

Abstract
Objective
Several lines of evidence show that selenium has potential protective effects in osteoarthritis (OA), however the exact mechanism is still unclear. As IL-1β is one of the key proinflammatory cytokines contributing to the progression in OA, we investigated the effect of selenium in neutralizing the inflammatory effects of IL-1β on nitric oxide (NO) and prostaglandin E (PGE2) production, and the signaling pathways involved.
Methods
Isolated primary human chondrocytes were pretreated with selenomethionine (0.5 μM SeMet) for 24 hours then co-treated without or with IL-1β (10 pg/ml or 50 pg/ml) for another 24 hours followed by RNA isolation. Gene expression of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX2) was determined by quantitative Real Time-Polymerase Chain Reaction. Culture media concentrations of NO and PGE2 were determined by nitrite assay and immunoassay respectively. For analysis of cell signaling pathways, chondrocytes were pretreated with SeMet then stimulated with IL-1β for 0 – 45 minutes. The activity of IL-1β signaling pathways was determined by Western blot screening of phosphorylation states of signal transduction proteins.
Results
SeMet inhibited chondrocyte gene expression of IL-1β induced iNOS (31–54%, p=0.031) and COX2 (50–65%, p=0.031) with corresponding reductions in both NO (19–47%, p=0.031) and PGE2 (24–32%, p=0.031) production. Pretreatment with SeMet attenuated IL-1β induced activation of p38 MAPK (39%, p=0.039) but not the ERK, JNK or NFkB pathways.
Conclusions
This study elucidates one potential protective mechanism of selenium, namely through the alteration of cell signaling and downstream transcription of pro-inflammatory effects of IL-1β.
Keywords: Chondrocyte, selenomethionine, iNOS, COX2, NO, PEG2, inflammation
Introduction
Selenium (Se) is an essential trace element involved in several key metabolic activities: protection against oxidative damage; regulation of immune and thyroid function; and fertility [1–2]. Recently, a growing interest in the potentially protective role of Se in Osteoarthritis (OA) has been generated based on epidemiology [3–5], genetic [6] and transgenic animal studies [7]. Profound Se deficiency is associated with the severe osteoarthropathy known as Kashin-Beck Disease that affects individuals in China (as many as 7 million) and neighboring regions [3]. Evidence for a role for Se in OA comes from a large population based study in the US in which a low but non-deficiency level of Se has been shown to be associated with OA presence and severity [4–5]. Moreover, a recent genetic study showed that a variant of Deiodinase 2 (DIO2), which encodes a selenoprotein involved in thyroid hormone activation, is associated with risk for developing OA [6]. While the exact mechanism is still unclear, DIO2 is hypothesized to play a role in bone remodeling in OA progression [6]. In mice, conditional knockout of the selenocysteine tRNA gene, which is required for incorporation of Se into selenoproteins, results in skeletal abnormalities and severe chondronecrosis of articular cartilage resembling Kashin-Beck Disease, as well as chondronecrosis of auricular and tracheal cartilages [7].
In vitro studies have also suggested a protective effect of Se. For instance, Se alters iNOS and COX2 gene expressions in response to lipopolysaccharide (LPS) stimulation in cultured macrophages [8–9]. Lack of Se in vitro has been associated with elevated Prostaglandin E2 (PGE2) production in calcium ionophore stimulated endothelial cells [10] and LPS induced macrophages [11]. A recent study showed that exposure of chondrocytes in vitro to selenomethionine (SeMet) could block IL-1 mediated inhibition of cartilage matrix macromolecule (collagen II and aggrecan) synthesis [12]. However, in the same study, SeMet did not have significant effects on iNOS or COX2 gene expression in the presence of high doses of IL-1β in bovine chondrocytes.
Taken together, these studies suggest an important role of Se in maintaining normal cartilage metabolism and potentially preventing OA. A better understanding of the mechanisms underlying these selenium-mediated protective effects could lead to the development of novel therapeutic approaches for the prevention and management of OA progression. We hypothesized that SeMet could block proinflammatory gene expression induced by physiological doses of IL-1β. In this study we chose to investigate potential mechanisms of Se mediated protective effects using physiological rather than supra-physiological doses of IL-1β as used in the majority of prior experiments[12–16]. Under these conditions we investigated the magnitude of selenium effects and the potential signaling pathways involved in primary human chondrocytes.
Materials and Methods
Chondrocyte Isolation and Culture
The samples used for this project were collected under approval of the Duke IRB. The IRB deemed these samples surgical waste tissues meeting the definition of research not involving human subjects as described in (45CFR46.102(f)) and was not subject to HIPAA (45CFR164.514(b)) as no information relating to patient identity was obtained with the sample. Articular cartilage samples were obtained as surgical waste tissues from 19 patients undergoing total knee replacement surgery [mean age, 61.8 +/− 7.7 years]. The isolated chondrocytes were used for experiments within the first three passages. Cartilage was harvested from non-lesional areas, further minced, and subjected to pronase and collagenase digestion to isolate primary chondrocytes, similar to previously published methods [17]. The SW-1353 chondrosarcoma cell line was obtained from the American Type Culture Collection (ATCC Manassas, VA).
IL-1β (R & D systems, Minneapolis, MN), concentrations of 10 pg/ml and 50 pg/ml were chosen on the basis of evidence for physiological relevance (equivalent to concentrations in human OA synovial fluid) [18–19]. Selenomethionine (SeMet) (Sigma, St Louis, MO), an organic form of selenium, was chosen as it is the primary dietary source of Se for human [20]. A concentration of 0.5 μM SeMet was chosen for chondrocyte cultures based on evidence of physiological relevance (equivalent to concentrations in normal and OA human synovial fluid [21]).
MTT ASSAY
Cell viability was assessed with the MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay based on the ability of mitochondria of viable cells to convert soluble MTT into an insoluble purple formazan reaction product. MTT (Sigma) solution (5 mg/ml in DMEM without phenol red) was added to cells in tissue culture for 2 hours. The MTT solution was aspirated, dimethyl sulfoxide (DMSO) was added (200 μl per each well of 12 well plate) to solubilize formazin and detection occurred by addition of 100 μl of the reaction mixture to a 96 well plate format and reading at O.D. 540 nm. MTT assay results for the SW-1353 cell line were derived from 2 independent experiments, performed in duplicate. MTT assay results for primary chondrocytes were derived from 2 independent experiments, performed in triplicate using 2 separate primary chondrocyte cell lines.
