

Abstract
Background:
Multiple sclerosis (MS) in the pediatric age group is being increasingly recognized. In adults, complex interactions between genetic and environmental factors contribute to risk and the major genetic component of MS susceptibility localizes to the major histocompatibility complex (human leukocyte antigen [HLA]). Whether HLA alleles predict MS in at-risk children presenting with acquired demyelinating syndromes (ADS) of the CNS is unknown.
Methods:
HLA-DRB1 alleles were typed using an allele-specific PCR amplification method on samples from 266 children presenting with ADS enrolled in the prospective Canadian Pediatric Demyelinating Disease Study and from 196 healthy controls.
Results:
Sixty-four of 266 children with ADS met established criteria for a diagnosis of MS during a mean follow-up of 3.2 ± 1.5 years. Children harboring DRB1*15 alleles were more likely to be diagnosed with MS (χ2 = 12.2, p < 0.001; OR = 2.7), an observation strengthened by children of European ancestry (χ2 = 10.5, p = 0.001; OR = 3.3). DRB1*15 allele frequencies in children with ADS of European ancestry subsequently diagnosed with MS were greater than in children with monophasic ADS (χ2 = 10.7, p = 0.001) or healthy controls (χ2 = 12.5, p < 0.001). The proportion of children with non-European ancestry diagnosed with MS was not influenced by DRB1*15 status.
Conclusion:
DRB1*15 alleles confer increased susceptibility to pediatric-onset MS, supporting a fundamental similarity in genetic contribution to MS risk in both pediatric- and adult-onset disease. The specificity of the DRB1*15 risk allele for children with subsequent MS diagnosis, but not for all children with ADS, indicates that the risk conveyed by DRB1*15 relates to chronic CNS disease (MS), rather than acquired demyelination in general.
Multiple sclerosis (MS) is being increasingly diagnosed in the pediatric age group.1 In children, an initial attack of demyelination (acquired demyelinating syndrome [ADS] of the CNS) often remains a monophasic illness, but in at least 20% of children it represents the first clinical attack of MS.2 This contrasts with adult-onset disease, where most individuals presenting with acute demyelination are subsequently diagnosed with MS. Whether pediatric MS shares the same biological underpinnings (including environmental and genetic risk factors) implicated in adult-onset MS is an area of active investigation.
In adult-onset MS, the best established haplotypic susceptibility resides in DRB1 alleles of the major histocompatibility complex (MHC).3 In particular, susceptibility to adult-onset MS in white Northern European populations has been fine mapped to the extended HLA class II haplotype, HLA-DQA1*0102-DQB1*O6O2-DRB1*1501-DRB5*0101.4
While a single study has reported a higher frequency of HLA-DRB alleles in pediatric MS,5 and another reported the absence of an association of HLA in a cohort of children with acute disseminated encephalomyelitis (ADEM),6 little is known regarding the HLA status in the unselected pediatric population presenting with a first demyelinating attack who are at high risk for developing MS. In particular, it is not known whether certain HLA alleles confer risk of developing ADS in general (including children who remain with monophasic ADS), or more specifically confer the risk of recurrent disease (MS). We hypothesize that the presence of HLA-DRB1*15 alleles will be associated with MS outcome in children at risk.
METHODS
Participants.
Children age 16 years, 0 months, or younger presenting within 30 days of onset of ADS at 1 of 23 participating pediatric health care centers across Canada were recruited into the 8-year prospective Canadian National Pediatric Demyelinating Disease Study (Coinvestigators). Comprehensive structured clinical history and neurologic examinations were performed at study entry and at all study visits (baseline, 3, 6, and 12 months and annually, and at the time of recurrent demyelination).
Presenting features at ADS were characterized by clinical history and examination (without MRI) as clinically isolated (all clinical symptoms and signs localizable to a single site) optic neuritis (ON), clinically isolated transverse myelitis (TM), or clinically isolated–other (features localizable to a single CNS site separate from the optic nerve or spine). Patients with findings localized to multiple CNS sites with specific recording of optic nerve or spine involvement were delineated as polyfocal ADS. ADEM was diagnosed using predefined criteria which require polyfocal deficits accompanied by encephalopathy.7 MS was diagnosed when either subsequent examination confirmed new neurologic deficits persisting for 24 hours or longer in the absence of fever or infection8,9 and occurring more than 30 days from ADS or if serial MRI scans revealed new lesions meeting criteria for dissemination in time.10 For children with an initial diagnosis of ADEM, 2 non-ADEM subsequent attacks (the first of which occurred more than 3 months from the incident ADEM event)8 were required.
