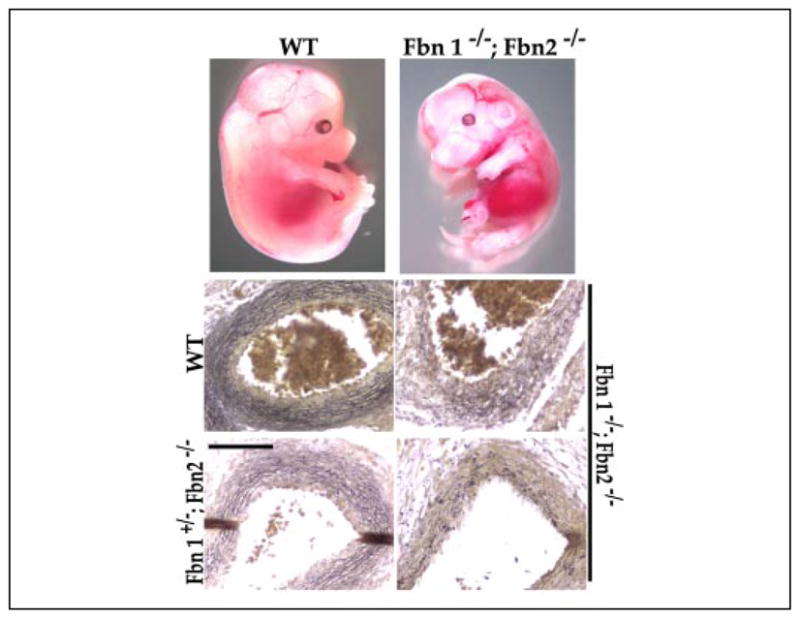
Top, illustrative examples of wild-type (WT) and Fbn1−/−;Fbn2 −/− embryos at E14.5. Bottom, Weigert-stained cross-sections of ascending aortas from the embryos shown above as well as from an additional double null embryo and a Fbn1 +/−;Fbn2 −/− embryo documenting poor organization of the vessel wall in the mutant samples. Scale bar, 50 μm.
In contrast to Fbn+/−;Fbn2+/− and Fbn1−/−;Fbn2+/− mice, a statistically significant deviation from the expected Mendelian frequency was observed for the Fbn1+/−;Fbn2−/− genotype at birth (Table 2). This unexpected finding suggested that functional redundancy between the fibrillins during fetal development is a gene dosage-dependent phenomenon in which Fbn2 plays a critical role. Indeed analysis of the medial layer of E14.5 Fbn1+/−;Fbn2−/− embryos showed that the elastic fibers are less intensively stained than control samples (Fig. 7). Such visual evidence was at least consistent with the notion of impaired/delayed matrix assembly in the aorta of Fbn1+/−;Fbn2 −/− embryos. One possible explanation for these observations is that elastic fiber formation is driven by the total amount of microfibrils deposited at any given time into the extracellular space irrespective of the actual identity of the fibrillins. This would imply a quantitatively critical contribution of fibrillin-2 molecules at mid-gestation when expression of Fbn2 is the highest and when expression of Fbn1 is significantly less than it is perinatally (8, 20). Another potential explanation is that fibrillin-2-rich microfibrils play a functionally distinct role in organizing the embryonic matrix. To establish whether or not there is at least a structural basis for the potentially unique function of fibrillin-2, microfibrils were isolated from wild-type, mgN/+, and mgN/mgN neonatal fibroblasts and analyzed by electron microscopy. Analysis of mgN/mgN cultures provided the first opportunity to visualize the ultrastructure of fibrillin-2 homopolymers. In contrast to the polymers extracted from wild-type and heterozygous mutant cultures, rotary shadowing and negative staining of microfibrils extracted from mgN/mgN fibroblast cultures showed that fibrillin-2 homopolymers appear as periodically beaded structures containing an electron-dense structure just to one side of each bead (Fig. 8). Rotary-shadowed wild-type extracted microfibrils, which contain fibrillin-1 and fibrillin-2, did not display any clear asymmetry or direction (Fig. 8A), whereas rotary-shadowed homozygous microfibrils, which contain fibrillin-2 but not fibrillin-1, appeared asymmetric and directional (Fig. 8B). Negative staining revealed a prominent electron density just to the side of each bead in the fibrillin-2-containing homozygous mutant microfibril (Fig. 8D). This unusual electron density has not been previously observed in negative stains of microfibrils from wild-type cultures or from fibroblasts producing structurally abnormal fibrillin-1 molecules (22), and it was only occasionally noted here in negative staining of microfibrils from mg/N fibroblast cultures (Fig. 8C). The unique ultrastructure of the fibrillin-2 homopolymers again raised the possibility of a specialized function of this microfibril component during the early phase of elastogenesis.
