

The expression, purification and crystallization of the SARS coronavirus nsp16 RNA-cap AdoMet-dependent (nucleoside-2′O)-methyltransferase in complex with its activating factor nsp10 are reported.
Keywords: SARS coronavirus, nsp10, nsp16
Abstract
To date, the SARS coronavirus is the only known highly pathogenic human coronavirus. In 2003, it was responsible for a large outbreak associated with a 10% fatality rate. This positive RNA virus encodes a large replicase polyprotein made up of 16 gene products (nsp1–16), amongst which two methyltransferases, nsp14 and nsp16, are involved in viral mRNA cap formation. The crystal structure of nsp16 is unknown. Nsp16 is an RNA-cap AdoMet-dependent (nucleoside-2′-O-)-methyltransferase that is only active in the presence of nsp10. In this paper, the expression, purification and crystallization of nsp10 in complex with nsp16 are reported. The crystals diffracted to a resolution of 1.9 Å resolution and crystal structure determination is in progress.
1. Introduction
In 2003, an outbreak of a novel virus occurred in China and spread through several countries. The identified agent, a previously unknown coronavirus, was named severe acute respiratory syndrome coronavirus (SARS-CoV). Much like other coronaviruses, SARS-CoV has an ∼30 kb RNA genome with a 5′ cap and a 3′ poly(A) tail (Snijder et al., 2003 ▶). The genome of SARS-CoV codes for two large polyproteins 1a and 1ab, which are autoproteolytically processed by at least two viral proteases, producing 16 nonstructural proteins (nsps). These nsps are thought to form a huge protein complex that is responsible for viral RNA replication and transcription. In addition, a set of subgenomic mRNAs encoding structural and accessory proteins is produced by a sophisticated mechanism involving nested mRNA synthesis using the full-length genomic RNA as template. These mRNAs are thought to be capped and polyadenylated (Lai & Stohlman, 1981 ▶; van Vliet et al., 2002 ▶).
Since 2003, interest in SARS-CoV has greatly promoted structural and functional studies of its nsps. In recent years, many three-dimensional structures of replication proteins have appeared in the literature, including those of nsp1 (Almeida et al., 2007 ▶); several domains of nsp3 [including (i) an N-terminal Glu-rich acidic domain (AD; Serrano et al., 2007 ▶), (ii) an X domain (XD; Egloff et al., 2006 ▶), (iii) the SUD domain (SARS-CoV unique domain; Chatterjee et al., 2009 ▶) and (iv) the papain-like protease PLP2 (Ratia et al., 2006 ▶)]; nsp5 (Anand et al., 2002 ▶); a complex consisting of nsp7–nsp8 (Zhai et al., 2005 ▶); the RNA-binding protein nsp9 (Egloff et al., 2004 ▶); the zinc-binding protein nsp10 (Joseph et al., 2006 ▶, Su et al., 2006 ▶); and the hexameric RNA endonuclease nsp15 (Ricagno et al., 2006 ▶). In many cases, the crystal structures allowed scientists to suggest or to ascertain a biochemical function for the nsps, such as nsp7–nsp8, which acts as an RNA-dependent RNA primase (Imbert et al., 2006 ▶), and the nsp15 endonuclease, the active site of which has been found to share structural homology to the active site of RNAse A (Ricagno et al., 2006 ▶; Joseph et al., 2007 ▶). The process of RNA capping, however, has not yet benefitted from this structural work. Structural data is lacking for the enzymes that are putatively involved in mRNA capping.
In eukaryotes, mRNA capping results from a series of three to four canonical reactions acting on the 5′-end of the mRNA. The nascent mRNA transcript endures a limited dephosphorylation in which the last 5′-phosphate is removed by a 5′-RNA triphosphatase. A guanylyltransferase (also named capping enzyme) attaches a GMP molecule onto the 5′-diphosphate RNA in a 5′-to-5′ orientation. The capped RNA is then methylated at the N7 position of the cap guanine nucleotide by an N7-guanine methyltransferase. This yields a cap-0 structure (m7GpppNN…) as found mainly in yeasts and lower eukaryotes. In higher eukaryotes and plants, a second methyltransferase acts on the first transcribed nucleotide at its 2′-O position to yield a cap-1 structure (m7GpppN2′OmN…), such as that found on coronavirus mRNAs (Lai & Stohlman, 1981 ▶; van Vliet et al., 2002 ▶).