RNA Isolation and Real Time RT-PCR
Cell lysates, prepared by RNeasy Lysis Buffer (Qiagen Valencia, CA) from each experimental condition, were first homogenized by passing them through a QIAshredder spin column (Qiagen, Valencia, CA). The total DNA and RNA fractions were further isolated using the AllPrep DNA/RNA/Protein Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. The isolated total RNA was reverse transcribed into cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) for Real Time RT-PCR analysis. The ABI Prism 7000 sequence detection system and relative quantification software (Applied Biosystems, Foster City, CA) were used for real-time analyses. The amplification for real-time RT-PCR used the following Applied Biosystems primer and probe sets: 18S rRNA endogenous control, Hs01075527_m1(iNOS), and Hs01573474_g1(COX2). The real-time reactions were each performed in triplicate in a final volume of 25 μl.
mRNA Quantification and Statistical Analysis
Raw mRNA expression values were computed by 2−ΔCt formula [22] with values normalized to 18S rRNA, where ΔCt represents the difference in Ct (threshold cycle) number of the 18S rRNA gene and the iNOS or COX2 genes. Results were derived from a total of 6 independent experiments for each dose of IL-1β for COX2 and iNOS respectively, performed in triplicate, using a total of 13 separate primary chondrocyte cell lines. The relative fold changes in mRNA expression levels of iNOS and COX2 were calculated by the 2 −ΔΔCt formula [22], between three different treatments (SeMet, IL-1β, SeMet and IL-1β treatment) and control without treatment (no SeMet, no IL-1β). For the purposes of graphical presentation, the relative mRNA level in cells without treatment was set at 100%.
Raw mRNA expression data were evaluated by two tailed Wilcoxon matched pairs test comparing subgroups (n=6 in each group from 13 separate cell lines): 1) the control group and SeMet pretreated group, 2) the IL-1β treatment group and IL-1β with SeMet pretreatment group, and 3) the control group and IL-1β treatment group. The nonparametric Wilcoxon matched pairs test was chosen as it is appropriate for comparing two paired groups allowing for the non-symmetrical distribution of the raw mRNA expression data [23].
Nitrite and PGE2 Assays
Nitrite (NO2-, one of the stable end products of NO) concentrations were determined by chemiluminescence using an Ionics/Sievers nitric oxide analyzer (NOA 280, Sievers Instruments, Boulder, CO), per the manufacturer’s instructions. Potassium iodide in acetic acid was used as a reductant for nitrite analysis because of its specificity for nitrite. Nitrite concentrations were determined by nitrite standards prepared from sodium nitrite (Sigma, St Louis, MO) and normalized to total DNA isolated from the corresponding chondrocytes. The total DNA concentration was quantified by Nanodrop 1000 (Thermo Scientific, Wilmington, DE).
The PGE2 concentration of the collected culture medium was determined using a competitive enzyme immunoassay based Prostaglandin E2 Parameter Assay Kit (R&D Systems, Minneapolis, MN). The data were normalized to total DNA isolated from the corresponding chondrocytes. For samples with undetectable PGE2 levels (from the control groups without any treatment and the SeMet pretreated alone groups), a value was assigned (13.5 pg/ml) which is equal to half the lowest detection limit of the kit.
Nitrite and PGE2 concentration Quantification and Statistical Analysis
Results of nitrite and PGE2 analyses were derived from a total of 6 independent experiments for each dose of IL-1β for nitrite and PGE2 respectively, using a total of 14 separate primary chondrocyte cell lines. The fold changes in nitrite and PGE2 concentrations were calculated for purposes of graphical presentation of nitrite and PGE2 concentration normalized to total DNA. The relative concentration in cells without treatment (no SeMet, no IL-1β) was set at 100%. Nitrite and PGE2 concentration data were evaluated by two tailed Wilcoxon matched pairs test comparing subgroups (n=6 in each group from 14 separate cell lines): 1) the control group and SeMet pretreated group, 2) the IL-1β treatment group and IL-1β with SeMet pretreatment group, and 3) the control group and IL-1β treatment group. All analyses were performed using GraphPad version 5.0 (GraphPad Software, LA Jolla, CA). The nonparametric Wilcoxon matched pairs test was chosen as it is appropriate for comparing two paired groups allowing for the non-symmetrical distribution of the nitrite and PGE2 data [23]
Western Blot Screening of Signal Transduction Proteins
Chondrocytes from each experimental condition were collected and homogenized in lysis buffer (10 mM HEPES, pH 7.5, 0.5% Triton X-100, 5 mM EDTA, 5 mM EGTA and 1 M NaCl), supplemented with protease inhibitor cocktail (Sigma, St. Louis, MO) and phosphatase inhibitors (5 mM PMSF, 10 mM NaF, 25 mM B-glycerophosphate, 0.5 M DTT, 1 mM Na3V04 ). Whole cell lysates, were further separated by SDS-PAGE, and transferred to nitrocellulose for immunoblotting. Membranes were blocked with 5% BSA in TBS/0.1% Tween 20 (TBS-T).
Polyclonal primary antibodies against Phospho-p38 MAPK (Thr180/Tyr182), Total p38 MAPK, Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), Phospho-SAPK/JNK (Thr183/Tyr185), Phospho-IKKα/β (Ser176/180) and Phospho-NFkB p65(Ser536) were obtained from Cell Signaling Technology (Danvers, MA) and used at 1:1000 dilution. A monoclonal antibody against α-tubulin (Sigma) was used as a normalization control at 1:10,000 dilution. Anti-rabbit and anti-mouse IgG-HRP (Jackson ImmunoResearch, West Grove, PA) secondary antibodies were used at a 1:5,000 dilution. The resulting films were scanned using CanoScan LiDE 70 (Canon, Lake Success, NY) and the band intensities were quantified using Adobe Photoshop CS and Image J (National Institutes of Health, Bethesda, MD). The mean and SEM intensities of the phosphosignaling proteins were normalized to the internal protein control (α-tubulin).
