

Abstract
Co-axial electrospun fibers can offer both topographical and biochemical cues for tissue engineering applications. In this study, we demonstrate the sustained treatment of hemophilia through a non-viral, tissue engineering approach facilitated by growth factor-releasing co-axial electrospun fibers. FVIII-producing skeletal myotubes were first engineered on aligned electrospun fibers in vitro, followed by implantation in hemophilic mice with or without a layer of core-shell electrospun fibers designed to provide sustained delivery of angiogenic or lymphangiogenic growth factors, which serves to stimulate the lymphatic or vascular systems to enhance the FVIII transport from the implant site into systemic circulation. Upon subcutaneous implantation into hemophilic mice, the construct seamlessly integrated with the host tissue within one month, and specifically induced either vascular or lymphatic network infiltration in accordance with the growth factors released from the electrospun fibers. Engineered constructs that induced angiogenesis resulted in sustained elevation of plasma FVIII and significantly reduced blood coagulation time for at least two months. Biomaterials-assisted functional tissue engineering was shown in this study to offer protein replacement therapy for a genetic disorder such as hemophilia.
Keywords: Hemophilia, skeletal muscle engineering, angiogenesis, lymphangiogenesis, electrospinning, protein replacement therapy
INTRODUCTION
Skeletal myoblasts, a population of muscle-specific progenitor cells, hold attractive therapeutic potential for gene therapy [1–5]. Normally responsible for the growth, repair and regeneration of skeletal muscle, myoblasts have long life span and are amenable to expansion and genetic engineering in vitro [6, 7]. However, injected myoblasts typically experience poor survival upon injection into the host [2]. We, as well as other groups, have demonstrated that aligned fibrous topography can induce the cultured myoblasts to stretch along the fibers, upregulate the membrane stretch-activated ion channels and facilitate differentiation into multi-nucleated myotubes [8–11]. These myotubes align and integrate with the scaffold according to the architecture of the fibrous substrate, which is mimicry of native skeletal myofiber structure [10]. However, simply implanting the biomimetic engineered tissue into the host is unlikely to be sufficient to encourage full tissue integration in vivo because of the lack of vascular and lymphatic, not to mention neural networks. In this study, we attempt to evaluate the tissue integration of engineered skeletal muscle in the presence of functional growth factor delivery from co-axial electrospun fibers [12, 13], with the ultimate goal of defining the potential of such tissue engineering-mediated nonviral gene therapy. We tested this concept in a clinically relevant hemophilic mouse model.
Hemophilia A is an X-linked genetic disorder that leads to a deficiency of plasma factor VIII (FVIII), a crucial cofactor in the proteolytic activation of factor X to factor Xa in the blood clotting cascade. Currently, hemophilic patients are required to take regular injections of recombinant plasma FVIII concentrates throughout their lifetime. The high cost of protein replacement therapy and the inconvenience of frequent intravenous injections have fueled the development of gene therapy. An attractive aspect of using gene therapy approach is the relatively low threshold for success: slight elevation of plasma FVIII level can offer measurable clinical benefits [14, 15]. While non-viral gene transfer strategies are relatively safe, they have shown low and transient improvement in plasma FVIII level [16–19]. Viral gene therapies have produced promising results in pre-clinical and phase I clinical trials, but faced limitations with T-cell mediated immunity [20–23]. Cell-based therapies have thus been pursued as alternatives for the treatment of hemophilia [24–26]. Recently, FVIII-transduced endothelial progenitor cells or endothelial cells derived from induced pluripotent stem cells have been transplanted into the liver to result in the phenotype correction of hemophilia A over a period of two months [27, 28]. In parallel, there have been approaches that investigate subcutaneous cell engraftment to treat hemophilia, involving the use of engineered endothelial progenitors or engineered myoblasts [29, 30]. The engrafted cells survived well at the implant site, but only resulted in limited increase in plasma FVIII level due to instability and poor transport of the protein from the implant site into circulation [30]. We seek to demonstrate a synergism of functional tissue engineering and controlled drug delivery to render the tissue-based approach of treating hemophilia viable.
We propose to engineer FVIII-producing skeletal muscle engineered construct (from here onwards, referred to as SMEC) that can integrate with the host and stimulate local lymphatic or vascular systems to improve FVIII transport, thereby providing prolonged treatment for hemophilia A. Adhering to a non-viral approach, skeletal myoblasts were isolated from mice, and engineered to stably produce FVIII via electroporation and clonal selection. The FVIII-producing skeletal myoblasts clones were differentiated into myotubes on aligned electrospun fibers, and implanted subcutaneously into the abdomen of hemophilic mice. Since protein transport into circulation is possible via both vascular or lymphatic systems, this study sets out to compare the efficacy of both approaches. Therefore, in addition to serving as a scaffold the aligned fibers also provide a controlled delivery of growth factors to stimulate local lymphatic (VEGFC) or vascular systems (VEGFA and PDGF-bb). The selection of the growth factors and dosage is based on careful consideration of published literature on vascular and lymphatic vessel development [31–34]. We hypothesize that the SMECs would enjoy excellent host-donor engraftment, and the FVIII protein produced by the donor tissue can efficiently transport into circulation via the infiltrating networks to ultimately reduce the clinical manifestation of hemophilia in a murine model.
MATERIALS AND METHODS
Fibrous scaffold engineering
In co-axial electrospinning, two needles of different gauge size were arranged in a concentric manner to dispense two different solutions concurrently (Sup Fig 1 a). 1 μg of growth factors (VEGFC (R&D systems), or VEGFA (VEGF-A165, Prospecbio) and PDGF-bb (Cordis Corporation)) dissolved in 100 μL phosphate buffered saline solution (PBS) was used as the core solution. Poly(urethane) dissolved in 75:25 v/v ratio of chloroform:ethanol (PU, AL 80, Cardiotech International, 10% w/v) was used as the shell solution. The solutions were set to dispense at a rate of 1mL/hr (core) and 6 mL/hr (shell) while being exposed to a high voltage gradient between the needles and a rotating wheel (4,000 RPM). The voltage gradient (15 kV over 5 cm) sheared the dispensing solution into fibers with core-shell feature, and electrospinning continued until the core solution was completely encapsulated. Small molecular porogen (0.7% wt/wt, polyethylene glycol, Mw - 3,400, Union Carbide Corporation, USA) was mixed into the shell solution (0.7% w/v) to facilitate sustained release as previously described[12, 13]. The released proteins were quantified via ELISA (R&D systems). The scaffolds were placed in 2 mL PBS at 37°C. At predetermined time points, the solutions were collected for quantification and replenished with fresh PBS. To demonstrate the co-axial feature of the electrospun fibers, the fibrous scaffolds were cut cross-sectionally in liquid nitrogen, sputter-coated with gold and viewed under scanning electron microscope (FEI XL30 SEM).