We have recently identified the RNA cap 2′-O-methyltransferase of the SARS-CoV (Bouvet et al., 2010 ▶; Lugari et al., 2010 ▶). This activity is harboured by nsp16 as predicted by signature-sequence analysis; however, the enzyme is only active in the presence of nsp10. The latter has been shown to interact strongly with nsp16 using the yeast two-hydrid method, indicating that the enzymes associate into a complex that is stable enough to withstand purification (Imbert et al., 2008 ▶).
Here, we report the cloning of nsp10 and nsp16 in a prokaryotic expression vector. Upon expression of both genes, a stable complex consisting of nsp10 and nsp16 (nsp10–nsp16) can be purified and crystallized. We present X-ray diffraction data from these SARS-CoV nsp10–nsp16 crystals.
2. Materials and methods
2.1. Tandem cloning of the nsp10 and nsp16 SARS-CoV genes
The Frankfurt 1 isolate of SARS-CoV (GenBank accession No. AY291315; Thiel et al., 2003 ▶) was amplified in Vero-E6 cells and used for production of the nsp10–nsp16 complex as follows: genes coding for SARS-CoV nsp10 (139 amino acids, 14.84 kDa) and nsp16 (298 amino acids, 33.5 kDa) were cloned into the pmCOX Escherichia coli dual expression plasmid kindly provided by Dr Bruno Coutard (AFMB, France) containing two separate promoters. In this backbone, SARS-CoV nsp10 is expressed under a tetA promoter and encodes a protein fused with an N-terminal Strep-Tag (eight amino acids; WSHPQFEK) and nsp16 is expressed under a T7lac promoter and encodes a protein fused with an N-terminal hexahistidine tag (Bouvet et al., 2010 ▶). This system allows independent regulation of the expression levels of the two genes by the addition of different concentrations of tetracycline and IPTG (isopropyl β-d-1-thiogalactopyranoside).
2.2. Expression and purification of the nsp10–nsp16 complex
SARS-CoV nsp10–nsp16 co-expression was performed in E. coli strain C41 (DE3) (Avidis SA, France) harbouring the pLysS plasmid (Novagen). Cultures were grown at 310 K until the OD600nm reached 0.6. Expression was induced by adding 50 µM IPTG and 200 µg l−1 anhydrotetracycline and the cells were incubated for 16 h at 297 K. Bacterial cell pellets were collected by centrifugation at 6000g, frozen and resuspended in lysis buffer (50 mM HEPES pH 7.5, 300 mM NaCl, 5 mM MgSO4) supplemented with 10 µg ml−1 DNase I. After cell disruption at 100 MPa and 277 K (Constant Cell, UK) and clarification by centrifugation at 20 000g for 30 min, the soluble protein fraction was incubated with Strep-Tactin Sepharose (IBA BioTAGnology). After three washes in buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 1 mM TCEP, 5 mM MgCl2), bound proteins were eluted in wash buffer supplemented with 2.5 mM d-desthiobiotin.