Phosphosignaling Protein Quantification and Statistical Analysis
Raw intensity data of phosphosignaling proteins were normalized to the internal protein control (α-tubulin). Results were generated for three time points 5, 30 and 45 minutes in the presence and absence of SeMet, and were derived from 3 independent experiments from 3 separate primary chondrocyte cell lines. The total area under each curve (from 5 to 45 minutes) was determined. The phosphosignaling protein intensities were evaluated by two-tailed Wilcoxon matched pairs test comparing the response to IL-1β induction at 5, 30 and 45 minutes between control (n=9, without Se pretreatment) and the Se pretreated (n=9, 0.5 μM for the prior 24 hours). The Wilcoxon matched pairs test was chosen as it is appropriate for comparing two paired groups allowing for the non-symmetrical distribution of the phosphosignaling data.
Results
Inhibitory Effect of Selenomethionine on IL-1β Induced iNOS Gene Expression and Nitrite Production
For cell toxicity assays we initially compared two forms of selenium, the organic selenomethionine (SeMet) form, and the inorganic selenite. Based on the MTT toxicity assay, SeMet showed no evidence of cellular toxicity at concentrations up to 1 μM in SW-1353 cells (Figures 1A) or isolated primary chondrocytes. (Figure 1B) while selenite showed cell toxicity beyond 1 μM. We subsequently selected SeMet at a concentration of 0.5 μM for all subsequent experiments. Twenty four hour treatment with IL-1β at 10 pg/ml and 50 pg/ml significantly induced iNOS mRNA (~70 fold and ~630 fold respectively) compared to the control without IL-1β (Figures 2A and 2B). SeMet alone had no effect on the basal iNOS mRNA level. Twenty four hour pretreatment with SeMet significantly repressed IL-1β induced iNOS steady state mRNA level by ~54% at 10 pg/ml IL-1β (P=0.031), and by ~31% at 50 pg/ml IL-1β ( P=0.031). We further examined the effect of SeMet on nitric oxide (NO), the enzymatic product of iNOS. Because of the short half-life of reactive NO, we measured nitrite (NO2−), which is a stable downstream product of NO. SeMet pretreatment inhibited IL-1β induced nitrite production, by 47% at 10 pg/ml IL-1β (P=0.031), and by 19% at 50 pg/ml IL-1β (P=0.031) (Figures 2C and 2D).
Figure 1. MTT cell toxicity assay for optimization of in vitro chondrocyte culture conditions.
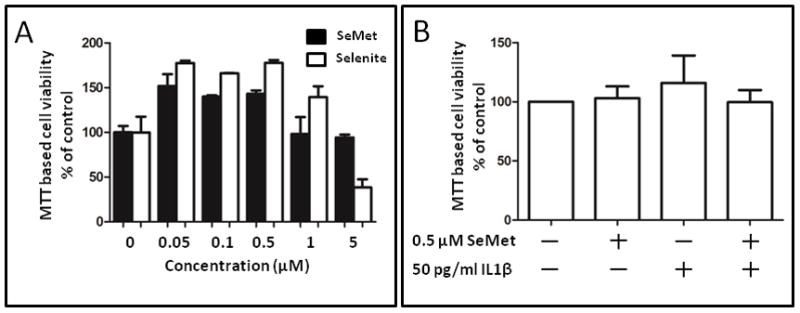
Effects of selenium on cell viability were assessed with the MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. (A) SW-1353 cells were cultured with different concentrations (0 μM to 5 μM) of selenite or selenomethionine (SeMet) for 48 hours. (B) Primary human chondrocytes were cultured for 24 hours in the absence (control) or presence of 0.5 μM SeMet, followed by 24 hours co-treatment without or with 50 pg/ml IL-1β. Values shown are mean and SEM of cell viability as a percentage of control (set at 100%).
Figure 2. SeMet inhibited IL-1 β induced iNOS gene expression and NO production in primary human chondrocytes.
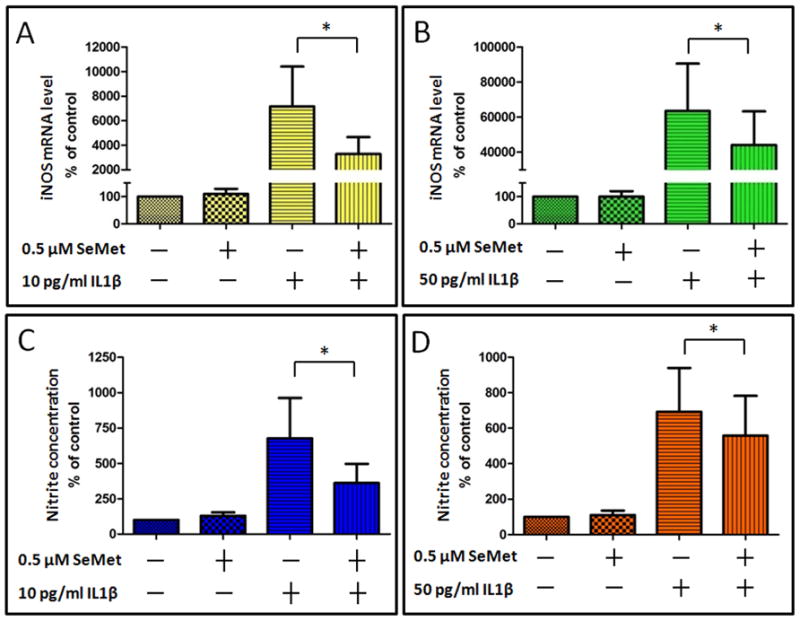
Primary human chondrocytes were cultured for 24 hours in the absence (control) or presence of 0.5 μM SeMet, followed by 24 hour co-treatment without IL-1β (control) or with either 10 pg/ml (A and C) or 50 pg/ml (B and D) IL-1β. Gene expression for iNOS (A and B) was determined by RT-PCR normalized to 18S rRNA (average of triplicates for six independent experiments). Corresponding culture media were analyzed for nitrite concentration (C and D). Data were normalized to total DNA of the corresponding chondrocytes. Values shown are mean and SEM of iNOS or nitrite as a percentage of control (set at 100%). *P=0.031
Inhibitory Effect of Selenomethionine on IL-1β Induced COX2 Gene Expression and PEG2 Production
Next we investigated if pretreatment of SeMet could also affect COX2 gene expression in the presence of IL-1β. Twenty four hour treatment with IL-1β at 10 pg/ml and 50 pg/ml significantly induced COX2 mRNA (~ 53 fold and 120 fold respectively) compared to the control without IL-1β (Figures 3A and 3B). SeMet had no effect on the basal COX2 mRNA level. Twenty four hour pretreatment with SeMet significantly repressed IL-1β induced COX2 steady state mRNA level by ~65% at 10 pg/ml IL-1β (p=0.031) and by ~50% at 50 pg/ml IL-1β (P=0.031). We further examined the effect of SeMet on PGE2 production, the downstream enzymatic product of COX2. SeMet pretreatment inhibited IL-1β induced PGE2 production, by 32% at 10 pg/ml IL-1β (P=0.031), and by 24% at 50 pg/ml IL-1β (P=0.031) (Figures 3C and 3D).
