

Abstract
Poly(ethylene oxide) (PEO) and low molecular weight poly(ethylene glycol) (PEG) were covalently immobilized on silicon wafers and gold films by way of the CH insertion reaction of perfluorophenyl azides (PFPAs) by either photolysis or thermolysis. The immobilization does not require chemical derivatization of PEO or PEG, and polymers of different molecular weights were successfully attached to the substrate to give uniform films. Microarrays were also generated by printing polymer solutions on PFPA-functionalized wafer or Au slides followed by light activation. For low molecular weight PEG, the immobilization was highly dependent on the quality of the film deposited on the substrate. While the spin-coated and printed PEG showed poor immobilization efficiency, thermal treatment of the PEG melt on PFPA-functionalized surfaces resulted in excellent film quality, giving, for example, a grafting density of 9.2 × 10−4/Å2 and an average distance between grafted chains of 33 Å for PEG 20,000. The anti-fouling property of the films was evaluated by fluorescence microscopy and surface plasmon resonance imaging (SPRi). Low protein adsorption was observed on thermally-immobilized PEG whereas the photoimmobilized PEG showed increased protein adsorption. In addition, protein arrays were created using polystyrene (PS) and PEG based on the differential protein adsorption of the two polymers.
Keywords: PEG, covalent immobilization, non-fouling surface, polymer arrays, protein patterning
Introduction
Microarrays and lab-on-a-chip devices have gained increasing popularity, and are now frequently used in bioassays and diagnoses. Miniaturization allows for high-density of ligands and materials to be fabricated on a single chip, drastically increasing the throughput and speed of analyses and biological evaluations.[1–14] A major challenge in bioanalyses is the assay sensitivity, ie, the signal/noise ratio. To increase assay sensitivity, two general strategies can be envisioned: amplification of the specific signal or reduction of the background noise. Proteins are frequently the targets in bioanalysis, and therefore, the development of protein-resistant surfaces to minimize non-specific protein adsorption is critical in the reduction of background noises.[15–18]
A limited number of non-fouling materials have been used to fabricate the protein-resistant surfaces which meet the requirements of practical biosensors.[15] PEO and low molecular weight PEG are water-soluble, nontoxic and non-immunogenic polymers. They are considered as effective protein-resistant materials and have been frequently used in micro-devices for biomedical and analytical applications.[15, 19–26] Several theories, including the large excluded volume of PEO and low molecular weight PEG, the entropic and osmotic repulsion resulting from protein approaching PEG/PEO, have been used to explain their low protein adsorption property.[23, 26, 27] Covalent immobilization of PEO and low molecular weight PEG has been achieved using the “graft-from” and “graft-to” methods. In the “graft-from” approach, an initiator is covalently attached to the substrate and the polymer is synthesized in situ by polymerizing monomers from the surfaces. In this case, high density polymer brushes are often produced giving thick films that offer excellent anti-fouling property.[28] The “graft-to” approach uses a functionalized polymer and the polymer is immobilized by a surface-specific coupling reaction. A number of different PEG derivatives can be used in this approach,[26, 29, 30] including PEG-silane,[31–34] PEG-aldehyde,[35] tresyl-PEO,[21] and PEG-thiol.[15, 36] Alternatively, specific methods such as plasma polymerization can also be used to graft PEG/PEO on the surfaces.[37]
Herein, we report an alternative approach to the immobilization of PEO and PEG films on silicon wafers and gold slides. The method utilizes a functionalized PFPA, which act as a coupling agent to covalently attach PEO and PEG thin films to substrate surface. The immobilization is a result of CH insertion reaction to the polymers by perfluorophenyl nitrene generated by either photolysis [38–41] or thermolysis[42] of the PFPA. The process is fast, occurring in minutes instead of hours as for most conventional conjugation procedures. The method is also versatile, applicable to PEO as well as low molecular weight PEG, and no specific functional groups on the polymer are needed. Importantly, the process can be readily integrated with the microarray fabrication process where the polymer can be positioned at specific locations via coating, printing, or lithography. In this article, we report the detailed procedures for the covalent immobilization of PEO and PEG films and their characterization by ellipsometry, AFM, and contact angle goniometry. The antifouling property of the films was tested where the non-specific protein adsorption was evaluated by fluorescence microscopy and surface plasmon resonance imaging (SPRi). Results from the studies were applied to create a protein array by a simple adsorption process using a hybrid polymer array.
Experimental Procedures and Methods
Materials
Milli-Q water for contact angle measurements as well as for gold slides and silicon wafer cleaning was obtained from a Millipore Milli-Q system with at least 18.2 MΩ resistivity. Concentrated H2SO4, H2O2 (35%), toluene, chloroform, dichloromethane and methanol were purchased from Fisher. Ethanol (95%) was purchased from Aaper Alcohol & Chemical Co. (Shelbyville, KY). Dichloromethane was dried by refluxing in CaH2 for 3 h and was distilled before use. Other solvents were used as received. PS ( ca 280,000), PEO ( 100,000, 300,000, 600,000, 1,000,000), PEG 20,000 ( 16,000–24,000) and poly(2-ethyl-2-oxazoline) (PEOX, ca 50,000 and 200,000) were used as received from Aldrich. PEG 6,000 (approx. Mol. Wt. 6,000), fluorescein isothiocyanate-conjugated bovine serum albumin (BSA-FITC) and phosphate buffered saline (PBS, pH 7.4) were purchased from Sigma. PEG 400 was purchased from Polyscience Inc. (Warrington, PA). The long-pass optical filter (280-nm) and high index N-SF10 glass slides (18 mm × 18 mm × 1 mm) were purchased from Schott Glass Technologies, Inc. (Fullerton, CA). PFPA-silane[41, 43] and PFPA-disulfide[44] (Scheme 1) were synthesized following previously published procedures.[42–44]
Scheme 1.