2.3. Crystallization and diffraction analysis
Stura Footprint Screen, JCSG+ Screen and Structure Screens I and II (Molecular Dimensions) were tried in CrystalQuick plates with three wells per reservoir (Greiner Bio-One) using 400, 300 and 200 nl drops. The drops contained increasing volumes (100, 200 and 300 nl) of protein solution at a concentration of 5.4 mg ml−1 in elution buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 1 mM TCEP, 5 mM MgCl2, 2.5 mM d-desthiobiotin) and constant volumes of precipitant. A hit condition was observed in 0.1 M bicine pH 9, 2 M MgCl2 after one week. Crystallization optimization was performed by vapour diffusion using the hanging-drop method at 293 K in Linbro plates (Hampton Research). 2 µl drops (consisting of 1.5 µl protein solution and 0.5 µl precipitant solution) were equilibrated against 1 ml reservoir solution. Optimization of crystal-growth conditions led to the following condition: 0.1 M CHES pH 9, 1.52 M MgCl2. Crystals appeared after 24 h and visible growth stopped after 48–72 h. Before cooling the crystals to 100 K in a nitrogen-gas stream (Oxford Cryosystems), the crystals were briefly soaked in a cryoprotectant consisting of 15%(v/v) glycerol added to the mother liquor. Native diffraction data were recorded using either an ADSC Quantum 315 3 × 3 array detector on beamline PROXIMA 1 at the SOLEIL Synchrotron Radiation Facility or an ADSC Quantum 210 2 × 2 array detector on beamline ID14-EH1 at the European Synchrotron Radiation Facility. Data were processed using XDS (Kabsch, 2010 ▶) and scaled with SCALA (Collaborative Computational Project, Number 4, 1994 ▶). Crystal parameters and data-collection statistics are summarized in Table 1 ▶.
Table 1. Data-collection and processing statistics.
Values in parentheses are for the highest resolution shell.
| No. of crystals | 1 |
| Beamline | PROXIMA 1 (SOLEIL synchrotron) |
| Wavelength (Å) | 0.979 |
| Detector | ADSC Q315r |
| Crystal-to-detector distance (mm) | 256.35 |
| Rotation range per image (°) | 1 |
| Total rotation range (°) | 90 |
| Exposure time per image (s) | 1 |
| Resolution range (Å) | 37.52–2.00 (2.11–2.00) |
| Space group | C2221 |
| Unit-cell parameters (Å, °) | a = 68.1, b = 184.6, c = 128.8, α = β = γ = 90.00 |
| Mosaicity (°) | 0.097 |
| Total no. of measured intensities | 273149 |
| Unique reflections | 54947 (7963) |
| Multiplicity | 3.7 (3.7) |
| Mean I/σ(I) | 7.5 (3.2) |
| Completeness (%) | 99.7 (100) |
| Rmerge† | 0.114 (0.430) |
| Rmeas | 0.136 (0.500) |
| Overall B factor from Wilson plot (Å2) | 37.3 |
R
merge = 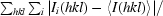
 , where Ii(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations of reflection hkl.
, where Ii(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations of reflection hkl.
2.4. Cross-linking of the purified nsp10–nsp16 complex
Purified nsp10–nsp16 complex (4 µg) in 50 mM HEPES pH 7.5, 150 mM NaCl, 5 mM MgCl2, 1 mM TCEP and 5% glycerol was incubated overnight at 277 K with a solution of suberic acid bis(N-hydroxysuccinimide ester) (SAB; Sigma) at a concentration of 0.005%. Samples were then denatured with an equal volume of 2× dissociation buffer [100 mM Tris–HCl pH 6.8, 20% glycerol and 200 mM DTT, 4% sodium dodecyl sulfate (SDS) and 0.2% bromophenol blue]. After 5 min of heating at 368 K, the proteins were separated and analyzed on an SDS NuPAGE 4–12% gel (Invitrogen).
3. Results and discussion
3.1. Expression, purification and crystallization of a stable nsp10–nsp16 complex
Many attempts in the laboratory to crystallize nsp16 on its own remained unsuccessful. Although significant amounts (on a milligram scale) of nsp16 can be obtained when expressed alone, the protein is unstable in a variety of buffers and precipitates under various storage conditions. Moreover, although signature-sequence analyses unambiguously identified a SAM-dependent methyltransferase fold (von Grotthuss et al., 2003 ▶; Decroly et al., 2008 ▶), SARS-CoV nsp16 alone does not exhibit this enzymatic activity. In contrast, nsp10 is expressed to high levels in E. coli and can readily be purified and crystallized. Two structures of nsp10 crystal forms were published in 2006: one revealed monomers and dimers (Joseph et al., 2006 ▶) at 1.8 Å resolution (PDB entry 2fyg), whereas a complex dodecameric structure at 2.1 Å resolution (PDB entry 2g9t) was observed when nsp10 was expressed and crystallized as a fusion with nsp11 (Su et al., 2006 ▶). Nevertheless, no function has been either predicted or demonstrated for nsp10, which is a zinc-binding protein.