Figure 3. SeMet inhibited IL-1 β induced COX2 gene expression and PGE2 production in primary human chondrocytes.
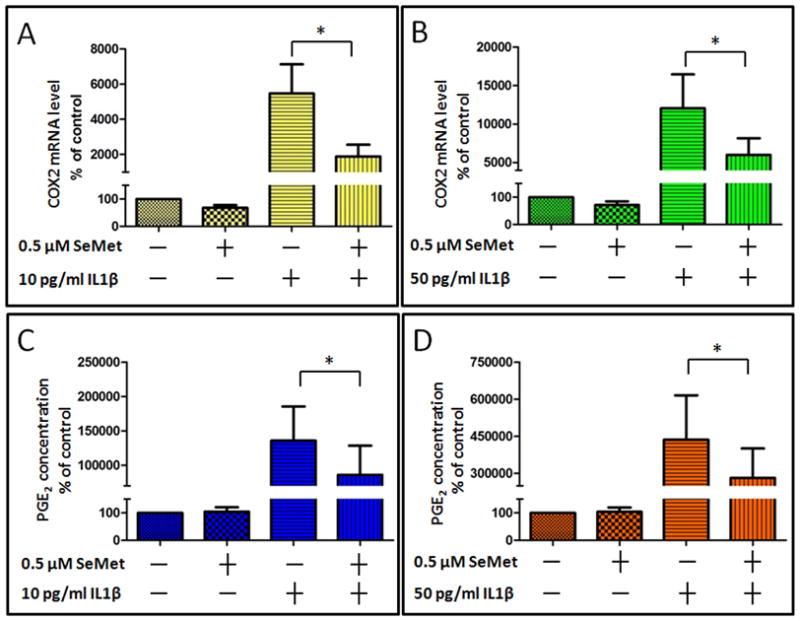
Primary human chondrocytes were cultured for 24 hours in the absence (control) or presence of 0.5 μM SeMet, followed by 24 hour co-treatment without IL-1β (control) or with either 10 pg/ml (A and C) or 50 pg/ml (B and D) IL-1β. Gene expression for COX2 (A and B) was determined by RT-PCR normalized to 18S rRNA (average of triplicates for six independent experiments). Corresponding culture media were analyzed for PGE2 concentration (C and D). Data were normalized to total DNA of the corresponding chondrocytes. Values shown are mean and SEM of COX2 or PGE2 as a percentage of control (set at 100%). *P=0.031
Effect of Selenomethionine on IL-1β Induced Signaling Pathways
To determine whether the inhibitory effect of SeMet could be associated with alterations of IL-1β induced signaling pathways, we examined the phosphorylation status of signaling proteins in IL-1β stimulated primary chondrocytes in the presence and absence of SeMet. Stimulation of chondrocytes with IL-1β resulted in activation of MAPK kinases, p38 MAPK, ERK1/2 and JNK as well as NFkB pathway signaling molecules, IKKα/β and NFkB p65, with phosphorylation peaking approximately 30 minutes after stimulation. SeMet pretreatment alone did not affect the basal phosphorylation level of any of the signaling molecules at time zero (Figure 4A, lane 5 versus lane 1). In contrast, SeMet attenuated IL-1β induced phosphorylation of p38 MAPK (Figure 4A, top row) but not the ERKs, JNK, IKKα/β or NFkB p65 (Figure 4A). The total basal level of p38 MAPK was unaffected by either IL-1β or SeMet.
Figure 4. SeMet selectively blocked IL-1β induced p38 MAPK.
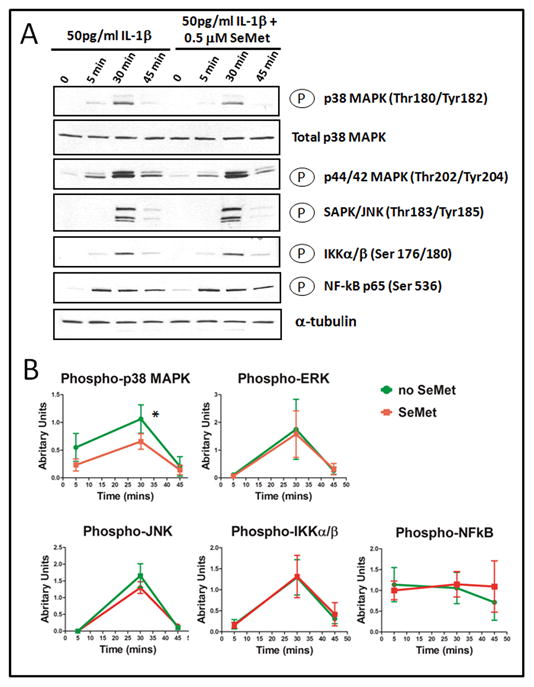
(A) Western blot analysis. Primary human chondrocytes were cultured for 24 hours in the absence (control) or presence of 0.5 μM SeMet, and then co-treated with 50 pg/ml IL-1β treatment for 0, 5, 30 and 45 minutes. Equal amounts of total cell lysate were separated by SDS-PAGE and analyzed by Western blot for the following proteins: Phospho-p38 MAPK (Thr180/Tyr182), Total p38 MAPK, Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), Phospho-SAPK/JNK (Thr183/Tyr185), Phospho-IKKα/β (Ser176/180) and Phospho-NFkB p65 (Ser536); α tubulin (bottom row) was used as a control for normalization. The results shown are representative of three independent experiments.