FIGURE 8. Ultrastructural morphology of fibrillin polymers.

Microfibrils purified from wild-type (A), mgN/+ (C), or mgN/mgN (B and D) neonatal fibroblast cultures are compared after rotary shadowing and electron microscopy. Wild-type and mgN/+ microfibrils contain the normal and half the normal amount of fibrillin-1, respectively, and significantly less fibrillin-2. Instead, mgN/mgN microfibrils are composed exclusively of fibrillin-2. A density just to one side of each globular domain was seen only in the fibrillin-2 microfibrils and best visualized after negative staining of the fibrillin-2 microfibrils (D). Similar electron densities were occasionally observed in microfibrils from mgN/+ fibroblast cultures (C) Arrows point to the globular beads, and arrowheads mark some of the densities. Scale bar, 100 nm.
DISCUSSION
Genetic studies in mice were used to delineate the diverse contributions of two structurally related matrix components, fibrillins 1 and 2, to the development and function of the aorta. There are two main findings of these studies. First, we have shown that loss of Fbn1 gene expression in mgN/mgN mice results in neonatal death due to structural impairment of elastic tissues that are normally subjected to stretching and expansile forces, such as the aorta, diaphragm, and lungs. These results are consistent with the conclusion from previous analyses of mice with other Fbn1 mutations that the main role of fibrillin-1 microfibrils is to impart force-bearing structural support to elastic tissues during life ex utero (17, 18). However, we demonstrated here that the aortic phenotype of mgN/mgN mice is accounted for by abnormal maturation of the vessel wall even in the presence of normal levels of elastin cross-links and that tissue repair processes are induced during aneurysm progression. These findings significantly extend the previous conclusion that fibrillin-1 is predominantly involved in tissue homeostasis during postnatal life. Second, by generating mutant mice with graded deficiency of fibrillin proteins, we documented that loss of one or both Fbn1 alleles in an Fbn2 null background causes embryonic death. This genetic finding rectifies the previous conclusion that fibrillin-2 is dispensable for initiation of matrix assembly and embryonic development (3, 21). In addition, histological evidence from these mutant mice demonstrates for the first time that elastic fiber formation requires fibrillin.
The wall of the aorta best exemplifies the interdependent relationship that exists between resident cells and the elastic matrix during morphogenesis and organ growth (7, 8). Our data show that the absence of fibrillin-1 microfibrils perturbs maturation of the aortic wall during early neonatal life. Similar to lysyl oxidase-deficient mice (15, 16), loss of fibrillin-1 dramatically impacts the performances of other elastic tissues that are subject to physiological stresses during neonatal growth, such as the diaphragm and lungs. Although microfibrils can by themselves support hemodynamic load in the circulatory systems of invertebrates and lower vertebrates, the evolutionary emergence of elastin and the development of an elastic fiber system closely integrated with the surrounding VSMC (the elastic unit) were the main factors that enabled higher vertebrates to meet the demand for non-linear elasticity of the arterial wall (32). The fact that both elastin and Fbn1 null mice can only complete fetal development further emphasizes the difference in mechanical compliance and, thus, matrix composition of elastic vessels in low and high blood pressure systems (12). Importantly, our finding of normal levels of elastin cross-links in mgN/mgN mice demonstrates that stabilization of the elastic unit in the aorta is not a process requiring fibrillin-1 microfibril assembly.