Engineering of minicircle cFVIII plasmid and stable FVIII clones
To transfect the isolated myoblasts to produce FVIII protein, a canine FVIII minicircle was constructed based on a previously published B-domain removed cFVIII sequence[35]. The gene was codon-optimized for the cDNA sequence expressed in canine cells, cloned into the pCA_Luc plasmid backbone via SalI and EcoRV sites to generate pCA_cFVIII, digested with SpeI and XmnI, and subsequently blunt ligated with p2fC31. Final pMCA_cFVIII construct was obtained by plasmid amplification and processing. Isolated myoblasts were induced to exhibit long term transgene expression by electroporation followed by antibiotic selection for stable clones. One million cells were co-transfected using the Amaxa Nucleofector instrument (Program B32, Lonza) with four μg of green fluorescent protein (pMaxGFP, Lonza) plasmid and two μg of hygromycin plasmid. Two days post nucleofection, hygromycin (2000× dilution) was added to provide selection against non-hygromycin resistant cells. GFP positive clones were selected using cloning discs (Scienceware). The selected cell clones were expanded and checked for their transgene expression and capability to differentiate into skeletal muscle. The engineering of myoblasts to produce FVIII was performed by screening for positive clones with supernatant secretion of canine FVIII via ELISA (Affinity Biologicals). The medium were supplemented with 50 ng/mL of von Willebrand Factor (Haematologic Technologies) to help stabilize the produced protein.
Isolation of myoblasts and cell culture
Skeletal myoblasts were isolated from the quadriceps of mice (B6129SF2/J, Jackson Laboratories) and purified through a series of preplating. The purity of isolated cell culture was assessed by immunostaining and flow cytometry with muscle markers such as α-actinin, MyoD and desmin (Abcam). Cell growth and expansion were maintained in 1:1 mixture of high glucose Dulbecco’s Modified Eagle Medium (DMEM, Gibco) and F12 mixture (Gibco), supplemented with 8% fetal bovine serum (Sigma), 8% bovine calf serum (Sigma), 0.5% chick embryo extract (Accurate chemicals) and 1% Penicillin/Streptomycin (Gibco). Cell differentiation was induced with DMEM supplemented with 8% horse serum (Sigma) and 1% Penicillin/Streptomycin. No myoblast cultures were used beyond fifteen passages.
In vivo experimental design and surgical procedure
The tissue-engineered skeletal muscle constructs (SMECs) were prepared for in vivo studies in the following procedure. Prior to cell seeding, the fibrous scaffolds were sterilized for one hour under UV light and coated with 50 μg/mL of fibronectin solution. Engineered myoblasts (GFP or FVIII) were seeded on fibronectin-coated aligned fibrous scaffolds at a density of 3*104 cells/cm2. The cell-seeded constructs were maintained in growth medium for five days before switching to differentiation medium for seven days. During the differentiation phase, the aligned fibrous scaffold was rolled onto a pre-sterilized silicone rod. This procedure enhanced cell-cell contact and extracellular matrix deposition to result in a multiple layered tissue structure (final dimension is 1.5 cm by 1 cm with a thickness of 200 μm). The cross sections of the tissue structure were evaluated with hematoxylin and eosin staining (H&E) and SEM. For the animal groups that received scaffolds with releasing angiogenic (VEGFA + PDGF-bb) or lymphangiogenic (VEGFC) growth factors, the tissue engineered constructs would consist of an additional layer of controlled release fibers produced by co-axial electrospinning. The controlled release portion of the scaffold was wrapped on the outside of the cell seeded portion and has an average thickness of 10 μm.
Mice were anesthetized with oxygen (2L/min) and isoflurane (2%). The animal’s abdomen was sterilized and a 2 cm incision was created in the skin layer of the abdominal area of the mice. The GFP+ construct was sutured on four corners to the abdominal wall with 4/0 PGA sutures. The wound was immediately closed with wound clips. The wound clips were removed seven days post implantation. To limit potential blood loss in the hemophilic mice, 100 μL of human FVIII protein (at a concentration of 10 U/mL) was injected through tail vein prior to surgery. B6129SF2/J mice (n = 3 per group, age ranging from 3–4 weeks) from Jackson Laboratories were used in this study. 4 animal groups were evaluated at one week and one month time points:
Myoblasts: Mice received a subcutaneous injection of 3*105 skeletal myoblasts suspended in 100 μL PBS.
Construct: SMECs derived from 3*105 GFP+ cells.
C+VEGFC: SMECs releasing 1 μg of VEGFC
C+VEGFA: SMECs releasing 1 μg of VEGFA and PDGF-bb
Histology and immunostaining
At predetermined time points, Mice that received implants were euthanized through CO2 inhalation and the tissues surrounding the implant site were surgically removed, fixed in 4% paraformaldehyde solution, embedded in paraffin and sectioned for analysis. The presence of implants could be identified by the persistence of GFP signal. The evaluation of inflammation and host-donor tissue response was based on qualitative H&E staining and immunostaining for fibroblasts (5B5, Abcam) and macrophages (RM0029-11H3, Abcam). Tissue integration was evaluated via immunostaining for skeletal muscle specific markers (sarcomeric α-actinin [EA-53], fast skeletal myosin heavy chain [MY-32] and desmin, Abcam). The same sections were also stained for Ki-67 (Abcam) and CD-8 (Abcam) to probe for proliferating donor cells and cytotoxic T-cells, respectively.
Changes in the lymphatic and vascular networks surrounding the implants were evaluated based on immunostaining for lymphatic endothelial cells (lymphatic vessel endothelial receptor 1, Abcam) and smooth muscle (smooth muscle actin, Abcam). All immunostaining images were taken with an inverted confocal microscope (Zeiss 510). The evaluation of diameter size of lymphatic and vascular vessels was performed using Image-J software, and the values reported are the mean±SD values based on a minimum of 300 blood vessels and 50 lymphatic vessels per group. Only positively stained vessels within one mm proximity from the implant site were included in the analysis. The reported values are based on three mice per group.
FVIII bioavailability and hemophilia animal study
A bioavailability study was performed based on construct alone, C+VEGFA and C+VEGFC groups. A group of untreated mice (n = 3) served as the control. All the mice in this study received 100 μL injection of human FVIII (at a concentration of 10U/mL in PBS) in the subcutaneous space above the implant site at six days post implantation. 24 hours post injection, 200 μL of blood samples were taken to determine the plasma hFVIII level via by ELISA (Affinity Biologicals). Changes in protein bioavailability were determined by the fraction of FVIII that ended up in circulation from the total amount of injected FVIII.