Immobilization of PEO or PEG via PFPA.
Instrumentation
Spin coating was performed on a P6204 spin-coater (Specialty Coating Systems, Inc., Indianapolis, IN). Sonication was carried out in a Branson 1510 sonicator (Fisher). The quartz mask used in photolithography, containing 5.5-micron in diameter circular openings, was a gift from Electro Scientific Industries, Inc. (Portland, OR). Irradiation of the polymer films was executed at ambient temperature with a medium pressure Hg lamp (450 W, Hanovia Ltd.). The lamp reached its full power after ~3 min warm-up to an intensity of 2.5 mW/cm2 at 21 cm from the source as measured by an OAI 306 UV powermeter (Optical Associates Inc. Milpitas, CA) with a 260-nm sensor.
IR spectra were recorded on a Nicolet Magna 550 FT-IR spectrometer (Thermo Fisher Scientific, Inc.) equipped with a Smart iTR attenuated total reflectance (ATR) accessory and a FT-80 Fixed 80° grazing angle accessory. Contact angles were measured on a Ramé-Hart model 250 standard goniometer (Ramé-Hart Instrument Co., Netcong, NJ). The advancing contact angle (θA) was determined by placing a drop of milli-Q water from a syringe dispenser attached to the instrument, advancing the periphery of the drop by adding water at the rate of 0.05 μL/s of at a time interval of 1.0 s, and recording the contact angle as well as the diameter of the droplet. The receding contact angle (θR) was measured by withdrawing water from the drop at the same rate and time interval, and recording the contact angle and the diameter of the droplet. The needle was kept within the water drops throughout the measurements. Data were recorded and analyzed using the DROPimage Advanced v2.2 software. Film thickness measurements were made on a Gaertner Model L116A ellipsometer (Gaertner Scientific Co.) with He/Ne laser (2 mW, Melles Griot) at an incident angle of 70° in the manual mode. The following refractive indices were used to determine the thickness of various film layers: SiO2 1.465, PFPA-silane 1.503, PS 1.592, PEOX 1.520 and PEO/PEG 1.459. Atomic force microscopy (AFM) images were collected on a Nanoscope III system (Veeco Instruments, Inc.) using a 125-μm cantilever equipped with a silicon nitride tip in the tapping mode at an oscillating frequency of 300 kHz. SEM images were obtained on an ISI SS40 scanning electron microscope (International Scientific Instruments, Inc.) equipped with the Oxford ISIS EDX system. The fluorescence image was taken on a Leica DMRB microscope (Leica Microsystems, Inc.) in upright mode with 450–490 nm excitation and 515 nm emission. The polymer microarrays on the surface of SPR chips were fabricated using a BioOdyssey™ Calligrapher™ MiniArrayer (Bio-Rad Laboratories Inc.). The capillary pin of 360 μm in diameter (MCP360, Bio-Rad) was used to generate spot sizes of ca. 400 μm. SPRi experiments were conducted using a SPRimager® II system (GWC Technologies, Inc.). Images were analyzed using the Digital Optics V++ Version 4 software.
Immobilization of PEO and PEOX on Silicon Wafers
Silicon wafers with a native oxide layer were cleaned in the piranha solution (3:1v/v conc. H2SO4/H2O2) at 80–90 °C for 2 hours (Caution: the piranha solution reacts violently with organic solvents.), washed with boiling water for 1–2 h, and then dried under nitrogen. Cleaned wafers were soaked in a solution of PFPA-silane in toluene (3 mg/mL) for 12 hours, rinsed with toluene followed by ethanol, and dried under nitrogen. The wafers were allowed to cure at room temperature overnight. The cured wafers were spin-coated at 2000 rpm for 60 seconds with a solution of PEO or PEOX in chloroform (10 mg/mL). The films were irradiated with the medium pressure Hg lamp for 5 min. A 280-nm optical filter was placed on the film surface during irradiation. The films were then sonicated in chloroform for 5 min, washed in Milli-Q water for 1 h, and dried under nitrogen. For PEO 600,000 and 1,000,000, additional 10 min sonication in water was included in the cleaning procedure after washing in water.
Immobilization of PEG on Silicon Wafers
Piranha-cleaned wafers were soaked in a solution of PFPA-silane in toluene (5 mg/mL) for 4 hours, rinsed in a gentle stream of ethanol, dried under nitrogen, and allowed to cure at room temperature for 12 hours. The wafers were dipped into melt PEG at ~70 °C, removed from the melt, and were then heated at 140 °C for various lengths of time. The samples were soaked in H2O while shaking until no visible PEG particles were observed on the surface. The wafers were then sonicated in Milli-Q water for 5 min, and dried under a stream of nitrogen.
Samples that required additional UV irradiation after heating were prepared as follows. PFPA-functionalized wafers were immersed in melt PEG, heated at 140 °C for the specified length of time, and irradiated immediately for 5 min. The unbound polymer was removed by shaking the sample in Milli-Q water until no visible PEG particles were observed. The wafers were then sonicated in Milli-Q water for 5 min, and dried with nitrogen.
Fabrication of hybrid PS/PEG structures
To fabricate PS/PEG hybrid structures, patterned PS films were first created as follows. A PFPA-functionalized wafer was spin-coated with a solution of PS in toluene (10 mg/mL) at 2000 rpm for 60 s. The sample was placed in a home-made photolithography apparatus with the photomask held in close contact with the wafer surface by applying vacuum. The 280-nm filter was then placed on top of the mask, and the entire assembly was irradiated with the medium pressure Hg lamp for 20 min. The wafer was sonicated in toluene for 5 min, rinsed with toluene and dried with nitrogen. The wafer, having patterned PS structures on the surface, was then dipped in melted PEG at 70 °C, and heated at 140 °C for 15 min. The sample was extracted in Milli-Q water to remove PEG particles, sonicated in Milli-Q water and chloroform for 5 min, respectively, and dried with nitrogen.