The mgN mouse lacks the histopathological manifestations that characterize the natural history of aneurysm progression in the mgR mouse, including evidence for a fibroproliferative response to vascular injury (18, 33). In contrast to the absence of histological findings, tissue repair processes are apparently induced in the aorta of mgN/mgN mice during aneurysm progression. The evidence includes stimulation of PAI-1 production and Smad2 phosphorylation, both of which are activated by transforming growth factor-β to promote matrix formation (31). Up-regulation of activin-A is also consistent with the role of this signaling molecule in the fibroproliferative response to vascular injury (29). Similarly, CYR61 induction is in line with its known function to support cell adhesion and regulate matrix remodeling as well as with the recent report of compromised integrity of large vessels in Cyr61 null mice (30, 34, 35). Although there may be other explanations, we hypothesize that the apparent discrepancy between activation of tissue repair and lack of histological evidence for it in mgN/mgN null mice may likely reflect the very rapid progression of vascular collapse in this particular mouse model of dissecting aneurysm. We further extrapolate from this neonatal lethal model of MFS that the lengthy process of elastic fiber fragmentation in the adult lethal model of MFS (the mgR mouse) may involve both the contribution of a structurally impaired tissue, which is expected to respond poorly to cyclic mechanical strain, and the progressive activation of processes of matrix remodeling, which are predicted to exacerbate disease progression and may be triggered by transforming growth factor-β misactivation (18, 33, 36). The potential involvement of hemodynamic stress in initiating the fibroproliferative response is consistent with preliminary data showing that homozygous and heterozygous mgN aortas display higher than normal levels of the angiotensin II receptor 1,4 which has been proposed to act as a “mechanical sensor” converting environmental stress into biochemical signals (37). An issue left unresolved by our study is the significance of dissecting aneurysm in the ascending aorta of mgN/mgN null mice rather than in the aortic root, which sustains most of the hemodynamic stress. It is interesting to note that this particular manifestation of mgN/mgN mice phenocopies human bicuspid aortic valve with ascending aortic aneurysm, a congenital heart malformation of unknown cause in which the intrinsic pathology of the aortic matrix and VSMC is believed to be responsible for aortic enlargement beyond that predicted by hemodynamic factors (38).
Early morphological findings of embryonic elastic fiber formation contributed to the long-held belief that microfibrils regulate elastogenesis by providing the scaffold that guides tropoelastin deposition (1). The subsequent demonstration that fibrillin-2 biosynthesis generally precedes that of fibrillin-1 and coincides with early morphogenesis identified fibrillin-2 microfibrils as the most likely regulators of elastic fiber assembly (20). Contrary to this prediction, however, Fbn2 null mice were later found to be viable and to display only a skeletal patterning defect due to altered BMP signaling (21). Thus, it was concluded that either fibrillin-2 is dispensable in matrix assembly or fibrillin-1 may compensate for its loss during embryonic development (3, 21). The finding that virtually all Fbn1−/−;Fbn2−/− embryos and half of the Fbn1+/−;Fbn2 −/− embryos fail to complete fetal development strongly supports the latter hypothesis. This genetic evidence also indicates that fibrillin-1 compensation for the absence of fibrillin-2 is exquisitely dependent on the amount of fibrillin-1 molecules deposited into the matrix. There are two not mutually exclusive models that explain this gene dosage effect. Elastic tissue formation may be driven by the total amount of extracellular microfibrils, or fibrillin-2 containing microfibrils may play a functionally distinct role in organizing the embryonic matrix. Because fibrillin-1 and fibrillin-2 are structurally homologous molecules with few distinguishing features between them (2, 3, 5), the ultrastructural findings presented here were surprising and at variance with the absence of asymmetric microfibrils in previous negative stains of wild-type cultures and fibroblasts producing only structurally abnormal fibrillin-1 molecules (22). Moreover, two additional findings suggest that the relative amounts of fibrillin-1 and fibrillin-2 may also influence microfibril structure and, thus, tissue development. They include the occasional presence of asymmetric microfibrils in mgN/+ cultures (Fig. 8C) and sub-clinical abnormalities in the architecture of the aortic wall in mgN/+ mice.4 We, therefore, propose from these correlative and indirect lines of evidence that fibrillin-2 performs discrete functions during matrix formation and tissue development by mediating distinct interactions with other matrix components, surrounding cells, and/or soluble signaling molecules. Fibrillin-2 may do this either by assembling a unique microfibrillar structure, which displays an apparent asymmetric direction, or by directing a unique protein-protein interaction, which is visualized as the electron densities to the side of each bead.
In summary, a model is beginning to emerge from ours and additional gene function studies in mice with regard to how different matrix components may participate in the development and function of the aortic wall (9–18). Fibrillin (either fibrillin-1 or fibrillin-2) is absolutely required for the initial assembly of the elastic fiber, conceivably by mediating critical interactions with cells, other matrix components, and/or signaling molecules. Fibrillin-2 is dispensable under conditions of increasing mechanical demands. The role of fibrillin-2 overlaps only in part with that of fibrillin-1, the key function of which is to guide the maturation of the aortic wall during fetal development and neonatal growth. Fibrillin-1 is also important to impart force-bearing structural support to the adult tissue. Fibulin-5 and lysyl oxidase-like protein-1 contribute to later developmental stages of elastogenesis than fibrillin, whereas elastin plays the same dual role as fibrillin in regulating the development and maturation of the elastic wall and in conferring structural integrity. Finally, lysyl oxidase stabilizes the aortic matrix during fetal development. Ongoing studies of mice with combined deficiencies of different elastic fiber components along with the biochemical characterization of molecular interactions among these extracellular proteins will further test the validity of these conclusions and other hypotheses.