In order to demonstrate treatment efficacy for hemophilia, FVIII-producing SMECs (producing 1U of cFVIII per day) were implanted subcutaneously into hemophilic mice (n = 5). The surgery procedure was the same as previously detailed. Blood samples (100 μL) were collected from the animals every week via the submandibular vein. Plasma FVIII level and neutralizing antibody production were determined based on cFVIII ELISA and Bethesda assay, respectively. At two months post implantation, the blood coagulation times of various experimental groups were determined using partial thromboplastin time (PTT) assay with a coagulometer (BMD M1 Coatron Single-Channel Portable Coagulometer, Biomedica Diagnostics Inc.).
Statistical analysis
The data presented herein is expressed as mean±SD. Statistical analysis of local vascular and lymphatic networks, plasma FVIII level and blood coagulation time was performed using one-way ANOVA followed by Tukey’s test, with p<0.05 considered significant. Power analysis was used to calculate the proposed group size in the hemophilia mice study.
RESULTS
Engineering of core-shell fibers to provide controlled growth factor delivery
Co-axial electrospun fibrous scaffold was designed to serve as a tissue engineering substrate as well as a controlled release device to stimulate angio/ lymphangiogenesis. TEM and SEM images of the fiber cross sections demonstrated the definitive core feature in greater than 90% of the electrospun fibers, compared to none in fibers produced using monoaxial electrospinning (Fig 1a–c). The co-axial fibers could be engineered to align during electrospinning process to provide uniaxial surface topography for the differentiation of myoblasts (Fig 1d). Equally as important, fluorescently labeled protein (FITC-BSA) encapsulated within the co-axial fibers was distributed evenly within the core, as opposed to discrete aggregation in the monoaxially electrospun fibers (Fig 1e–f). Release of growth factors from the core-shell fibers was controlled by the nanoporous structure of the shell (Sup Fig 1 b). Eighty percent of the encapsulated VEGFA and PDGF were released in a near-linear fashion within two weeks, while sixty percent of VEGFC protein was released within ten days (Fig 1g–h). Our previous study has established that proteins released from these co-axial fibers maintained the same level of bioactivity as fresh growth factors [13].
Figure 1. Core-shell electrospun fibers to provide sustained protein delivery.
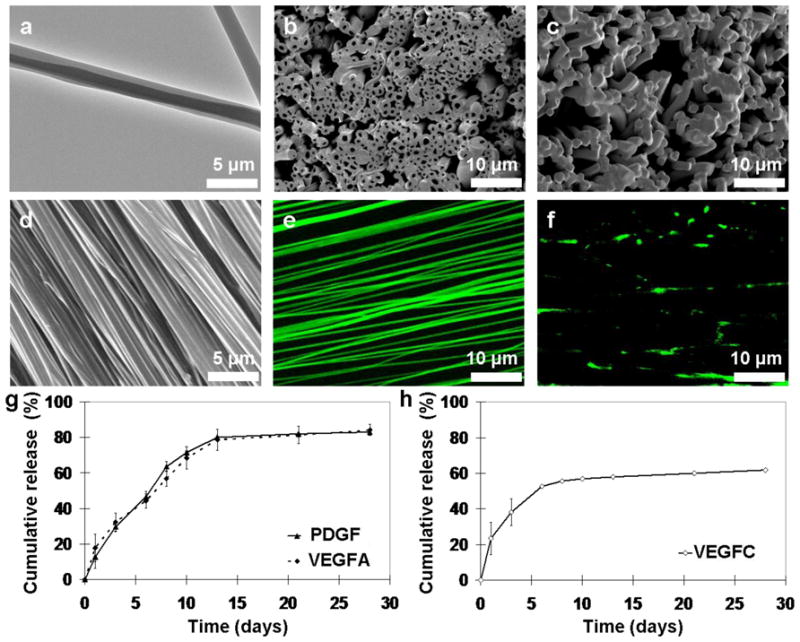
TEM (a) and SEM (b) images of the co-axially electrospun fibers. (c) Mono-axial fibers serve as a comparison to illustrate the core-shell structure. SEM (d) and confocal (e) images of encapsulated FITC-BSA within aligned co-axial fibers demonstrated uniform protein distribution within the fibers, compared to clusters of precipitated proteins in mono-axially electrospun fibers (f). Controlled release study of the encapsulated growth factors (PDGF and VEGFA in (g) and VEGFC in (h)) demonstrated that proteins were released in a sustained manner over a period of 30 days.
Primary skeletal muscle tissue engineering
Skeletal myoblasts were isolated and characterized using immunostaining and flow cytometry. Ninety percent of the purified myoblasts was stained positive for α-actinin, MyoD and desmin (Sup Fig 2 a–d). Stable GFP+ clones were derived from the isolated myoblasts (94% GFP positive after eight weeks, Fig 2j, Sup Fig 2 e) and used to engineer skeletal muscle on electrospun fibers. GFP+ myoblasts aligned and elongated on the uniaxial direction of the electrospun fibers (Fig 2a). When the cells were cultured in differentiation medium, they began to undergo cell-cell fusion and formed aligned, multi-nucleated myotubes over seven days (Fig 2b–d). Spontaneous contraction of the skeletal muscle engineered constructs (SMECs) was observed starting from four days post differentiation. The underlying thin fibrous scaffold (~10 μm) was engulfed by the cell cell-produced extracellular matrix after seven days in differentiation medium (Fig 2g-i). SMECs were produced by rolling layers of the cell-seeded constructs into thick 3-D tissue-like structures (Fig 2f). FVIII-producing SMECs were generated from myoblasts transfected with a cFVIII minicircle plasmid (Sup Fig 2 f). After an initial drop of FVIII production during the cell differentiation, the FVIII production level maintained at approximately 1,000 mU per day (Fig 2k) for over thirty days.
Figure 2. Skeletal myotubes engineered on electrospun fibers.