Protein Adsorption
The BSA-FITC solution (0.3 mg/mL) was prepared in a pH 7.4 PBS buffer (0.01 M). Wafers with attached polymer films or patterned structures were incubated in the solution at 4 °C for 1 hour, rinsed with PBS, and dried with nitrogen. The samples were imaged with a fluorescence microscope at 450–490 nm excitation and 515 nm emission.
Fabrication of Polymer Arrays on SPR Chips
SPR chips were fabricated as follows. High index N-SF10 glass slides were cleaned in piranha solution (3:1 v/v conc. H2SO4/H2O2) at r.t. for 60 min. The slides were thoroughly washed in boiling water three times for 60 minutes each. The slides were then coated with a 2 nm thick Ti followed by a 45 nm gold film in an electron beam evaporator (SEC-600, CHA Industries, Fremont, CA) at the Microfabrication Lab, Washington Technology Center (University of Washington).
Before they were chemically functionalized, these SPR chips were again cleaned with the piranha solution for 1 min, washed thoroughly 3 times with boiling water for 30 min each, and dried under a stream of nitrogen. Cleaned SPR chips were soaked in a solution of PFPA-disulfide in chloroform (10 mM) for 24 hours. The chips were then rinsed gently with chloroform and dried under nitrogen. Polymer samples of PEG, PEO, and PEOX were prepared at the concentration of 30 mg/mL in Milli-Q water, and each solution (200 μL) was then pipetted to the well of the 96-well source plate according to the microarray layout designed for the experiment. The source plate was immediately placed into the MiniArrayer for printing the designed array on functionalized SPR chip. The pin approach speed to the source plate and SPR chip was 10 and 5 mm/s, respectively, and the dwell time in the source plate and on SPR chip was 500 and 100 ms, respectively. Milli-Q water and 80% ethanol were used as the wash solutions for 3000 and 5000 ms, respectively, followed by drying in vacuum. A constant humidity of 60% was maintained throughout the printing process. PS spots were generated by manually spotting a solution of PS in toluene (30 mg/mL) onto the SPR chip using a pipette tip. The printed SPR chip was then irradiated for 15 min with the medium pressure Hg lamp. A 280-nm optical filter was placed on the film surface during irradiation. The chip was rinsed thoroughly in toluene, Milli-Q water and chloroform for 2 h in each solvent, respectively, and was finally dried with nitrogen.
The SPR sensor chips with a UV-immobilized PEG and PEOX microarray, and a thermally immobilized PEG film were prepared as follows. PEG and PEOX solutions were printed on half of the SPR chip functionalized with PFPA-disulfide. The other half without spotted polymer was covered by a piece of aluminum foil, and the chip was irradiated for 15 min with a 280-nm optical filter, washed in Milli-Q water and chloroform (1 h in each solvent), and dried with nitrogen. The chip was then dipped into melted PEG and was heated at 140 °C for 30 min to thermally immobilize PEG on the gold surface. The excess PEG was removed by thoroughly washing the chip in Milli-Q water and chloroform, respectively (2 h in each solvent), and the chip was dried with nitrogen.
SPR Imaging
SPRi experiments were carried out at an angle of incidence in the range of 42–55° at room temperature. The polymer microarray was primed in pH 7.4 PBS buffer until a stable baseline was reached. A BSA solution in the PBS buffer (10 μg/mL or 100 μg/mL) was then injected and SPR responses recorded. The flow rate was kept at 100 μL/min. Data acquisition was conducted by selecting the area within printed spots on a microarray image, i.e., region of interest (ROI). An average of 30 images/frames was utilized and SPR signals converted to normalized percentage in reflectivity (%ΔR) following the protocol provided by GWC. All SPR images were collected using the V++ image analysis software package.
Results and discussion
Covalently immobilized polymer films were prepared by coating the polymer on PFPA-functionalized surface followed by light or thermal activation (Scheme 1). The covalent bond formation was established by way of the CH insertion reaction between the polymer and the perfluorophenyl nitrene generated by photolysis or thermolysis.[40, 45–47] Silicon oxide (silicon wafers, glass slides) and gold films were used as the substrates, and were treated with PFPA-silane or PFPA-disulfide to introduce the perfluorophenyl azido groups on the substrate surfaces. The functionalization can be conveniently monitored by FTIR, especially the characteristic azido peak at around 2130 cm−1 (Fig. 1).
Figure 1.

FTIR spectrum of PFPA-disulfide (top), and GAATR-IR spectrum of PFPA on Au film (bottom). The arrows indicate the azido absorption at 2138 cm−1.
High molecular weight PEO was immobilized by spin coating a solution of PEO in chloroform on PFPA-functionalized substrate followed by light activation. The unattached polymer was removed by solvent washing, leaving a thin film of PEO on the substrate surface. The film thicknesses were measured, and results are shown in Table 1. The thickness of the immobilized PEO film increased with the molecular weight of PEO, a result that is consistent with the “graft-to” mechanism that higher molecular weight polymers, having larger radii of gyration, would yield thicker films.[43]
Table 1.
Thickness of immobilized PEO films with respect to PEO molecular weight.
| Molecular Weight | Film Thicknessa |
|---|---|
| 100,000 | 20 ± 1 Å |
| 300,000 | 23 ± 2 Å |
| 600,000 | 28 ± 1 Å |
| 1,000,000 | 36 ± 4 Å |
each data was the average of three samples.