We have reported that nsp10 and nsp16 interact in vitro when co-expressed in yeast (Imbert et al., 2008 ▶) or bacteria (Bouvet et al., 2010 ▶). This observation prompted us to co-express the two proteins in the prokaryotic expression vector and to attempt purification of the complex. Nsp10 was cloned under the control of the tetA promoter in fusion with a Strep-Tag peptide at its N-terminus and nsp16 tagged with a hexahistidine tag at its N-terminus was inserted into the same plasmid under the T7lac promoter. After transformation into bacterial strain C41, protein expression was induced by the addition of IPTG and tetracycline for 16 h at 297 K. The bacterial lysate was clarified and nsp10 was adsorbed onto Strep-Tactin Sepharose beads. After several washes, the proteins bound to Strep-Tactin were eluted with 2.5 mM of the biotin analogue desthiobiotin. Upon SDS–PAGE analysis, we detected the presence of Strep-nsp10 and His6-nsp16 proteins migrating as expected at around 15 and 35 kDa, respectively, indicating that the two proteins had co-purified (Fig. 1 ▶). The overall yield of the purified complex was typically 1 mg per litre of E. coli culture. Estimation of protein concentration and normalization with regard to molecular mass indicated an approximate 1:1 ratio of the proteins.
Figure 1.
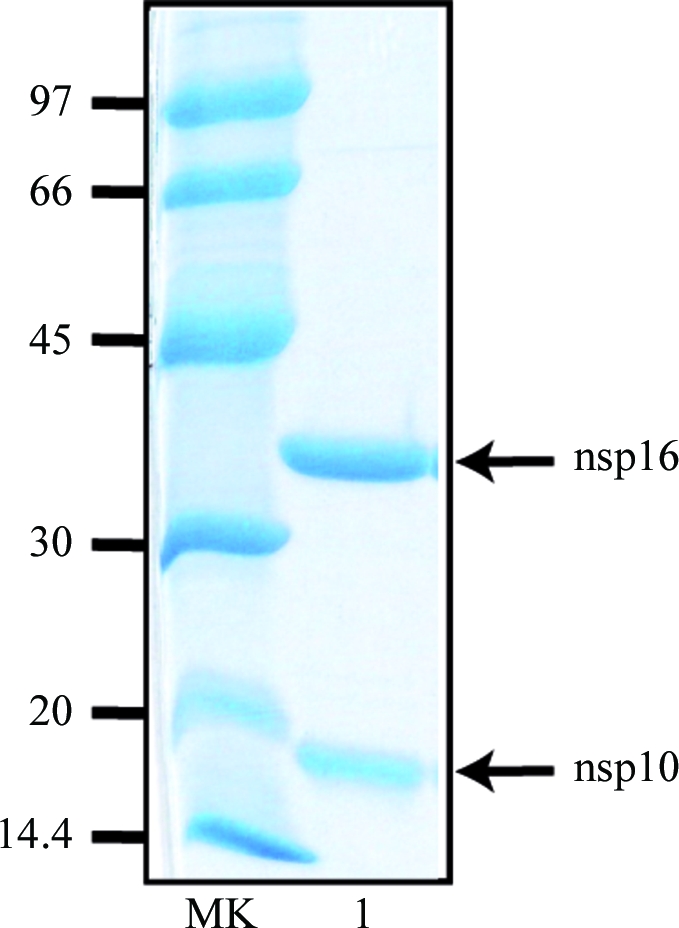
Purification of SARS-CoV nsp10 in complex with nsp16. The purified SARS-CoV nsp10–nsp16 complex was analyzed by 12% SDS–PAGE and stained using Coomassie Blue. Lane MK, molecular-weight markers; lane 1, 2 µg nsp10–nsp16 protein complex eluted from the Strep-Tactin column.