(B) Quantification of the effects of SeMet on IL-1β induced phosphorylation of signaling proteins. Phosphoprotein signaling intensity was quantified by Image J analysis of Western blots for up to 3 time points from three independent experiments (as described in Figure 3) with normalization to the internal control, α-tubulin. The normalized mean and SEM levels of phosphosignaling protein intensity are shown for Phospho-p38 MAPK (Thr180/Tyr182), Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204), Phospho-SAPK/JNK (Thr183/Tyr185), Phospho-IKKα/β (Ser176/180) and Phospho-NFkB p65(Ser536). SeMet inhibited IL-1β activation of p38 MAPK significantly but not the generation of the other phosphoproteins. *P=0.039.
The mean phosphorylation level for each signaling molecule was determined by Image J analyses (normalized to the α-tubulin control) from three separate experiments (Figure 3B). SeMet pretreatment only modestly (39%) but significantly (p=0.039) reduced the IL-1β induced activation of p38 MAPK based on the total area under the curve (from 5 to 45 minutes), compared to the IL-1β treated condition (Figure 4B). Pretreatment of SeMet did not affect the IL-1β activation of the other molecules (Figures 4B) nor total p38 MAPK.
Discussion
Elevated concentrations of the proinflammatory cytokine, IL-1, are found in the synovial fluid of OA joints [24]. IL-1 stimulates several key proinflammatory mediators such as PGE2 and NO that have been implicated in the pathogenesis of OA [25–26]. IL-1 stimulates NO and PGE2 through regulation of iNOS and COX2 gene expression respectively [27–29]. Our data demonstrated that pretreatment of chondrocytes with the antioxidant SeMet significantly inhibited both production of NO and PGE2 in response to IL-1β. These results have potential clinical significance based on the known involvement of PGE2 and NO in joint metabolism summarized below.
PGE2 has protean manifestations - some catabolic and some anabolic. This is thought to be mediated by different EP receptors such as EP2 and EP4 [30–31]. PGE2 upregulates matrix metalloproteinases (MMPs) that cause joint cartilage degradation [32]. PGE2 has been shown to positively modulate type II collagen gene expression in cultured chondrocytes [33]. Furthermore, PGE2 promotes differentiation and proliferation of growth plate chondrocytes [34–35]. Sensitization of peripheral nociceptors by PGE2 is believed to contribute to pain associated with inflammation [36].
NO, another key mediator downstream of IL-1, has been demonstrated to have an impact on cartilage homeostasis by regulating the balance of anabolic and catabolic metabolism. NO inhibits collagen and proteoglycan synthesis of cartilage in vitro [37], and also induces matrix metalloproteinase synthesis in articular chondrocytes [38]. In addition, NO has been shown to reduce IL-1 receptor antagonist (IL-1ra) synthesis by chondrocytes [39]. Incubation of human articular chondrocytes with the NO donor, sodium nitroprusside (SNP), activates apoptotic gene expression [40]. NO can trigger apoptosis by a mitochondria dependent mechanism that could be mediated through regulation of expression of apoptotic related genes such as caspase- 3 and 7 [40–41]. Furthermore, 3-nitrotyrosine, a stable product formed by reaction of NO and reactive oxygen species, has correlated with IL-1β induced oxidative stress in aging and osteoarthritic cartilage tissue [42].
The Se mediated reduction in the NO and PGE2 production was associated with corresponding reductions in iNOS and COX2 transcript levels. The direction and magnitude of the effect of SeMet on the IL-1β induced NO production was similar to the effect on iNOS mRNA expression. Therefore, the full effect of SeMet to modulate NO production is likely mediated at a transcriptional level. The inhibitory effect of SeMet on IL-1β induced NO production found in our study was similar to the inhibitory effect shown previously by others for the inorganic form of Se, namely selenite (5–10μM), on LPS-induced iNOS gene expression and NO production in murine macrophages [43–44].
In the case of PGE2 production, the magnitude of the inhibition of PGE2 protein production was ~ 50% less than the magnitude of inhibition of COX2 gene expression. It is known that inhibition of NO may lead to a corresponding compensatory release in PGE2 [45]. Therefore, we believe the PGE2 results represent a combination of downstream effects: SeMet inhibition of COX2 gene expression; and SeMet inhibition of NO leading to increase in PGE2. These NO and PGE2 data provide a plausible mechanism whereby Se may play a protective role in OA.
Signaling pathways induced by IL-1β, that regulate NO and PGE2 production, are sensitive to the redox state of cells, and can be altered by natural antioxidants such as curcumin and capparis spinosa [15, 46]. Cytokines, including IL-1, have been shown to induce NO and PGE2 production through MAPK and NFkB pathways in human and rat chondrocytes [16, 47–48]. We demonstrated that Se partially inhibited the IL-1β induced phosphorylation of p38 MAPK, but not other MAPKs that are commonly involved in IL-1β cell signaling. This is consistent with a past study that also showed selective targeting of p38 MAPK by selenite, an inorganic form of Se [43]. However, we cannot exclude the fact that Se may alter IL-1β induced iNOS or COX2 gene expression through other signaling pathways since our study was limited by having investigated only a single form of Se, SeMet. Selenate (another inorganic form of Se) has been shown to activate the PI3K/AKT pathway [49], which negatively regulates LPS induced COX2 [50]. In contrast to our results, generated with human chondrocytes and physiologically relevant concentrations of SeMet and IL-1β (10 and 50 pg/ml), a previous study utilizing bovine chondrocytes and supra-physiological doses of IL-1β (10 ng/ml), failed to observe an effect of SeMet on IL-1β induced p38 kinase activity [12]. Thus, the results are likely dependent on the dose and form of Se used. By the same token, the dose and form of Se we chose may also explain why SeMet did not have a significant effect on phosphorylation of NFkB p65 while high doses of selenite have been shown to inhibit NFkB activity in cells other than chondrocytes, such as macrophages [11, 43–44]. Finally, in contrast to other studies that measured the DNA binding activities of NFkB, we examined the phosphorylation level of NFkB p65 which might account for different results between studies.