Initial attempt to immobilize low molecular weight PEG by photochemical initiation was unsuccessful. Spin coating the chloroform solution of PEG on the PFPA-silane-functionalized silicon wafer followed by UV irradiation and solvent extraction yielded no detectable PEG films on the surface. In order to obtain covalently immobilized uniform films, the cast polymer film must cover the wafer without defects, and the polymer needs be in close proximity with the surface azido groups for the CH insertion to occur.[48] Low molecular weight PEG gave solutions of low viscosity, and spin-coated films were thin even when a saturated PEG solution was used. In addition, low molecular weight PEG tends to crystallize, resulting in uneven films which compromised the film quality as well as the contact with the substrate surface. Thermal activation was also attempted where the sample was heated. When the temperature was higher than the melting temperature of PEG (63 °C for PEG 20,000[49]), the spin-coated PEG films dewetted and formed droplets on the PFPA-functionalized wafer during heating. This is expected since the PFPA-functionalized surface was fairly hydrophobic; the static, advancing and receding water contact angles of the surface were 78.5±1.0°, 84.9±0.3° and 51.1±1.4°, respectively.
Successful immobilization was accomplished by dipping the PFPA-functionalized wafer in melt PEG to form a thicker polymer layer and heating the sample at 140 °C.[40, 42] At temperatures higher than the melting point of PEG, the polymer liquidified, forming a conformal contact with the wafer surface. Because the immobilization occurs at the interface between the surface azido groups and the polymer in the close proximity, a monolayer of polymer was covalently immobilized and the excess polymer could be removed by solvent extraction. The thickness of the immobilized PEG film increased with the heating time, and did not change after ~30 min of heating (open squares in Fig. 2). When the samples were heated for less than 30 min, thinner films were obtained due to incomplete reactions. The immobilization could also be accomplished by simultaneously heating the samples at 70 °C while irradiating. Thicker films of ~ 30 Å were obtained in this case, and were achieved at a shorter heating time (~10 min) (open triangles in Fig. 2). However, if the films were irradiated after the heated samples were cooled to room temperature, thinner films were obtained (solid squares in Fig. 2). These results demonstrated that a conformal contact of the polymer with the substrate, by heating PEG in this case, was critical to ensure efficient polymer immobilization.
Figure 2.

(a) Thicknesses of immobilized PEG 20,000 as a function of time by heating at 140 °C (□), heating at 140 °C followed by UV irradiation for 5 min (■), or heating at 70 °C while irradiating (△). Control samples were prepared in the similar manner except that the wafers were not functionalized with PFPA (▲). (b) AFM image of immobilized PEG 20,000 film by heating at 140 °C.
To confirm that the film immobilization was indeed caused by the presence of the surface azido groups, control samples were prepared with Piranha-cleaned wafers that were not functionalized with PFPA. No substantial amount of polymer was detected even after extended heat treatment (solid triangles in Fig. 2). Note that the polymer is not expected to decompose under the experimental conditions. The study by Conder et al. demonstrated that PEG was heat-stable, and no decomposition was observed when heating at below 160 °C in air.[50]
Dynamic contract angle measurements were carried out where the advancing and receding contact angles of the PEG films were determined. The contact angle hysteresis, the difference between advancing and receding contact angles, measures the adhesion of the liquid droplet to the surface and is influenced by surface roughness, chemical heterogeneity, and the extent of interactions between the water droplet and the surface constituents.[51] A small contact angle hysteresis is consistent with a surface that is homogeneous and smooth, and a large hysteresis implies a heterogenous surface or a surface that is rough or unstable. The PEG films generated in our studies were smooth, showing a root-mean-square (RMS) roughness value of 0.66 nm as measured by AFM (Fig. 2b). The static contact angle, θA, and θR of the immobilized PEG films were measured to be 32.4±1.5°, 34.3±2.4° and 23.0±2.7°, respectively. During the course of the advancing contact angle measurement, the contact angle remained more or less constant when additional water droplets were added, and the drop diameter increased steadily (Fig. 3a). Similar observation was reported by Mackel et al.,[52] likely due to the favorable interaction of PEG with water where PEG acts as a hydrogel uptaking water into its structure.
Figure 3.

Typical water contact angle and drop diameter graphs of immobilized PEG 20,000 film by heating at 140 °C during the course of advancing (a) and receding (b) contact angle measurements. The θA and θR values were computed from measurements of 10 samples each.
Analysis of dynamic contact angle data provides additional information on the structure and morphology of the attached polymer layer. The small hysteresis (11.3°) indicates a smooth and chemically homogeneous surface of the PEG film. In the work of Kaper et al., PEO brushes (MW 9,800) were synthesized on glass surface, and θA and θR of the resulting films were 54° and 22°, respectively.[53] The larger hysteresis (32°) than that of our films may be due to the higher mobility of the polymer brush structure than the monolayer film in our case. The polymer brush was attached to the surface at one end whereas our monolayer polymer was likely bound to the surface at multiple points along the polymer chain.
The surface concentration (τ), average distance between immobilized polymer chains (D) and grafting density (σ) were calculated, and results are shown in Table 2. The surface concentration τ, i.e., the amount of immobilized polymer per unit area (μg/cm2), can be calculated by applying Eq. 1,[54] where d is the thickness of the polymer film, and ρdry is the density of dry PEO or PEG layer which was assumed to be a constant value of 1 g/cm3.[21]
Table 2.
The surface concentration τ, average distance between polymer chains D, and grafting density σ for immobilized PEG and PEO films.
| Polymers | τ (μg/cm2) | D (Å) | σ (Å−2) |
|---|---|---|---|
| PEG 20,000 | 0.30 | 33 | 9.2 × 10−4 |
| PEO 100,000 | 0.20 | 91 | 1.2 × 10−4 |
| PEO 300,000 | 0.23 | 147 | 4.6 × 10−5 |
| PEO 600,000 | 0.28 | 189 | 2.8 × 10−5 |
| PEO 1,000,000 | 0.36 | 215 | 2.2 × 10−5 |
| (1) |
Another parameter, D, measures the average distance between the immobilized polymer coils (Scheme 2). Assuming that the unit surface area per single polymer is D2 (Scheme 2), the unit weight of a single polymer would be D2·d·ρdry, where d is the thickness of the polymer film. Considering that the weight of a single polymer also equals M/NA, where M is the molecular weight of the polymer and NA is the Avogadro’s number, Eq. 2 is derived where D can be computed. This equation is identical to that used to calculate the D value for linear PEG immobilized via single-point attachment.[21]
Scheme 2.