3.2. Characterization of the nsp10–nsp16 complex
The nsp10–nsp16 complex was further characterized before attempting to identify crystallization conditions. We first confirmed the identity of each recombinant protein by matrix-assisted laser-desorption ionization time-of-flight mass spectrometry after in-gel trypsin digestion (data not shown). We also analyzed the proteins eluted from the Strep-Tactin column by size-exclusion chromatography. Fig. 2 ▶(a) shows the gel-filtration elution profile. We observed that the stable nsp10–nsp16 complex elutes as a single peak corresponding to 50 kDa. The interaction between nsp10 and nsp16 was also confirmed by a cross-linking experiment. For this purpose, the nsp10–nsp16 complex was incubated with 0.005% SAB, a cross-linking reagent that specifically reacts with lysine residues. After SDS–PAGE separation, we detected the bands corresponding to nsp10 and nsp16 monomers as well as an additional band corresponding to a nsp10–nsp16 complex migrating around 50 kDa (lane Xtal in Fig. 3 ▶ b). The presence of nsp16 in the complex was also demonstrated by a methyltransferase assay (Selisko et al., 2010 ▶) which showed that the purified fraction containing the nsp10–nsp16 complex exhibited 2′-O-methyltransferase activity on short N7-methylated capped RNA substrates (Bouvet et al., 2010 ▶). Together, these results indicate that we have developed a method allowing the production and purification of an active nsp10–nsp16 complex by a one-step procedure. This protein preparation was used without additional treatment for crystal growth.
Figure 2.

Characterization of SARS-CoV nsp10 in complex with nsp16. (a) Gel-filtration chromatogram of the SARS-CoV nsp10–nsp16 complex. The nsp10–nsp16 complex eluted from the Strep-Tactin column was analyzed on a 16/60 S200 gel-filtration column and the elution of protein and nucleic acid was followed by measuring the absorption at 280 nm (blue) and 260 nm (orange), respectively. The main peak eluting after 90 ml corresponds to elution of a 50 kDa protein. (b) Cross-linking experiment. The purified SARS-CoV nsp10–nsp16 complex was loaded onto a 4–12% NuPAGE gel and stained using Coomassie Blue. Lane MK, molecular-weight markers; lane 1, 4 µg non-cross-linked nsp10–nsp16 complex, lane 2, 4 µg of the nsp10–nsp16 complex incubated overnight at 277 K with 0.005% SAB cross-linker.
Figure 3.

(a) Optimized crystal of the SARS-CoV nsp10–nsp16 complex. The scale bar is 100 µm in length. (b) NuPAGE analysis of the nsp10–nsp16 complex from optimized crystals. Ten optimized crystals were loaded onto a 4–12% NuPAGE gel and stained using Coomassie blue. Lane MK, molecular-weight markers; lane Xtal, nsp10–nsp16 complex. (c) X-ray diffraction pattern from a crystal of the SARS-CoV nsp16–nsp10 protein complex. Resolution arcs are shown. Reflections are observed to below 2 Å (an enlargement is shown in the inset).
3.3. Crystal growth and data collection
The complex was initially crystallized using 0.1 M bicine pH 9, 2 M MgCl2 (condition E2 fom Structure Screens I and II). Crystal growth was then optimized and the best condition was finally chosen as 0.1 M CHES pH 9, 1.52 M MgCl2. Crystals typically appeared overnight and visible growth stopped after 48–72 h (Fig. 3 ▶ a). To confirm that the crystal contained the nsp10–nsp16 complex, we collected ten crystals, which were analyzed by SDS–PAGE after two washes. Fig. 2 ▶(b) shows that both proteins, nsp10 and nsp16, were detected on Coomassie Blue staining. This confirms that the crystals consist of the nsp10–nsp16 complex with a similar stoichiometry as observed prior to crystallization.