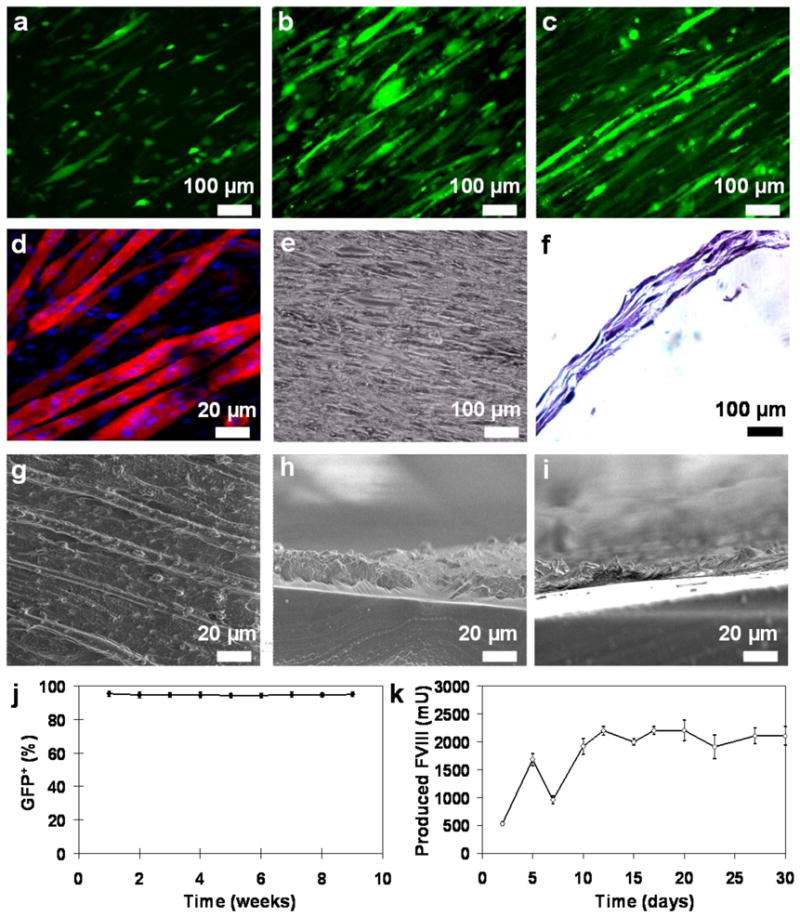
GFP+ myoblasts were seeded on aligned fibers and cultured under differentiation medium for 0 (a), 2 (b) and 7 days (c). (d) The differentiated skeletal muscle constructs were stained with α-actinin (red) and DAPI (blue) to illustrate myotube assembly. (e) Bright field image of the skeletal muscle construct. (f) H&E cross sectional view of the multi-layered SMEC. Top view (g) and cross sectional SEM images (h) revealed that cell-produced ECM (average thickness of 20 μm) encased the underlying scaffold. (i) Prior to cell culture, the scaffold had an average thickness of 10 μm. (j) Transfected myoblasts maintained stable GFP expression over nine weeks. (k) FVIII production from the FVIII engineered skeletal muscle construct averaged around 1U per day. The seeded cells were cultured in proliferation medium between days 0–5 and switched to differentiation medium between days 5–30. The transient drop off in production on day seven is likely due to the initiation of cell differentiation.
Host-donor tissue integration
The SMECs were implanted subcutaneously into the abdomen of mice). At one week, there was mild inflammation around the implants (Fig 3a, d). Higher magnification of the implanted SMEC revealed very high cell-to-scaffold ratio, with noticeable number of developing myofibers (Fig 3d). Immunostaining for fibroblasts and macrophages revealed significant amount of fibroblast infiltration but limited amount of activated macrophages (Fig 3e). At one month, the implanted tissue reconstructed and was well integrated into the host skeletal muscle with nearly seamless host to donor tissue interface (Fig 3b, f). The infiltration of fibroblasts and macrophages subsided at the one-month time point (Fig 3g). Two months post implantation, the regenerating tissue structure begin to resemble host skeletal muscle (Figure 3c). The implanted construct was stained positive for Ki 67, fast myosin heavy chain, desmin and α-actinin, indicating the resemblance of regenerating skeletal muscle (Fig 3l-m, o-q). Since the implanted tissue was derived from homologous cell source, there was very little recruitment of cytotoxic T-cells (Fig 3n). As a comparison, GFP+ myoblasts injected intramuscularly induced an intense level of macrophage activation at one week (Fig 3hi). At one month, the injected GFP+ myoblasts were cleared and only small number of macrophages was left at the injected site (Fig 3j–k). Such poor engraftment of injected myoblasts was widely documented [2]. There were two additional noteworthy observations made based on histology and immunostaining: transgene expression was still detectable one month post implantation (Fig 3g & l), and the underlying fibrous scaffold was undetectable, suggesting its embedment into the ECM of the donor tissue.
Figure 3. Host-donor tissue integration.
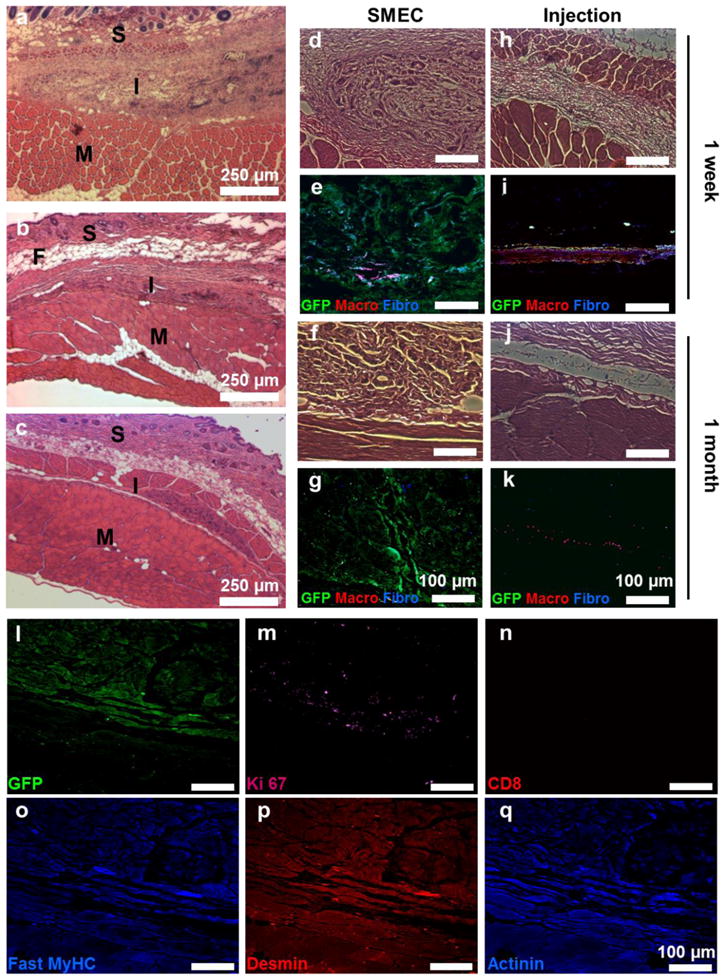
(a–c) Histology images of the tissue cross-section at one week (a), one month (b) and two month time points (c). (d–g) 20x histology and immunofluorescence images of the tissue cross-section at one week (d–e) and one month (f–g) time points. The tissues were labeled for GFP (green), fibroblasts (blue) and macrophages (red) to evaluate the host-donor response and integration. (h–k) As a control, the induced host response to injected GFP+ myoblasts was evaluated at one week (h–i) and one month (j–k). One month post implantation, the implanted tissue showed persistence of GFP signal (l), and was stained positive for Ki67 (m) and negative for cytotoxic T-cells (n). The implanted tissue was also stained positive for skeletal muscle specific markers such as fast skeletal myosin heavy chain (o), desmin (p) and α-actinin (q). In panels b–c, S = skin, M = muscle, I = implant and F = fat.