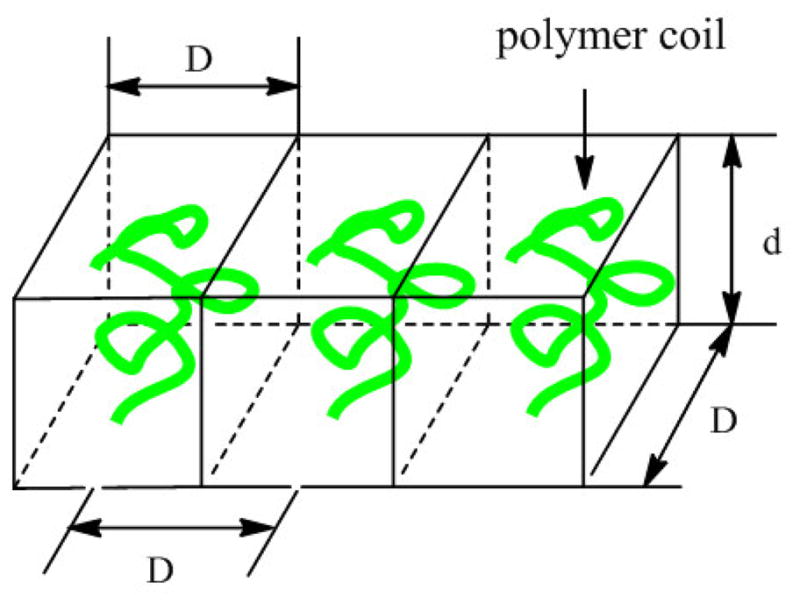
Polymer chains immobilized on surface.
| (2) |
The grafting density σ, defined as the number of molecules per unit area, can be calculated according to Eq. 3.[55, 56]
| (3) |
For PEG 20,000, films of ~30 Å in thickness were obtained by either thermal immobilization or a combination of light activation and heating. The corresponding grafting density, 9.2 × 10−4, is significantly higher than those of photochemically immobilized PEO films (Table 2). In addition, the average distance between PEG 20,000 chains grafted on the surface is around 33 Å, which is considerably smaller than those of the photoimmobilized PEO films (Table 2), implying a higher packing density. These numbers compare well with those reported for end-grafted polymers, for example by Sofia and coworkers, where the D value and the grafting density of PEG 20,000, calculated from Eqs. 2 and 3, were ~55 Å and 3.3 × 10−4 Å−2, respectively.[21] (Note that the grafting density in ref. 21 was defined as (a/D)2 where a is the size of the monomer unit.)
A preliminary test on the non-fouling property of the prepared PEG films was carried out by treating the films with BSA-FITC and the adsorbed protein was analyzed by fluorescence microscopy. In addition to PEG, PS, a well-known fouling polymer, and PEOX that is known to resist protein adsorption,[57] were also covalently immobilized using the same photocoupling chemistry. These polymers were used as the references together with the control samples of piranha-cleaned and PFPA-functionalized surfaces. Low fluorescence intensities were observed on PEG and PEOX films (Fig. 4a, 4b), consistent with the finding that PEG and PEOX effectively resist protein adsorption. The fluorescence intensity was the highest on the PS film (Fig. 4c), confirming that the protein was efficiently adsorbed on the PS surface non-specifically. The piranha-cleaned and the PFPA-functionalized surfaces showed moderate protein adsorption as evidenced by the increased fluorescence intensities on these surfaces (Fig. 4d, 4e). Bright fluorescence spots were obvious on the clean wafer, unlike the PEG and PEOX films that were uniform and even.
Figure 4.

Fluorescence microscopy images after incubating thermally immobilized PEG 20,000 (a); PEOX 200,000 (b); PS 280,000 (c); piranha-cleaned silicon wafer (d); and PFPA-functionalized wafer (e) in BSA-FTIC. Scale bars: 100 μm.
The anti-fouling property of the immobilized PEG films was further evaluated using SPRi. SPR is a label-free and real-time sensing technique, and SPRi offers an additional benefit of being able to detect multiple receptor-ligand interactions in an array format by simultaneously taking real-time measurements of each spot on the array.[58–64] Polymer microarrays consisting of six different polymer samples, including PS 280,000, PEG 400, PEG 6,000, PEG 20,000, PEOX 50,000, and PEOX 200,000, were fabricated on PFPA-functionalized SPR sensor chip (Fig. 5). PEG and PEOX were printed from the aqueous solutions using a robotic printer whereas PS was spotted from the toluene solution using a micropipettor. The polymers were immobilized by light activation and excess polymers were removed by solvent extraction. The BSA solution was then introduced to the flow cell having the sensor chip, and the SPR responses for each polymer recorded simultaneously. As expected, the PS films showed the highest BSA adsorption in comparison to PEG and PEOX (Fig. 5). However, the PEG films exhibited consistently higher BSA adsorption than PEOX at all BSA concentrations, contradicting with its excellent non-fouling property demonstrated by many others. As discussed above, PEG tends to crystallize, and when spotting on the surface, the rapid solvent evaporation may cause the polymer to crystallize. The poor contact of the polymer with the PFPA-functionalized surface would compromise the covalent bond formation leading to defects in the films.
Figure 5.