Acknowledgments
We thank Drs. H. Dietz and D. Rifkin for the critical reading of the manuscript and many invaluable suggestions, Drs. F. Laub and S. Smaldone for technical insights, B. Bertucci, C. Dragomir, and N. Mor for excellent assistance with the experiments, and K. Johnson for organizing the manuscript.
Footnotes
This work was supported by National Institutes of Health Grants AR42044, AR48111, and AR049698, by the Fundação de Amparo à Pesquisa do Estado de São Paulo and Conselho Nacional de Desenvolvimento Cientifico e Tecnoló gico, the Shriners Hospitals for Children, and the James D. Farley family and the St. Giles Foundation.
The abbreviations used are: MFS, Marfan syndrome; CCA, congenital contractural arachnodactyly; CYR61, cysteine-rich angiogenic protein 61; Fbn1 and Fbn2, the mouse genes coding for fibrillin-1 and fibrillin-2, respectively; PAI-1, plasminogen activator inhibitor-1; VSMC, vascular smooth muscle cells; MMP, matrix metalloproteinase.
L. Carta, unpublished data.
References
- 1.Mecham RP, Davis E. In: Extracellular Matrix Assembly and Structure. Yurchenco PD, Birk DE, Mecham RP, editors. Academic Press, Inc; New York: 1994. pp. 281–314. [Google Scholar]
- 2.Handford PA, Downing AK, Reinhardt DP, Sakai LY. Matrix Biol. 2000;19:457–470. doi: 10.1016/s0945-053x(00)00100-1. [DOI] [PubMed] [Google Scholar]
- 3.Kielty CM, Sherratt MJ, Shuttleworth CA. J Cell Sci. 2002;115:2817–2828. doi: 10.1242/jcs.115.14.2817. [DOI] [PubMed] [Google Scholar]
- 4.Ramirez F. Curr Opin Genet Dev. 1996;6:309–315. doi: 10.1016/s0959-437x(96)80007-4. [DOI] [PubMed] [Google Scholar]
- 5.Charbonneau NL, Ono RN, Corson GM, Keene DR, Sakai LY. Birth Defects Res C Embryo Today. 2004;72:37–50. doi: 10.1002/bdrc.20000. [DOI] [PubMed] [Google Scholar]
- 6.Pyeritz RE. In: Principles and Practice of Medical Genetics. 4. Rimoin DL, Connor JM, Pyeritz RE, Korf BR, editors. Churchill Livingstone; New York, NY: 2002. pp. 3977–4020. [Google Scholar]
- 7.Brooke BS, Karnik SK, Li DY. Trends Cell Biol. 2003;13:51–56. doi: 10.1016/s0962-8924(02)00007-7. [DOI] [PubMed] [Google Scholar]
- 8.Kelleher CM, McLean SE, Mecham RP. Curr Top Dev Biol. 2005;62:153–188. doi: 10.1016/S0070-2153(04)62006-0. [DOI] [PubMed] [Google Scholar]
- 9.Yanagisawa H, Davis EC, Starcher BC, Ouchi T, Yanagisawa M, Richardson JA, Olson EN. Nature. 2002;415:168–171. doi: 10.1038/415168a. [DOI] [PubMed] [Google Scholar]
- 10.Nakamura T, Lozano PR, Ikeda Y, Iwanaga Y, Hinek A, Minamisawa S, Cheng CF, Kobuke K, Dalton N, Takada Y, Tashiro K, Ross J, Jr, Honjo T, Chien KR. Nature. 2002;415:171–175. doi: 10.1038/415171a. [DOI] [PubMed] [Google Scholar]
- 11.Liu X, Zhao Y, Gao J, Pawlyk B, Starcher B, Spencer JA, Yanagisawa H, Zuo J, Li T. Nat Genet. 2004;36:178–182. doi: 10.1038/ng1297. [DOI] [PubMed] [Google Scholar]
- 12.Li DY, Faury G, Taylor DG, Davis EC, Boyle WA, Mecham RP, Stenzel P, Boak B, Keating MT. J Clin Investig. 1998;102:1783–1787. doi: 10.1172/JCI4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karnik SK, Brooke BS, Bayes-Genis A, Sorensen L, Wythe JD, Schwartz RS, Keating MT, Li DY. Development. 2003;130:411–423. doi: 10.1242/dev.00223. [DOI] [PubMed] [Google Scholar]