The mechanism whereby Se modifies the phosphorylation state of p38 MAPK is unknown. Se may regulate the redox state and redox state may alter the activity of upstream kinases or phosphatases that in turn control the phosphorylation status of p38 MAPK and hence the downstream signaling. Sulfhydryl groups of protein tyrosine phosphatase can be reversibly oxidized and the enzymatic activity regulated [51]. By analogy, regulation of redox state by the antioxidant Se, might explain the Se related modulation of IL-1β signaling. Further work remains to be done to elucidate the mechanisms of Se mediated regulation of IL-1β signaling.
In conclusion, we showed that the antioxidant SeMet inhibited IL-1β induced NO and PGE2 production through modulation of iNOS and COX2 gene expression in primary chondrocytes. Our study also showed that Se partially blocked IL-1 activation of the p38 MAPK pathway. These results suggest that one mechanism whereby Se may exert a protective effect is through regulation of the expression of inflammation related genes, possibly mediated in part through inhibition of IL-1β cell signaling.
Supplementary Material
Acknowledgments
This study was funded by Center for Biomolecular and Tissue Engineering International Fellowship at Duke (to AWMC), Osteoarthritis Research Society International (OARSI) Scholarship (to AWMC), NIH/NIA Claude D. Pepper OAIC 5P30 AG028716 and NIH/NIAMS grant UO1 AR050898.
Footnotes
Contributions
AWMC participated in the conception and design of the study, isolating of primary chondrocytes, performing gene expression studies, PGE2 assays, statistical analysis and drafted the manuscript. TVS performed the nitrite assay and edited the manuscript. MB coordinated and organized surgical waste tissue collection and revised the manuscript. VBK conceived of the study, supervised the project, and participated in statistical analysis and manuscript preparation and editing. All authors read and approved the final manuscript.
Competing interest
The authors declare that they have no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rayman MP. The importance of selenium to human health. Lancet. 2000;356(9225):233–241. doi: 10.1016/S0140-6736(00)02490-9. [DOI] [PubMed] [Google Scholar]
- 2.Ryan-Harshman M, Aldoori W. The relevance of selenium to immunity, cancer, and infectious/inflammatory diseases. Can J Diet Pract Res. 2005;66 (2):98–102. doi: 10.3148/66.2.2005.98. [DOI] [PubMed] [Google Scholar]
- 3.Allander E. Kashin-Beck disease. An analysis of research and public health activities based on a bibliography 1849–1992. Scand J Rheumatol Suppl. 1994;99:1–36. doi: 10.3109/03009749409117126. [DOI] [PubMed] [Google Scholar]
- 4.Jordan J, Fang F, Arab L, Morris J, Renner J, Ngwenyama R, Helmick CG. Low selenium levels are associated with increased odds of radiographic knee and multijoint OA. Osteoarthritis & Cartilage. 2005;13(Suppl 1):S35. [Google Scholar]
- 5.Jordan J, Fang F, Schwartz T, Morris J, Renner J, Chen J, He K, Nelson A, Hochberg M, Helmick C. Low selenium levels are assoicated with increased odds of radiographic hip osteoarthritis in African American and White women. Osteoarthritis & Cartilage. 2007;15(Suppl 3 ):C33. [Google Scholar]
- 6.Meulenbelt I, Min JL, Bos S, Riyazi N, Houwing-Duistermaat JJ, van der Wijk HJ, Kroon HM, Nakajima M, Ikegawa S, Uitterlinden AG, et al. Identification of DIO2 as a new susceptibility locus for symptomatic osteoarthritis. Hum Mol Genet. 2008;17(12):1867–1875. doi: 10.1093/hmg/ddn082. [DOI] [PubMed] [Google Scholar]
- 7.Downey CM, Horton CR, Carlson BA, Parsons TE, Hatfield DL, Hallgrimsson B, Jirik FR. Osteo-chondroprogenitor-specific deletion of the selenocysteine tRNA gene, Trsp, leads to chondronecrosis and abnormal skeletal development: a putative model for Kashin-Beck disease. PLoS Genet. 2009;5 (8):e1000616. doi: 10.1371/journal.pgen.1000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vunta H, Belda BJ, Arner RJ, Channa Reddy C, Vanden Heuvel JP, Sandeep Prabhu K. Selenium attenuates pro-inflammatory gene expression in macrophages. Mol Nutr Food Res. 2008;52(11):1316–1323. doi: 10.1002/mnfr.200700346. [DOI] [PubMed] [Google Scholar]
- 9.Prabhu KS, Zamamiri-Davis F, Stewart JB, Thompson JT, Sordillo LM, Reddy CC. Selenium deficiency increases the expression of inducible nitric oxide synthase in RAW 264.7 macrophages: role of nuclear factor-kappaB in up-regulation. Biochem J. 2002;366(Pt 1):203–209. doi: 10.1042/BJ20020256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao YZ, Reddy CC, Sordillo LM. Altered eicosanoid biosynthesis in selenium-deficient endothelial cells. Free Radic Biol Med. 2000;28(3):381–389. doi: 10.1016/s0891-5849(99)00251-8. [DOI] [PubMed] [Google Scholar]
- 11.Zamamiri-Davis F, Lu Y, Thompson JT, Prabhu KS, Reddy PV, Sordillo LM, Reddy CC. Nuclear factor-kappaB mediates over-expression of cyclooxygenase-2 during activation of RAW 264.7 macrophages in selenium deficiency. Free Radic Biol Med. 2002;32(9):890–897. doi: 10.1016/s0891-5849(02)00775-x. [DOI] [PubMed] [Google Scholar]
- 12.Andriamanalijaona R, Kypriotou M, Bauge C, Renard E, Legendre F, Raoudi M, Boumediene K, Gatto H, Monginoux P, Pujol JP. Comparative effects of 2 antioxidants, selenomethionine and epigallocatechin-gallate, on catabolic and anabolic gene expression of articular chondrocytes. J Rheumatol. 2005;32 (10):1958–1967. [PubMed] [Google Scholar]