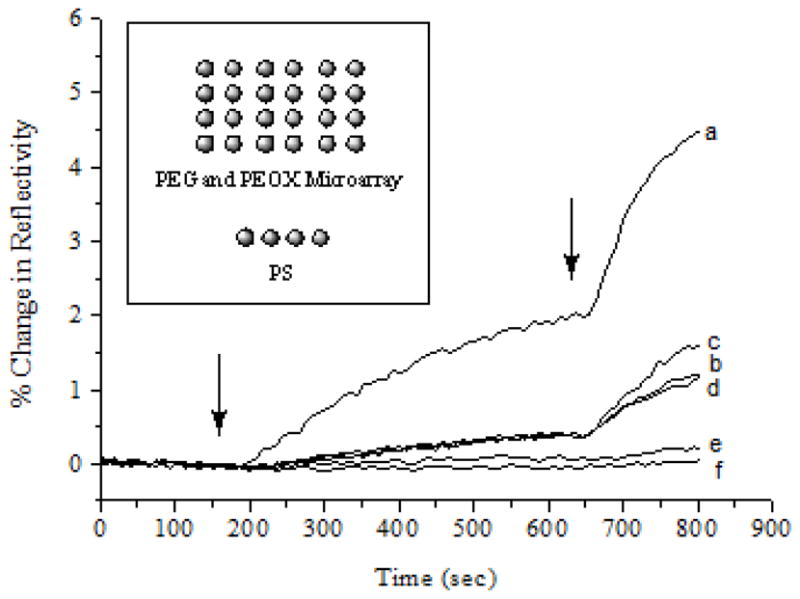
SPR responses of polymer microarray to BSA. The sensor chip was treated with pH 7.4 PBS buffer followed by BSA in PBS at the concentration of 10 μg/mL and 100 μg/mL, and injected at 150 s and 630 s, respectively. For clarity, only one response curve was shown for each polymer. Insert: Layout of the polymer microarray, consisting of six different polymers including (a) PS 280,000, (b) PEG 400, (c) PEG 6,000, (d) PEG 20,000, (e) PEOX 50,000, and (f) PEOX 200,000. All polymers were printed from an aqueous solution of 30 mg/mL concentration except for PS which was spotted from a toluene solution using a micropipettor. Each polymer was spotted in quadruplicate.
To test this hypothesis, thermally immobilized PEG films were included in the polymer microarray and the protein adsorption experiments were repeated using SPRi. Results in Fig. 6 were obtained from an array containing UV-immobilized PEG and PEOX by robotic printing, and thermally-immobilized PEG from melt PEG. Low BSA adsorption was observed on thermally immobilized PEG films, similar to that of UV-immobilized PEOX, whereas UV-immobilized PEG again showed higher BSA adsorption. Since the thermal activation gave smooth and thicker PEG films (Fig. 2), the higher protein adsorption on the UV-immobilized films was likely due to defects in the films resulting from the incomplete immobilization of PEG.
Figure 6.

SPR responses of the polymer array consisting of thermally-immobilized PEG 20,000 (a), UV-immobilized PEOX 200,000 (b), and UV-immobilized PEG 20,000 (c). The sensor chip was treated with pH 7.4 PBS buffer followed by BSA in PBS injected at 150 s (10 μg/mL) and 510 s (100 μg/mL), respectively. Each polymer was spotted in quadruplicate. Only one SPR curve was shown for clarity.
This immobilization chemistry was then applied to create protein arrays, taking advantage of the differential adsorption of proteins on different polymer surfaces. With a hybrid array of PS/PEG, proteins would be selectively adsorbed on PS films whereas PEG would provide boundaries resisting proteins from adhering in these regions, thus creating a protein array. The polymer thin film arrays were generated as shown in Figure 7a. Patterned PS was first fabricated by photolithography, where a PS film coated on PFPA-functionalized wafer was exposed to UV irradiation in the presence of a photomask having 5.5-μm circular openings. The PS in the un-exposed regions was removed by sonicating the wafer in toluene, exposing the un-reacted PFPA on the wafer surface. The sample was then dipped into melt PEG and was heated at 140 °C for 15 min. This resulted in the covalent attachment of PEG in the areas where there were no PS films. In regions where PS structures were present, no covalent bonds could be formed and thus the coated PEG layer on top of the PS structures was removed by solvent extraction. Array of covalently immobilized PS/PEG thin film were generated as shown in Figure 7b. The protein array was created by incubating the polymer array with BSA-FITC. The protein was adsorbed on PS films whereas areas covered with PEG had minimal protein adsorption (Fig. 7c).
Figure 7.

a) Fabrication of PS/PEG array. b) SEM image of PS/PEG array created. c) Fluorescence image of the array after treating with BSA-FITC.
Conclusions
An alternative approach was developed to covalently immobilize PEO and low molecular weight PEG on silicon wafers and gold films. The method relies on PFPA-functionalized surface, and PEO and PEG of various molecular weights were immobilized by either photochemical or thermal activation of the surface PFPA. For low molecular weight PEG, the immobilization was highly dependent on the quality of the film deposited. While the spin-coated and printed PEG showed poor immobilization efficiency, thermal treatment of the PEG melt on PFPA-functionalized surfaces with or without light activation resulted in excellent film quality and high immobilization efficiency. The grafting density of thermally immobilized PEG 20,000 was comparable to those reported for the end-grafted PEG films. The anti-fouling property of the immobilized films was evaluated by fluorescence microscopy and SPRi. Low protein adsorption was observed on thermally-immobilized PEG whereas the photoimmobilization of printed PEG showed increased protein adsorption, an observation that was consistent with the film quality. The advantage of the developed method is its generality and simplicity. No polymerization or chemical modification of the polymer is required, and the immobilization is carried out by a simple coating and fast activation procedure. The process can be readily applied to the fabrication of microarrays where the covalently immobilized polymer films serve as the non-fouling surface reducing the background noises and thus increasing the detection sensitivity. This strategy was successfully demonstrated in a simple experiment where the hybrid structures of PS/PEG were used to create protein microarrays taking advantage of the differential protein adsorption property of PS and PEG.