Angio/lymphangiogenesis induced by factors released from scaffold
Figure 4 illustrates the effect of the controlled release of growth factors on local vascular and lymphatic networks based on immunostaining for smooth muscle actin (SMA) and lymphatic vessel endothelial receptor 1 (LYVE-1), respectively. A baseline level of blood and lymphatic vessel infiltration around the implant without encapsulated growth factors can be seen at one week (Fig 4a) and one month (Fig 4d). SMECs encapsulated with VEGFA induced a two-fold increase in infiltrating blood vessel density over construct alone and VEGFC encapsulated SMECs (Fig 4b, i–j). The diameter of infiltrated blood vessels increased after one month, an indication of vascular network maturation (pink and red bars, Fig 4e, i). Similarly, VEGFC group induced significantly larger and higher density of LYVE-1+ vessels around the implants at one week and one month (Fig 4c, f–h). It is noteworthy that both VEGFA and VEGFC groups showed no significant cross influence on vascular or lymphatic networks (Fig 4g–j). Detectable higher concentration of VEGFA and VEGFC was observed in the respective groups at one week but in the construct alone group (Sup Fig 3). Taken together, the data from Figure 4 and Supplementary Figure 3 suggest that the induced angio/lymphangiogenesis is a result of higher local VEGFA or VEGFC concentration near the implant sites.
Figure 4. Angio/lymphangiogenesis induced by factors released from scaffold.
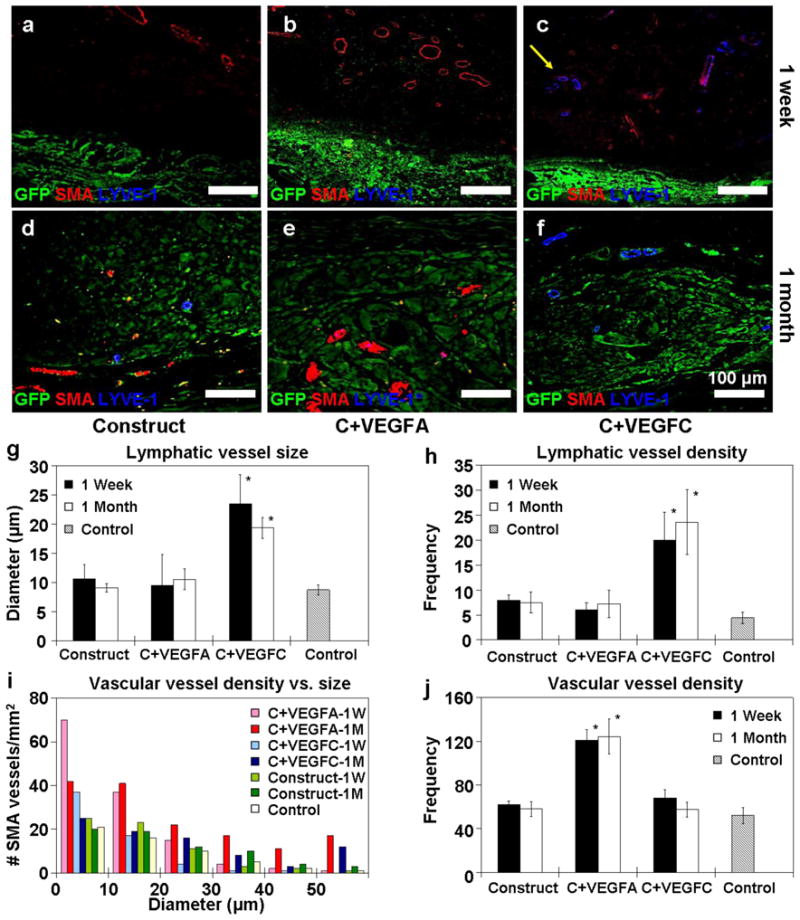
(a–f) Confocal microscopy images tracking localized angio/lymphangiogenesis around the GFP+ SMEC at one week (a–c) and one month (d–f). In the group encapsulated with VEGFA and PDGF-bb (b & e), there were significantly more developing blood vessels (labeled red for α-smooth muscle actin) in close proximity when compared to the construct alone group (a&d). Similarly, there was a significantly higher density of lymphatic vessels (labeled blue for LYVE-1) around the group encapsulated with VEGFC (c & f). The density and diameter of the developing blood and lymphatic vessels were quantified based on 3 mice (n=3) per group and reported in (g–j). The lymphatic vessels that developed around the VEGFC group remained larger in size (g) and higher in density (h) at 1 week and 1 month. Likewise, the VEGFA group induced a higher amount of microvessels (< 35 μm) at both 1 week and 1 month time points (i & j). In panels g–h, * denotes statistical significance over VEGFA, construct alone and control groups. Similarly, in panel j * represents statistical significance over VEGFC, construct alone and control groups.
Implanted tissue-engineered constructs result in phenotypic correction
A bioavailability assay was first conducted to evaluate how induced angio/lymphangiogenesis would influence protein transport from the implant site into circulation. Eight percent of the injected hFVIII protein near the implant site was detected in the circulation in VEGFA group, as opposed to approximately three percent in the VEGFC and construct alone groups, and no detectable hFVIII in the untreated control (Fig 5a). Subsequently, FVIII-producing SMECs were implanted into hemophilic mice to evaluate for phenotypic correction. In agreement with the bioavailability data, VEGFA group showed a statistically higher level of plasma FVIII (~10%) over VEGFC (~5%) and construct alone groups (Fig 5b). The enhanced level of plasma FVIII was sustained over one month and slowly declined over time, but still higher than the other two groups at the two month time point. There was undetectable level of anti-cFVIII antibody over the first month, but the construct alone group showed gradual increase in antibody production (Fig 5c). PTT assay performed at the two month time point revealed significant reduction in coagulation time in the VEGFA group over construct alone, VEGFC and untreated groups (Fig 5d). The observed coagulation time in the VEGFA group (47 seconds) resembles that of mild hemophilic manifestation.