Crystals were flash-cooled in the same buffer supplemented with 15%(v/v) glycerol before exposure to synchrotron X-ray radiation. A typical diffraction image is shown in Fig. 3 ▶(c), in which reflections are visible to 2 Å resolution. Data integration and reduction indicated that the crystals belonged to space group C2221, with one complex per asymmetric unit (Table 1 ▶). The crystals (space group C2221, unit-cell parameters a = 68.1, b = 184.6, c = 128.8 Å) contained one nsp10–nsp16 complex per asymmetric unit, with a solvent content of 70% and a V M value of 4.17 Å3 Da−1.
4. Conclusions
We have crystallized a complex of the SARS-CoV nsp10 and nsp16 proteins. The presence of nsp10 in the complex confers 2′-O-methyltransferase activity on nsp16. The crystal structure of nsp10 is known, whereas that of nsp16 is not. Given the quality of the crystals described here and the fact that nsp10 is a zinc-binding protein, it should be possible to determine the crystal structure using molecular-replacement techniques merged with phases obtained by SAD studies, taking advantage of the phasing power of the Zn atoms.
Acknowledgments
This work was supported in its initial phase by the VIZIER integrated project (LSHG-CT-2004-511960) of the European Union Sixth Framework and then by the French National Research agency under reference ANR-08-MIEN-032, the Fondation pour la Recherche Médicale (Programme équipe FRM) to BC and the Direction Générale de l’Armement (contract 07co404). MB has a fellowship from the Direction Générale de l’Armement.
References
- Almeida, M. S., Johnson, M. A., Herrmann, T., Geralt, M. & Wüthrich, K. (2007). J. Virol. 81, 3151–3161. [DOI] [PMC free article] [PubMed]
- Anand, K., Palm, G. J., Mesters, J. R., Siddell, S. G., Ziebuhr, J. & Hilgenfeld, R. (2002). EMBO J. 21, 3213–3224. [DOI] [PMC free article] [PubMed]
- Bouvet, M., Debarnot, C., Imbert, I., Selisko, B., Snijder, E. J., Canard, B. & Decroly, E. (2010). PLoS Pathog. 6, e1000863. [DOI] [PMC free article] [PubMed]
- Chatterjee, A., Johnson, M. A., Serrano, P., Pedrini, B., Joseph, J. S., Neuman, B. W., Saikatendu, K., Buchmeier, M. J., Kuhn, P. & Wüthrich, K. (2009). J. Virol. 83, 1823–1836. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Decroly, E., Imbert, I., Coutard, B., Bouvet, M., Selisko, B., Alvarez, K., Gorbalenya, A. E., Snijder, E. J. & Canard, B. (2008). J. Virol. 82, 8071–8084. [DOI] [PMC free article] [PubMed]
- Egloff, M. P., Ferron, F., Campanacci, V., Longhi, S., Rancurel, C., Dutartre, H., Snijder, E. J., Gorbalenya, A. E., Cambillau, C. & Canard, B. (2004). Proc. Natl Acad. Sci. USA, 101, 3792–3796. [DOI] [PMC free article] [PubMed]
- Egloff, M. P., Malet, H., Putics, A., Heinonen, M., Dutartre, H., Frangeul, A., Gruez, A., Campanacci, V., Cambillau, C., Ziebuhr, J., Ahola, T. & Canard, B. (2006). J. Virol. 80, 8493–8502. [DOI] [PMC free article] [PubMed]