- 14.Spencer JA, Hacker SL, Davis EC, Mecham RP, Knutsen RH, Li DY, Gerard RD, Richardson JA, Olson EN, Yanagisawa H. Proc Natl Acad Sci U S A. 2005;102:2946–2951. doi: 10.1073/pnas.0500058102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mäki JM, Räsänen J, Tikkanen H, Sormunen R, Mäkikallio K, Kari I, Kivirikko KI, Soininen R. Circulation. 2002;106:250–259. doi: 10.1161/01.cir.0000038109.84500.1e. [DOI] [PubMed] [Google Scholar]
- 16.Hornstra IK, Birge S, Starcher B, Bailey AJ, Mecham RP, Shapiro SD. J Biol Chem. 2003;278:14387–14393. doi: 10.1074/jbc.M210144200. [DOI] [PubMed] [Google Scholar]
- 17.Pereira L, Andrikopoulos K, Tian J, Lee SY, Keene DR, Ono R, Reinhardt DP, Sakai LY, Jensen-Biery N, Bunton T, Dietz HC, Ramirez F. Nat Genet. 1997;17:218–222. doi: 10.1038/ng1097-218. [DOI] [PubMed] [Google Scholar]
- 18.Pereira L, Lee SY, Gayraud B, Andrikopoulos K, Shapiro SD, Bunton T, Jensen-Biery N, Dietz HC, Sakai LY, Ramirez F. Proc Natl Acad Sci U S A. 1999;96:3819–3823. doi: 10.1073/pnas.96.7.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, Sakai LY, Dietz HC. J Clin Investig. 2004;114:172–181. doi: 10.1172/JCI20641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H, Hu W, Ramirez F. J Cell Biol. 1995;129:1165–1176. doi: 10.1083/jcb.129.4.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arteaga-Solis E, Garyraud B, Lee SY, Shum L, Sakai L, Ramirez F. J Cell Biol. 2001;154:275–281. doi: 10.1083/jcb.200105046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gayraud B, Keene DR, Sakai LY, Ramirez F. J Cell Biol. 2000;150:667–679. doi: 10.1083/jcb.150.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Isogai Z, Aspberg A, Keene DR, Ono RN, Reinhardt DP, Sakai LY. J Biol Chem. 2002;277:4565–4572. doi: 10.1074/jbc.M110583200. [DOI] [PubMed] [Google Scholar]
- 24.Starcher B, Condrad M. Connect Tissue Res. 1995;31:133–140. doi: 10.3109/03008209509028401. [DOI] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.Davis EC. J Cell Sci. 1994;107:727–736. doi: 10.1242/jcs.107.3.727. [DOI] [PubMed] [Google Scholar]
- 27.Kuro-o M, Nagai R, Nakahara K, Katoh H, Tsai RC, Tsuchimochi H, Yazaki Y, Ohkubo A, Takaku F. J Biol Chem. 1991;266:3768–3773. [PubMed] [Google Scholar]
- 28.Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Molloy CJ, Taylor DS, Pawlowski JE. J Endocrinol. 1999;161:179–185. doi: 10.1677/joe.0.1610179. [DOI] [PubMed] [Google Scholar]
- 30.Mo FE, Muntean AG, Chen CC, Stolz DB, Watkins SC, Lau LF. Mol Cell Biol. 2002;22:8709–8720. doi: 10.1128/MCB.22.24.8709-8720.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonewald LF. Crit Rev Eukaryotic Gene Expression. 1999;9:33–44. [PubMed] [Google Scholar]
- 32.Faury G. Pathol Biol. 2001;49:310–325. doi: 10.1016/s0369-8114(01)00147-x. [DOI] [PubMed] [Google Scholar]
- 33.Bunton TE, Jensen Biery N, Gayraud B, Ramirez F, Dietz HC. Circ Res. 2001;88:37–43. doi: 10.1161/01.res.88.1.37. [DOI] [PubMed] [Google Scholar]
- 34.Chen CC, Chen N, Lau LF. J Biol Chem. 2001;276:10443–10452. doi: 10.1074/jbc.M008087200. [DOI] [PubMed] [Google Scholar]
- 35.Chen CC, Mo FE, Lau LF. J Biol Chem. 2001;276:47329–47337. doi: 10.1074/jbc.M107666200. [DOI] [PubMed] [Google Scholar]
- 36.Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC. Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 37.Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I. Nat Cell Biol. 2004;6:499–506. doi: 10.1038/ncb1137. [DOI] [PubMed] [Google Scholar]
- 38.Gelb BD. Curr Opin Cardiol. 2004;19:110–115. doi: 10.1097/00001573-200403000-00007. [DOI] [PubMed] [Google Scholar]