Figure 5. Effects of FVIII-producing SMECs on phenotypic correction of hemophilic A mice.
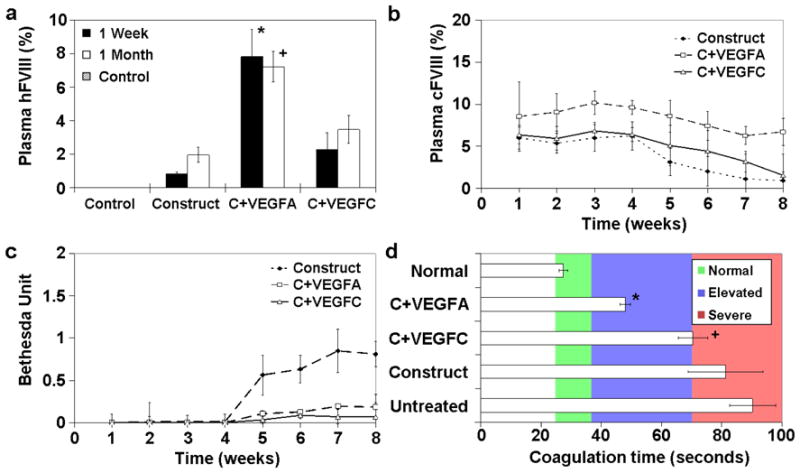
(a) FVIII was injected near the implant site to evaluate protein transport efficiency from the subcutaneous space into circulation. VEGFA group showed higher amount of plasma hFVIII (8%) compared to the other experimental groups. (b) Plasma cFVIII level of hemophilic mice receiving different SMEC implants over a period of two months. (c) Bethesda assay for development of anti-cFVIII antibodies over two months. (d) Blood coagulation time of different experimental groups at two month post SMEC implantation. Normal refers to non-hemophilic mice while untreated denotes untreated hemophilic mice. The reported values are based on 5 mice per group (n = 5). * denotes statistical significance over VEGFC, construct alone and control groups, while + represents statistical significance when compared to VEGFA, construct alone and control groups.
DISCUSSION
This study introduces the use of core-shell, controlled release electrospun fibers as substrates for the engineering of biomimetic skeletal muscle tissue, and its application towards the treatment of hemophilia. To our knowledge, this is the first study in which core-shell electrospun fibers have been tested in an animal model. The integration of the engineered tissue, development of local vascular or lymphatic networks and efficacy of implanted tissue in treating hemophilia were systematically investigated. Primary skeletal myoblasts were genetically engineered via electroporation and antibiotic selection to avoid any use of viral vectors in the cell engineering process. A minicircle plasmid was used to minimize any potential immune response. In vitro maturation of the genetically engineered myoblasts before implantation served to facilitate tissue integration. Controlled release of angiogenic factors from the scaffold enhanced the transport of the gene product from the implant site to the systemic circulation. This study validates the potential of applying functional engineered tissue for protein replacement therapy.
Electrospun fibrous scaffolds were used in this study to generate porous, biomimetic 3-D tissues that were rapidly integrated into the host tissue. The high cell-to-substrate ratio and the integration of underlying fibrous scaffold into the skeletal muscle ECM led to favorable host-donor tissue response by limiting the presentation of foreign materials (Fig 3). The lack of inflammation (macrophage activation and formation of fibrous capsule) and porous tissue layers enabled the seamless host-donor tissue integration into skeletal muscle as shown in Figure 3. Tissue integration was observed at a faster rate than previously findings that involved scaffolds producing acidic degradation byproducts (PLGA) [36, 37]. At two months, the implanted tissue began to closely resemble surrounding skeletal muscle (Figure 3c). Avoiding chronic inflammation and establishing rapid tissue integration are important for the development of infiltrating blood/lymphatic vessels, and thus optimizing the chance of success in the treatment of hemophilia.
Equally important is the aspect of localized angiogenesis versus lymphangiogenesis around the implant constructs in order to enhance the transport of produced FVIII protein. The use of engineered constructs to induce localized angiogenesis has been previously investigated [30, 38, 39]. The delivery of VEGFA alone induces leaky and non-functional blood vessels, and increases the potential of tissue edema and animal death at high dosage [30, 34]. This drawback can be countered by the co-delivery with PDGF-bb to stabilize the infiltrating vessels [32–34]. Therefore, the co-axial electrospun fibers were engineered to simultaneously deliver VEGFA and PDGF-bb to induce angiogenesis. This study also investigates the strategy of inducing lymphangiogenesis to enhance FVIII protein transport. Lymphatic system is generally responsible of regulating tissue osmosis and protein reabsorption, and the delivery of VEGFC can specifically stimulate local lymphangiogenesis [31, 40, 41]. However, the capacity of the lymphatic system to transport large protein such as FVIII is unreported.
Figure 4 suggests that there is significantly higher amount of vascular and lymphatic vessels around VEGFA and VEGFC implants, respectively. However, the bioavailability study shows that the induction of angiogenesis is significantly more effective in enhancing FVIII transport than lymphangiogenesis (Fig 5a). Although there is a higher level of lymphatic vessels developed around the VEGFC implants, it did not translate into significant improvement in FVIII transport (Fig 5b). This may be attributed to the tortuous path through the lymph nodes, where proteins are vulnerable to degradation. Induced local angiogenesis on the other hand allows the produce FVIII to access circulation directly. Another benefit of the VEGFA group is the increased level of von Willebrand Factor around the implants, which can help stabilize the produced FVIII protein in the subcutaneous space and reduce its immunogenicity through complexation [42].
In this study, the integrated FVIII-producing tissue-engineered constructs resulted in the sustained elevation of plasma FVIII protein and the correction of hemophilic phenotype. With induced local angiogenesis, plasma FVIII level reached 10% (a level considered as significantly therapeutic) over the first month. Blood coagulation time was significantly shortened in the VEGFA group at the 2 month time point. The encouraging results observed in this study validate the concept that functional tissue engineering would be a viable approach for protein replacement therapy.