- 13.Lazzerini PE, Capecchi PL, Nerucci F, Fioravanti A, Chellini F, Piccini M, Bisogno S, Marcolongo R, Laghi Pasini F. Simvastatin reduces MMP-3 level in interleukin 1beta stimulated human chondrocyte culture. Ann Rheum Dis. 2004;63(7):867–869. doi: 10.1136/ard.2003.009746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou PH, Liu SQ, Peng H. The effect of hyaluronic acid on IL-1beta-induced chondrocyte apoptosis in a rat model of osteoarthritis. J Orthop Res. 2008;26 (12):1643–1648. doi: 10.1002/jor.20683. [DOI] [PubMed] [Google Scholar]
- 15.Panico AM, Cardile V, Garufi F, Puglia C, Bonina F, Ronsisvalle G. Protective effect of Capparis spinosa on chondrocytes. Life Sci. 2005;77(20):2479–2488. doi: 10.1016/j.lfs.2004.12.051. [DOI] [PubMed] [Google Scholar]
- 16.Thomas B, Thirion S, Humbert L, Tan L, Goldring MB, Bereziat G, Berenbaum F. Differentiation regulates interleukin-1beta-induced cyclo-oxygenase-2 in human articular chondrocytes: role of p38 mitogen-activated protein kinase. Biochem J. 2002;362(Pt 2):367–373. doi: 10.1042/0264-6021:3620367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuettner KE, Pauli BU, Gall G, Memoli VA, Schenk RK. Synthesis of cartilage matrix by mammalian chondrocytes in vitro. I. Isolation, culture characteristics, and morphology. J Cell Biol. 1982;93(3):743–750. doi: 10.1083/jcb.93.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Westacott CI, Whicher JT, Barnes IC, Thompson D, Swan AJ, Dieppe PA. Synovial fluid concentration of five different cytokines in rheumatic diseases. Ann Rheum Dis. 1990;49(9):676–681. doi: 10.1136/ard.49.9.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kahle P, Saal JG, Schaudt K, Zacher J, Fritz P, Pawelec G. Determination of cytokines in synovial fluids: correlation with diagnosis and histomorphological characteristics of synovial tissue. Ann Rheum Dis. 1992;51 (6):731–734. doi: 10.1136/ard.51.6.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schrauzer GN. Selenomethionine: a review of its nutritional significance, metabolism and toxicity. J Nutr. 2000;130(7):1653–1656. doi: 10.1093/jn/130.7.1653. [DOI] [PubMed] [Google Scholar]
- 21.Yazar M, Sarban S, Kocyigit A, Isikan UE. Synovial fluid and plasma selenium, copper, zinc, and iron concentrations in patients with rheumatoid arthritis and osteoarthritis. Biol Trace Elem Res. 2005;106(2):123–132. doi: 10.1385/BTER:106:2:123. [DOI] [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23.Motulsky H. GraphPad Instat Version 3.0 The Instat guide to choosing and interpreting statistical tests. 1990. [Google Scholar]
- 24.Goldring SR, Goldring MB. The role of cytokines in cartilage matrix degeneration in osteoarthritis. Clin Orthop Relat Res. 2004;(427 Suppl):S27–36. doi: 10.1097/01.blo.0000144854.66565.8f. [DOI] [PubMed] [Google Scholar]
- 25.Jacques C, Gosset M, Berenbaum F, Gabay C. The role of IL-1 and IL-1Ra in joint inflammation and cartilage degradation. Vitam Horm. 2006;74:371–403. doi: 10.1016/S0083-6729(06)74016-X. [DOI] [PubMed] [Google Scholar]
- 26.Abramson SB. Osteoarthritis and nitric oxide. Osteoarthritis Cartilage. 2008;16 (Suppl 2):S15–20. doi: 10.1016/S1063-4584(08)60008-4. [DOI] [PubMed] [Google Scholar]
- 27.Vuolteenaho K, Moilanen T, Al-Saffar N, Knowles RG, Moilanen E. Regulation of the nitric oxide production resulting from the glucocorticoid-insensitive expression of iNOS in human osteoarthritic cartilage. Osteoarthritis Cartilage. 2001;9(7):597–605. doi: 10.1053/joca.2001.0431. [DOI] [PubMed] [Google Scholar]
- 28.Geng Y, Blanco FJ, Cornelisson M, Lotz M. Regulation of cyclooxygenase-2 expression in normal human articular chondrocytes. J Immunol. 1995;155(2):796–801. [PubMed] [Google Scholar]
- 29.Charles IG, Palmer RM, Hickery MS, Bayliss MT, Chubb AP, Hall VS, Moss DW, Moncada S. Cloning, characterization, and expression of a cDNA encoding an inducible nitric oxide synthase from the human chondrocyte. Proc Natl Acad Sci U S A. 1993;90(23):11419–11423. doi: 10.1073/pnas.90.23.11419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Attur M, Al-Mussawir HE, Patel J, Kitay A, Dave M, Palmer G, Pillinger MH, Abramson SB. Prostaglandin E2 exerts catabolic effects in osteoarthritis cartilage: evidence for signaling via the EP4 receptor. J Immunol. 2008;181(7):5082–5088. doi: 10.4049/jimmunol.181.7.5082. [DOI] [PubMed] [Google Scholar]
- 31.Li X, Ellman M, Muddasani P, Wang JH, Cs-Szabo G, van Wijnen AJ, Im HJ. Prostaglandin E2 and its cognate EP receptors control human adult articular cartilage homeostasis and are linked to the pathophysiology of osteoarthritis. Arthritis Rheum. 2009;60(2):513–523. doi: 10.1002/art.24258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tung JT, Arnold CE, Alexander LH, Yuzbasiyan-Gurkan V, Venta PJ, Richardson DW, Caron JP. Evaluation of the influence of prostaglandin E2 on recombinant equine interleukin-1beta-stimulated matrix metalloproteinases 1, 3, and 13 and tissue inhibitor of matrix metalloproteinase 1 expression in equine chondrocyte cultures. Am J Vet Res. 2002;63(7):987–993. doi: 10.2460/ajvr.2002.63.987. [DOI] [PubMed] [Google Scholar]
- 33.Goldring MB, Suen LF, Yamin R, Lai WF. Regulation of Collagen Gene Expression by Prostaglandins and Interleukin-1beta in Cultured Chondrocytes and Fibroblasts. Am J Ther. 1996;3(1):9–16. doi: 10.1097/00045391-199601000-00003. [DOI] [PubMed] [Google Scholar]