- Grotthuss, M. von, Wyrwicz, L. S. & Rychlewski, L. (2003). Cell, 113, 701–702. [DOI] [PMC free article] [PubMed]
- Imbert, I., Guillemot, J. C., Bourhis, J. M., Bussetta, C., Coutard, B., Egloff, M. P., Ferron, F., Gorbalenya, A. E. & Canard, B. (2006). EMBO J. 25, 4933–4942. [DOI] [PMC free article] [PubMed]
- Imbert, I., Snijder, E. J., Dimitrova, M., Guillemot, J. C., Lécine, P. & Canard, B. (2008). Virus Res. 133, 136–148. [DOI] [PMC free article] [PubMed]
- Joseph, J. S., Saikatendu, K. S., Subramanian, V., Neuman, B. W., Brooun, A., Griffith, M., Moy, K., Yadav, M. K., Velasquez, J., Buchmeier, M. J., Stevens, R. C. & Kuhn, P. (2006). J. Virol. 80, 7894–7901. [DOI] [PMC free article] [PubMed]
- Joseph, J. S., Saikatendu, K. S., Subramanian, V., Neuman, B. W., Buchmeier, M. J., Stevens, R. C. & Kuhn, P. (2007). J. Virol. 81, 6700–6708. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Lai, M. M. & Stohlman, S. A. (1981). J. Virol. 38, 661–670. [DOI] [PMC free article] [PubMed]
- Lugari, A., Betzi, S., Decroly, E., Bonnaud, E., Hermant, A., Guillemot, J. C., Debarnot, C., Borg, J. P., Bouvet, M., Canard, B., Morelli, X. & Lécine, P. (2010). J. Biol. Chem. 285, 33230–33241. [DOI] [PMC free article] [PubMed]
- Ratia, K., Saikatendu, K. S., Santarsiero, B. D., Barretto, N., Baker, S. C., Stevens, R. C. & Mesecar, A. D. (2006). Proc. Natl Acad. Sci. USA, 103, 5717–5722. [DOI] [PMC free article] [PubMed]
- Ricagno, S., Egloff, M. P., Ulferts, R., Coutard, B., Nurizzo, D., Campanacci, V., Cambillau, C., Ziebuhr, J. & Canard, B. (2006). Proc. Natl Acad. Sci. USA, 103, 11892–11897. [DOI] [PMC free article] [PubMed]
- Selisko, B., Peyrane, F. F., Canard, B., Alvarez, K. & Decroly, E. (2010). J. Gen. Virol. 91, 112–121. [DOI] [PubMed]
- Serrano, P., Johnson, M. A., Almeida, M. S., Horst, R., Herrmann, T., Joseph, J. S., Neuman, B. W., Subramanian, V., Saikatendu, K. S., Buchmeier, M. J., Stevens, R. C., Kuhn, P. & Wüthrich, K. (2007). J. Virol. 81, 12049–12060. [DOI] [PMC free article] [PubMed]
- Snijder, E. J., Bredenbeek, P. J., Dobbe, J. C., Thiel, V., Ziebuhr, J., Poon, L. L., Guan, Y., Rozanov, M., Spaan, W. J. & Gorbalenya, A. E. (2003). J. Mol. Biol. 331, 991–1004. [DOI] [PMC free article] [PubMed]
- Su, D., Lou, Z., Sun, F., Zhai, Y., Yang, H., Zhang, R., Joachimiak, A., Zhang, X. C., Bartlam, M. & Rao, Z. (2006). J. Virol. 80, 7902–7908. [DOI] [PMC free article] [PubMed]
- Thiel, V., Ivanov, K. A., Putics, A., Hertzig, T., Schelle, B., Bayer, S., Weissbrich, B., Snijder, E. J., Rabenau, H., Doerr, H. W., Gorbalenya, A. E. & Ziebuhr, J. (2003). J. Gen. Virol. 84, 2305–2315. [DOI] [PubMed]
- Vliet, A. L. van, Smits, S. L., Rottier, P. J. & De Groot, R. J. (2002). EMBO J. 21, 6571–6580. [DOI] [PMC free article] [PubMed]
- Zhai, Y., Sun, F., Li, X., Pang, H., Xu, X., Bartlam, M. & Rao, Z. (2005). Nature Struct. Mol. Biol. 12, 980–986. [DOI] [PMC free article] [PubMed]