CONCLUSIONS
In this study, we have used co-axial electrospun fibers in a non-viral, functional tissue engineering approach to derive a genetically engineered skeletal muscle implant for efficient protein replacement therapy. In a hemophilia-A mice model, FVIII protein producing skeletal myotubes were engineered on aligned electrospun fibers, while the accompanying core-shell electrospun fibers were designed to provide sustained delivery of VEGFA/PDGF or VEGFC. The SMECs implanted in the abdominal muscle of the mice integrated with the host tissue with one month and closely resembled host skeletal muscle in two months. At one month, the VEGF-A/PDGF-releasing SMECs significantly stimulated vascular ingrowth while the VEGFC-releasing SMECs encouraged lymphangiogenesis. The angiogenic SMECs produced higher level of plasma FVIII (10%) over the experimental period when compared to the lymphangiogenic SMECs (~5%) and control (~3%). Hemophilic mice implanted with angiogenic SMECs also experienced reduced blood coagulation time at the two month time point.
Supplementary Material
Acknowledgments
Support from NIH (HL89764) is acknowledged. The authors would like to thank Dr. Douglas Rouse for his assistance with surgical procedures, Dr. Yihua Loo for the gift of canine FVIII plasmid construct, and Andrew Yanchun Chou for his help with animal care.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
I-Chien Liao, Email: ichien.liao@duke.edu.
Kam W. Leong, Email: kam.leong@duke.edu.
References
- 1.Cossu G, Mavilio F. Myogenic stem cells for the therapy of primary myopathies: wishful thinking or therapeutic perspective? J Clin Invest. 2000;105(12):1669–74. doi: 10.1172/JCI10376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuang S, Rudnicki MA. The emerging biology of satellite cells and their therapeutic potential. Trends Mol Med. 2008;14(2):82–91. doi: 10.1016/j.molmed.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 3.Cao B, Deasy BM, Pollett J, Huard J. Cell therapy for muscle regeneration and repair. Phys Med Rehabil Clin N Am. 2005;16(4):889–907. doi: 10.1016/j.pmr.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 4.Regulier E, Schneider BL, Deglon N, Beuzard Y, Aebischer P. Continuous delivery of human and mouse erythropoietin in mice by genetically engineered polymer encapsulated myoblasts. Gene Ther. 1998;5(8):1014–22. doi: 10.1038/sj.gt.3300687. [DOI] [PubMed] [Google Scholar]
- 5.MacColl GS, Goldspink G, Bouloux PMG. Using skeletal muscle as an artificial endocrine tissue. J Endocrinol. 1999;162(1):1–9. doi: 10.1677/joe.0.1620001. [DOI] [PubMed] [Google Scholar]
- 6.Lorenzon P, Bernareggi A, Degasperi V, Nurowska E, Wernig A, Ruzzier F. Properties of primary mouse myoblasts expanded in culture. Exp Cell Res. 2002;278(1):84–91. doi: 10.1006/excr.2002.5562. [DOI] [PubMed] [Google Scholar]
- 7.Lu QL, Bou-Gharios G, Partridge TA. Non-viral gene delivery in skeletal muscle: a protein factory. Gene Ther. 2003;10(2):131–42. doi: 10.1038/sj.gt.3301874. [DOI] [PubMed] [Google Scholar]
- 8.Huang NF, Patel S, Thakar R, Wu J, Hsiao BS, Chu B, et al. Myotube assembly on nanofibrous and micropatterned polymers. Nano lett. 2006;6:537–42. doi: 10.1021/nl060060o. [DOI] [PubMed] [Google Scholar]
- 9.Engler AJ, Griffin MA, Sen S, Bonnemann CG, Sweeney HL, Discher DE. Myotube differentiate optimally on substrates with tissue-like stiffness:pathological implications for soft or stiff microenvironments. J Cell Biol. 2004;166:877–87. doi: 10.1083/jcb.200405004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liao IC, Liu JB, Bursac N, Leong KW. Effect of electromechanical stimulation on the maturation of myotubes on aligned electrospun fibers. Cell Mol Bioeng. 2008;1:133–45. doi: 10.1007/s12195-008-0021-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Formigli L, Meacci E, Sassoli C, Squecco R, Nosi D, Chellini F, et al. Cytoskeleton/stretch-activated ion channel interaction regulates myogenic differentiation of skeletal myoblasts. J Cell Physiol. 2007;211:296–306. doi: 10.1002/jcp.20936. [DOI] [PubMed] [Google Scholar]
- 12.Liao IC, Chen S, Liu JB, Leong KW. Sustained viral gene delivery through core-shell fibers. J Control Release. 2009;139(1):48–55. doi: 10.1016/j.jconrel.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liao IC, Chew SY, Leong KW. Aligned core-shell nanofibers delivering bioactive proteins. Nanomedicine. 2006;1(4):465–71. doi: 10.2217/17435889.1.4.465. [DOI] [PubMed] [Google Scholar]
- 14.Murphy SL, High KA. Gene therapy for haemophilia. Br J Haematol. 2008;140(5):479–87. doi: 10.1111/j.1365-2141.2007.06942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manno CS. Gene therapy for bleeding disorders. Curr Opin Hematol. 2002;9(6):511–5. doi: 10.1097/00062752-200211000-00008. [DOI] [PubMed] [Google Scholar]
- 16.Bowman K, Sarkar R, Raut S, Leong KW. Gene transfer to hemophilia A mice via oral delivery of FVIII-chitosan nanoparticles. J Control Release. 2008;132(3):252–9. doi: 10.1016/j.jconrel.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chao H, Walsh CE. RNA repair for haemophilia A. Expert Rev Mol Med. 2006;8(1):1–8. doi: 10.1017/S1462399406010337. [DOI] [PubMed] [Google Scholar]
- 18.Chao HJ, Mansfield SG, Bartel RC, Hiriyanna S, Mitchell LG, Garcia-Blanco M, et al. Phenotype correction of hemophilia A mice by spliceosome-mediated RNA trans-splicing. Nat Med. 2003;9(8):1015–9. doi: 10.1038/nm900. [DOI] [PubMed] [Google Scholar]
- 19.Long YC, Jaichandran S, Ho LP, Tien SL, Tan SY, Kon OL. FVIII gene delivery by muscle electroporation corrects murine hemophilia A. J Gene Med. 2005;7(4):494–505. doi: 10.1002/jgm.683. [DOI] [PubMed] [Google Scholar]
- 20.Powell JS, Ragni MV, White GC, Lusher JM, Hillman-Wiseman C, Moon TE, et al. Phase 1 trial of FVIII gene transfer for severe hemophilia A using a retroviral construct administered by peripheral intravenous infusion. Blood. 2003;102(6):2038–45. doi: 10.1182/blood-2003-01-0167. [DOI] [PubMed] [Google Scholar]