- 34.Schwartz Z, Gilley RM, Sylvia VL, Dean DD, Boyan BD. The effect of prostaglandin E2 on costochondral chondrocyte differentiation is mediated by cyclic adenosine 3′,5′-monophosphate and protein kinase C. Endocrinology. 1998;139(4):1825–1834. doi: 10.1210/endo.139.4.5919. [DOI] [PubMed] [Google Scholar]
- 35.Brochhausen C, Neuland P, Kirkpatrick CJ, Nusing RM, Klaus G. Cyclooxygenases and prostaglandin E2 receptors in growth plate chondrocytes in vitro and in situ--prostaglandin E2 dependent proliferation of growth plate chondrocytes. Arthritis Res Ther. 2006;8(3):R78. doi: 10.1186/ar1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dray A, Read SJ. Arthritis and pain. Future targets to control osteoarthritis pain. Arthritis Res Ther. 2007;9(3):212. doi: 10.1186/ar2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taskiran D, Stefanovic-Racic M, Georgescu H, Evans C. Nitric oxide mediates suppression of cartilage proteoglycan synthesis by interleukin-1. Biochem Biophys Res Commun. 1994;200(1):142–148. doi: 10.1006/bbrc.1994.1426. [DOI] [PubMed] [Google Scholar]
- 38.Sasaki K, Hattori T, Fujisawa T, Takahashi K, Inoue H, Takigawa M. Nitric oxide mediates interleukin-1-induced gene expression of matrix metalloproteinases and basic fibroblast growth factor in cultured rabbit articular chondrocytes. J Biochem. 1998;123(3):431–439. doi: 10.1093/oxfordjournals.jbchem.a021955. [DOI] [PubMed] [Google Scholar]
- 39.Pelletier JP, Mineau F, Ranger P, Tardif G, Martel-Pelletier J. The increased synthesis of inducible nitric oxide inhibits IL-1ra synthesis by human articular chondrocytes: possible role in osteoarthritic cartilage degradation. Osteoarthritis Cartilage. 1996;4(1):77–84. doi: 10.1016/s1063-4584(96)80009-4. [DOI] [PubMed] [Google Scholar]
- 40.Maneiro E, Lopez-Armada MJ, de Andres MC, Carames B, Martin MA, Bonilla A, Del Hoyo P, Galdo F, Arenas J, Blanco FJ. Effect of nitric oxide on mitochondrial respiratory activity of human articular chondrocytes. Ann Rheum Dis. 2005;64(3):388–395. doi: 10.1136/ard.2004.022152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu GJ, Chen TG, Chang HC, Chiu WT, Chang CC, Chen RM. Nitric oxide from both exogenous and endogenous sources activates mitochondria-dependent events and induces insults to human chondrocytes. J Cell Biochem. 2007;101(6):1520–1531. doi: 10.1002/jcb.21268. [DOI] [PubMed] [Google Scholar]
- 42.Loeser RF, Carlson CS, Del Carlo M, Cole A. Detection of nitrotyrosine in aging and osteoarthritic cartilage: Correlation of oxidative damage with the presence of interleukin-1beta and with chondrocyte resistance to insulin-like growth factor 1. Arthritis Rheum. 2002;46(9):2349–2357. doi: 10.1002/art.10496. [DOI] [PubMed] [Google Scholar]
- 43.Kim SH, Johnson VJ, Shin TY, Sharma RP. Selenium attenuates lipopolysaccharide-induced oxidative stress responses through modulation of p38 MAPK and NF-kappaB signaling pathways. Exp Biol Med (Maywood) 2004;229(2):203–213. doi: 10.1177/153537020422900209. [DOI] [PubMed] [Google Scholar]
- 44.Yun CH, Yang JS, Kang SS, Yang Y, Cho JH, Son CG, Han SH. NF-kappaB signaling pathway, not IFN-beta/STAT1, is responsible for the selenium suppression of LPS-induced nitric oxide production. Int Immunopharmacol. 2007;7(9):1192–1198. doi: 10.1016/j.intimp.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 45.Fermor B, Weinberg JB, Pisetsky DS, Guilak F. The influence of oxygen tension on the induction of nitric oxide and prostaglandin E2 by mechanical stress in articular cartilage. Osteoarthritis Cartilage. 2005;13 (10):935–941. doi: 10.1016/j.joca.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 46.Shakibaei M, John T, Schulze-Tanzil G, Lehmann I, Mobasheri A. Suppression of NF-kappaB activation by curcumin leads to inhibition of expression of cyclo-oxygenase-2 and matrix metalloproteinase-9 in human articular chondrocytes: Implications for the treatment of osteoarthritis. Biochem Pharmacol. 2007;73(9):1434–1445. doi: 10.1016/j.bcp.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 47.Masuko-Hongo K, Berenbaum F, Humbert L, Salvat C, Goldring MB, Thirion S. Up-regulation of microsomal prostaglandin E synthase 1 in osteoarthritic human cartilage: critical roles of the ERK-1/2 and p38 signaling pathways. Arthritis Rheum. 2004;50(9):2829–2838. doi: 10.1002/art.20437. [DOI] [PubMed] [Google Scholar]
- 48.Lianxu C, Hongti J, Changlong Y. NF-kappaBp65-specific siRNA inhibits expression of genes of COX-2, NOS-2 and MMP-9 in rat IL-1beta-induced and TNF-alpha-induced chondrocytes. Osteoarthritis Cartilage. 2006;14 (4):367–376. doi: 10.1016/j.joca.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 49.Heart E, Sung CK. Insulin-like and non-insulin-like selenium actions in 3T3-L1 adipocytes. J Cell Biochem. 2003;88(4):719–731. doi: 10.1002/jcb.10395. [DOI] [PubMed] [Google Scholar]
- 50.Monick MM, Robeff PK, Butler NS, Flaherty DM, Carter AB, Peterson MW, Hunninghake GW. Phosphatidylinositol 3-kinase activity negatively regulates stability of cyclooxygenase 2 mRNA. J Biol Chem. 2002;277(36):32992–33000. doi: 10.1074/jbc.M203218200. [DOI] [PubMed] [Google Scholar]
- 51.Monteiro HP, Ivaschenko Y, Fischer R, Stern A. Inhibition of protein tyrosine phosphatase activity by diamide is reversed by epidermal growth factor in fibroblasts. FEBS Lett. 1991;295(1–3):146–148. doi: 10.1016/0014-5793(91)81405-w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.