Research highlights.
A simple, fast, and general method for the immobilization of un-derivatized PEO and PEG films using perfluorophenyl azides.
High grafting density comparable to PEG films prepared by other graft-to approach
Lowest protein adsorption on thermally-immobilized films, corresponding to the highest film thickness and quality.
Acknowledgments
The authors thank Dr. Chunfei Li of the Physics Department for the technical assistance on SEM, Yin-Chu Chen of Oregon Medical Laser Center and Prof. Anna-Louise Reysenbach of the Biology Department for their assistance in fluorescence microscopy. This work was supported by the National Institutes of General Medical Science (NIGMS) under NIH Award Numbers R01GM080295 and R15GM066279.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brocchini S, James K, Tangpasuthadol V, Kohn J. J Biomed Mater Res. 1998;42:66. doi: 10.1002/(sici)1097-4636(199810)42:1<66::aid-jbm9>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 2.de Gans BJ, Schubert US. Macromol Rapid Commun. 2003;24:659. [Google Scholar]
- 3.Anderson DG, Levenberg S, Langer R. Nat Biotechnol. 2004;22:863. doi: 10.1038/nbt981. [DOI] [PubMed] [Google Scholar]
- 4.Anderson DG, Putnam D, Lavik EB, Mahmood TA, Langer R. Biomaterials. 2005;26:4892. doi: 10.1016/j.biomaterials.2004.11.052. [DOI] [PubMed] [Google Scholar]
- 5.Tweedie CA, Anderson DG, Langer R, Van Vliet KJ. Adv Mater. 2005;17:2599. [Google Scholar]
- 6.Tourniaire G, Collins J, Campbell S, Mizomoto H, Ogawa S, Thaburet JF, Bradley M. Chem Commun. 2006:2118. doi: 10.1039/b602009g. [DOI] [PubMed] [Google Scholar]
- 7.Urquhart AJ, Anderson DG, Taylor M, Alexander MR, Langer R, Davies MC. Adv Mater. 2007;19:2486. [Google Scholar]
- 8.Urquhart AJ, Taylor M, Anderson DG, Langer R, Davies MC, Alexander MR. Anal Chem. 2008;80:135. doi: 10.1021/ac071560k. [DOI] [PubMed] [Google Scholar]
- 9.Zhang R, Liberski A, Khan F, Diaz-Mochon JJ, Bradley M. Chem Commun. 2008:1317. doi: 10.1039/b717932d. [DOI] [PubMed] [Google Scholar]
- 10.Koehn M. J Pept Sci. 2009;15:393. doi: 10.1002/psc.1130. [DOI] [PubMed] [Google Scholar]
- 11.Tare RS, Khan F, Tourniaire G, Morgan SM, Bradley M, Oreffo ROC. Biomaterials. 2009;30:1045. doi: 10.1016/j.biomaterials.2008.10.038. [DOI] [PubMed] [Google Scholar]
- 12.Kurkuri MD, Driever C, Johnson G, McFarland G, Thissen H, Voelcker NH. Biomacromolecules. 2009;10:1163. doi: 10.1021/bm801417s. [DOI] [PubMed] [Google Scholar]
- 13.Mei Y, Gerecht S, Taylor M, Urquhart A, Bogatyrev SR, Cho SW, Davies MC, Alexander MR, Langer RS, Anderson DG. Adv Mater. 2009;21:2781. doi: 10.1002/adma.200803184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hook AL, Anderson DG, Langer R, Williams P, Davies MC, Alexander MR. Biomaterials. 2010;31:187. doi: 10.1016/j.biomaterials.2009.09.037. [DOI] [PubMed] [Google Scholar]
- 15.Prime KL, Whitesides GM. Science. 1991;252:1164. doi: 10.1126/science.252.5009.1164. [DOI] [PubMed] [Google Scholar]
- 16.Heyes CD, Groll J, Moller M, Nienhaus GU. Mol Biosyst. 2007;3:419. doi: 10.1039/b700055n. [DOI] [PubMed] [Google Scholar]
- 17.Krishnan S, Weinman CJ, Ober CK. J Mater Chem. 2008;18:3405. [Google Scholar]
- 18.Jain P, Baker GL, Bruening ML. Annu Rev Anal Chem. 2009;2:387. doi: 10.1146/annurev-anchem-060908-155153. [DOI] [PubMed] [Google Scholar]
- 19.Rabinow BE, Ding YS, Qin C, McHalsky ML, Schneider JH, Ashline KA, Shelbourn TL, Albrecht RM. J Biomater Sci Polym Ed. 1994;6:91. doi: 10.1163/156856295x00788. [DOI] [PubMed] [Google Scholar]
- 20.Prime KL, Whitesides GM. J Am Chem Soc. 1993;115:10714. [Google Scholar]
- 21.Sofia SJ, Premnath V, Merrill EW. Macromolecules. 1998;31:5059. doi: 10.1021/ma971016l. [DOI] [PubMed] [Google Scholar]
- 22.Alcantar NA, Aydil ES, Israelachvili JN. J Biomed Mater Res. 2000;51:343. doi: 10.1002/1097-4636(20000905)51:3<343::aid-jbm7>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 23.Andrade JD, Hlady V. Adv Polym Sci. 1986;79:1. [Google Scholar]
- 24.Gombotz WR, Guanghui W, Horbett TA, Hoffman AS. J Biomed Mater Res. 1991;25:1547. doi: 10.1002/jbm.820251211. [DOI] [PubMed] [Google Scholar]
- 25.Graham NB. Poly(ethylene Glycol) Chemistry. Plenum; New York, NY: 1992. Poly(ethylene Glycol) Gels and Drug Delivery. [Google Scholar]