- 21.Kay MA, High K. Gene therapy for the hemophilias. Proc Natl Acad Sci U S A. 1999;96(18):9973–5. doi: 10.1073/pnas.96.18.9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao GP, Alvira MR, Wang LL, Calcedo R, Johnston J, Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci U S A. 2002;99(18):11854–9. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mingozzi F, Meulenberg JJ, Hui DJ, Basner-Tschakarjan E, Hasbrouck NC, Edmonson SA, et al. AAV-1-mediated gene transfer to skeletal muscle in humans results in dose-dependent activation of capsid-specific T cells. Blood. 2009;114(10):2077–86. doi: 10.1182/blood-2008-07-167510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu D, Alipio Z, Fink LM, Adcock DM, Yang JC, Ward DC, et al. Phenotypic correction of murine hemophilia A using an iPS cell-based therapy. Proc Natl Acad Sci U S A. 2009;106(3):808–13. doi: 10.1073/pnas.0812090106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roth DA, Tawa NE, O’Brien JM, Treco DA, Selden RF, Factor VTT. Nonviral transfer of the gene encoding coagulation factor VIII in patients with severe hemophilia A. N Engl J Med. 2001;344(23):1735–42. doi: 10.1056/NEJM200106073442301. [DOI] [PubMed] [Google Scholar]
- 26.Wen JP, Xu N, Li A, Bourgeois J, Ofosu FA, Hortelano G. Encapsulated human primary myoblasts deliver functional hFIX in hemophilic mice. J Gene Med. 2007;9(11):1002–10. doi: 10.1002/jgm.1098. [DOI] [PubMed] [Google Scholar]
- 27.Follenzi A, Benten D, Novikoff P, Faulkner L, Raut S, Gupta A. Transplanted endothelial cells repopulate the liver endothelium and correct the phenotype of hemophilia A mice. J Clin Invest. 2008;118:935–45. doi: 10.1172/JCI32748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu D, Alipio Z, Fink LM, Adcock DM, Yang J, Ward DC, et al. Phenotypic correction of murine hemophilia A using an iPS cell-based therapy. Proc Natl Acad Sci U S A. 2009;106:808–13. doi: 10.1073/pnas.0812090106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsui H, Shibata M, Brown B, Labelle A, Hegadorn C, Andrews C, Hebbel RP, Galipeau J, Hough C, Lillicrap D. Ex vivo gene therapy for hemophilia A that enhances safe delivery and sustained in vivo Factor VIII expression from lentivirally engineered endothelial progenitors. Stem cells. 2007;25:2660–9. doi: 10.1634/stemcells.2006-0699. [DOI] [PubMed] [Google Scholar]
- 30.Thorrez L, Vandenburgh H, Callewaert N, Mertens N, Shansky J, Wang L, et al. Angiogenesis enhances factor IX delivery and persistence from retrievable human bioengineered muscle implants. Mol Ther. 2006;14(3):442–51. doi: 10.1016/j.ymthe.2006.03.019. [DOI] [PubMed] [Google Scholar]
- 31.Szuba A, Skobe M, Karkkainen MJ, Shin WS, Beynet DP, Rockson NB, et al. Therapeutic lymphangiogenesis with human recombinant VEGF-C. FASEB J. 2002;16(12):1985–7. doi: 10.1096/fj.02-0401fje. [DOI] [PubMed] [Google Scholar]
- 32.Richardson TP, Peters MC, Ennet AB, Mooney DJ. Polymeric system for dual growth factor delivery. Nat Biotechnol. 2001;19:1029–34. doi: 10.1038/nbt1101-1029. [DOI] [PubMed] [Google Scholar]
- 33.Ribatti D. The crucial role of vascular permeability factor/vascular endothelial growth factor in angiogenesis: a historical review. Br J Haematol. 2005;128(3):303–9. doi: 10.1111/j.1365-2141.2004.05291.x. [DOI] [PubMed] [Google Scholar]
- 34.Korpisalo P, Karvinen H, Rissanen TT, Kilpijoki J, Marjomaki V, Baluk P, et al. Vascular endothelial growth factor-A and platelet-derived growth factor-B combination gene therapy prolongs angiogenic effects via recruitment of interstitial mononuclear cells and paracrine effects rather than improved pericyte coverage of angiogenic vessels. Circ Res. 2008;103(10):1092–9. doi: 10.1161/CIRCRESAHA.108.182287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sabatino DE, Freguia CF, Toso R, Santos A, Merricks EP, Kazazian HH, et al. Recombinant canine B-domain-deleted FVIII exhibits high specific activity and is safe in the canine hemophilia A model. Blood. 2009;114(20):4562–5. doi: 10.1182/blood-2009-05-220327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levenberg S, Rouwkema J, Macdonald M, Garfein ES, Kohane DS, Darland DC, et al. Engineered vascularized skeletal muscle tissue. Nature Biotechnol. 2005;23:879–84. doi: 10.1038/nbt1109. [DOI] [PubMed] [Google Scholar]
- 37.Drewa T, Galazka P, Prokurat A, Wolski Z, Sir J, Wysocka K, Czajkowski R, et al. Abdominal wall repair using a biodegradable scaffold seeded with cells. J Pediatr Surg. 2005;40:317–21. doi: 10.1016/j.jpedsurg.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 38.Lu Y, Shansky J, Del Tatto M, Ferland P, Wang X, Vandenburgh H. Recombinant vascular endothelial growth factor secreted from tissue-engineered bioartificial muscles promotes localized angiogenesis. Circulation. 2001;104:594–9. doi: 10.1161/hc3101.092215. [DOI] [PubMed] [Google Scholar]
- 39.Borselli C, Storrie H, Benesch-Lee F, Shvartsman D, Cezar C, Lichtman JW, et al. Functional muscle regeneration with combined delivery of angiogenesis and myogenesis factors. Proc Natl Acad Sci U S A. 2010;107:3287–92. doi: 10.1073/pnas.0903875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swartz MA. The physiology of the lymphatic system. Advanced Drug Delivery Reviews. 2001;50:3–20. doi: 10.1016/s0169-409x(01)00150-8. [DOI] [PubMed] [Google Scholar]
- 41.Porter CJH, Charman SA. Lymphatic transport of proteins after subcutaneous administration. J Pharm Sci. 2000;89(3):297–310. doi: 10.1002/(SICI)1520-6017(200003)89:3<297::AID-JPS2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 42.Lacroix-Desmazes S, Repesse Y, Kaveri SV, Dasgupta S. The role of vWF in the immunogenicity of FVIII. Thromb Res. 2008;122:S3–S6. doi: 10.1016/S0049-3848(08)70002-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.