- 26.Harris JM, editor. Poly(ethylene glycol) chemistry. Plenum; New York, NY: 1992. Introduction to Biotechnical and Biomedical Applications of Poly(ethylene Glycol) [Google Scholar]
- 27.Jeon SIL, Andrade JH, Degennes JD, PG J Colloid Interface Sci. 1991;142:149. [Google Scholar]
- 28.Hucknall A, Rangarajan S, Chilkoti A. Adv Mater. 2009;21:2441. doi: 10.1002/adma.200803125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amiji M, Park K. J Biomater Sci Polym Ed. 1993;4:217. doi: 10.1163/156856293x00537. [DOI] [PubMed] [Google Scholar]
- 30.Llanos GR, Sefton MV. J Biomater Sci Polym Ed. 1993;4:381. doi: 10.1163/156856293x00069. [DOI] [PubMed] [Google Scholar]
- 31.Popat KC, Mor G, Grimes CA, Desai TA. Langmuir. 2004;20:8035. doi: 10.1021/la049075x. [DOI] [PubMed] [Google Scholar]
- 32.Zhang MQ, Desai T, Ferrari M. Biomaterials. 1998;19:953. doi: 10.1016/s0142-9612(98)00026-x. [DOI] [PubMed] [Google Scholar]
- 33.Yang ZH, Galloway JA, Yu HU. Langmuir. 1999;15:8405. [Google Scholar]
- 34.Papra A, Gadegaard N, Larsen NB. Langmuir. 2001;17:1457. [Google Scholar]
- 35.Kingshott P, Thissen H, Griesser HJ. Biomaterials. 2002;23:2043. doi: 10.1016/s0142-9612(01)00334-9. [DOI] [PubMed] [Google Scholar]
- 36.Unsworth LD, Sheardown H, Brash JL. Langmuir. 2005;21:1036. doi: 10.1021/la047672d. [DOI] [PubMed] [Google Scholar]
- 37.Gombotz WR, Guanghui W, Hoffman AS. J Appl Polym Sci. 1989;37:91. [Google Scholar]
- 38.Yan M, Harnish B. Adv Mater. 2003;15:244. [Google Scholar]
- 39.Liu L, Yan M. Angew Chem, Int Ed. 2006;45:6207. doi: 10.1002/anie.200602097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan M. Chem--Eur J. 2007;13:4138. doi: 10.1002/chem.200700317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu L, Engelhard MH, Yan M. J Am Chem Soc. 2006;128:14067. doi: 10.1021/ja062802l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan M, Ren J. Chem Mater. 2004;16:1627. [Google Scholar]
- 43.Bartlett MA, Yan M. Adv Mater. 2001;13:1449. [Google Scholar]
- 44.Wang X, Ramstrom O, Yan M. J Mater Chem. 2009;19:8944. doi: 10.1039/B917900C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soundararajan N, Platz MS. J Org Chem. 1990;55:2034. [Google Scholar]
- 46.Keana JFW, Cai SX. J Org Chem. 1990;55:3640. [Google Scholar]
- 47.Poe R, Schnapp K, Young MJT, Grayzar J, Platz MS. J Am Chem Soc. 1992;114:5054. [Google Scholar]
- 48.Graupner RK, Yan MD. Langmuir. 2004;20:8675. doi: 10.1021/la0494728. [DOI] [PubMed] [Google Scholar]
- 49.Data from Aldrich. In.
- 50.Conder JR, Fruitwala NA, Shingari MK. J Chromatogr. 1983;269:171. [Google Scholar]
- 51.Johnson REJ, Dettre RH. J Phys Chem. 1964;68:1744. [Google Scholar]
- 52.Mackel MJ, Sanchez S, Kornfield JA. Langmuir. 2007;23:3. doi: 10.1021/la0617762. [DOI] [PubMed] [Google Scholar]
- 53.Kaper HJ, Busscher HJ, Norde W. J Biomater Sci, Polym Ed. 2003;14:313. doi: 10.1163/156856203321478847. [DOI] [PubMed] [Google Scholar]
- 54.Cuypers PA, Corsel JW, Janssen MP, Kop JMM, Hermens WT, Hemker HC. J Biol Chem. 1983;258:2426. [PubMed] [Google Scholar]
- 55.Halperin A, Tirrell M, Lodge TP. Adv Polym Sci. 1992;100:31. [Google Scholar]
- 56.Brittain WJ, Minko S. J Polym Sci Part A: Polym Chem. 2007;45:3505. [Google Scholar]
- 57.Konradi R, Pidhatika B, Muhlebach A, Textor M. Langmuir. 2008;24 doi: 10.1021/la702917z. [DOI] [PubMed] [Google Scholar]
- 58.Boozer C, Kim G, Cong SX, Guan HW, Londergan T. Curr Opin Biotechnol. 2006;17:400. doi: 10.1016/j.copbio.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 59.Yeatman E, Ash EA. Electron Lett. 1987;23:1091. [Google Scholar]
- 60.Homola J, Yee SS, Gauglitz G. Sens Actuators, B. 1999;54:3. [Google Scholar]
- 61.Brockman JM, Nelson BP, Corn RM. Annu Rev Phys Chem. 2000;51:41. doi: 10.1146/annurev.physchem.51.1.41. [DOI] [PubMed] [Google Scholar]
- 62.Bally M, Halter M, Voros J, Grandin HM. Surf Interface Anal. 2006;38:1442. [Google Scholar]
- 63.Ray S, Mehta G, Srivastava S. Proteomics. 2010;10:731. doi: 10.1002/pmic.200900458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scarano S, Mascini M, Turner APF, Minunni M. Biosens Bioelectron. 2010;25:957. doi: 10.1016/j.bios.2009.08.039. [DOI] [PubMed] [Google Scholar]