A total of 316 children meeting inclusion criteria were enrolled between September 1, 2004, and June 1, 2010, in the Canadian National Demyelinating Disease Study, with June 30, 2010, serving as the date of data closure for the present article. We excluded from analysis 2 children who manifested with recurrent episodes of myelitis (alone or with ON) and confirmed neuromyelitis optica immunoglobulin seropositivity as per established criteria for the diagnosis of neuromyelitis optica.11 Four families withdrew from the study. Eighteen children were excluded after subsequent investigations revealed an alternative diagnosis (tumor n = 3, CNS vasculitis n = 5, CNS lupus n = 1, mitochondrial disease n = 2, CNS neuroborreliosis n = 1, retinitis n = 1, cerebellitis n = 1, acute necrotizing encephalitis n = 1, neurosarcoidosis n = 1, stroke n = 1, and migraine n = 1). Fourteen of the 292 eligible families declined genetic studies and in 12 children limited quantity or quality of DNA did not permit accurate genotyping. Thus, HLA-DRB1 alleles were typed in 266 (91%) eligible children.
A control group of 196 unrelated, healthy adults of Northern European ancestry was recruited through the longitudinal, population-based Canadian Collaborative Project on the Genetic Susceptibility to Multiple Sclerosis (CCPGSMS).12
Standard protocol approvals and patient consents.
Institutional ethical approval was obtained at all 23 sites, and written informed consent was obtained from all ADS study participant families. The control population was consented as part of the CCPGSMS.12
MRI analyses.
Brain MRIs were ordered at baseline and at months 3, 6, and 12, as well as at the time of a second demyelinating event, using a standardized research MRI protocol. Additional annual MRIs were carried out on participants enrolled at The Hospital for Sick Children (SickKids; n = 100), Toronto, Canada. All scans were analyzed centrally. For the purpose of the present study, scans were specifically evaluated for the presence of new lesions according to established criteria for dissemination in time.10 Although acquisition of spine MRIs was not a formal component of our national program, the MRI reports of all available spinal scans were reviewed centrally. Each spine lesion was scored as longitudinally extensive (LETM, lesion extending greater than 3 spinal segments in longitudinal extent) or as non-LETM (maximal longitudinal extent less than 3 spinal segments). Patients with either combinations of LETM and non-LETM lesions or a combination of one or more spinal cord lesions with brain lesions present on the same scan were considered “other.” If the report did not clearly indicate the longitudinal extent of the spinal lesion, the spine images were sent to the central site (SickKids) for review.
Genotyping.
Total genomic DNA, extracted from whole blood, was used to type HLA-DRB1 alleles by an allele-specific PCR amplification method,13 for all participants, with the exception of 10 children (2 of European ancestry) with limited DNA availability, for whom only the presence of the DRB1*15 allele was determined. Seventy-two (high-resolution) PCR reactions were performed to amplify allelotypes corresponding to alleles HLA-DRB1*01, 03, 04, 07, 08, 09, 10, 11, 12, 13, 14, 15, 16. Samples were genotyped blinded to clinical diagnosis.
Statistical analyses.
The likelihood of MS diagnosis in children with ADS was assessed as a function of HLA-DRB1*15 status (proportion of patients with one or both alleles, compared to those with no alleles). Frequency of DRB1 alleles (each allele contributing independently) was compared between groups (MS vs monophasic ADS; MS vs healthy controls; monophasic ADS vs healthy controls). As only 10 children with ADS and 6 with MS were homozygous for DRB1*15, we did not attempt to analyze MS risk as a function of homozygosity vs heterozygosity for HLA. Continuous summary statistics are presented as mean ± SD. Odds ratios, χ2, or Fisher exact test (2-tailed), where appropriate, assessed differences between groups. Survival analysis assessed the time to MS diagnosis as a function of HLA-DRB1*15 status. Logistic regression was used to control for effect of age and sex.
RESULTS
Of 266 children with ADS, 64 (24%) met criteria for the diagnosis of MS over an average follow-up duration of 3.2 ± 1.5 years. The average time from ADS presentation to MS diagnosis was 10.2 ± 9.0 months (median 6.3; range 1.2–37.3). Clinical and demographic characteristics of all children are presented in table 1. As previously reported, older age at ADS and female sex were associated with a greater likelihood of MS diagnosis.2,14
Table 1.
Clinical and demographic features of children with ADS diagnosed with MS vs those remaining monophasic ADS
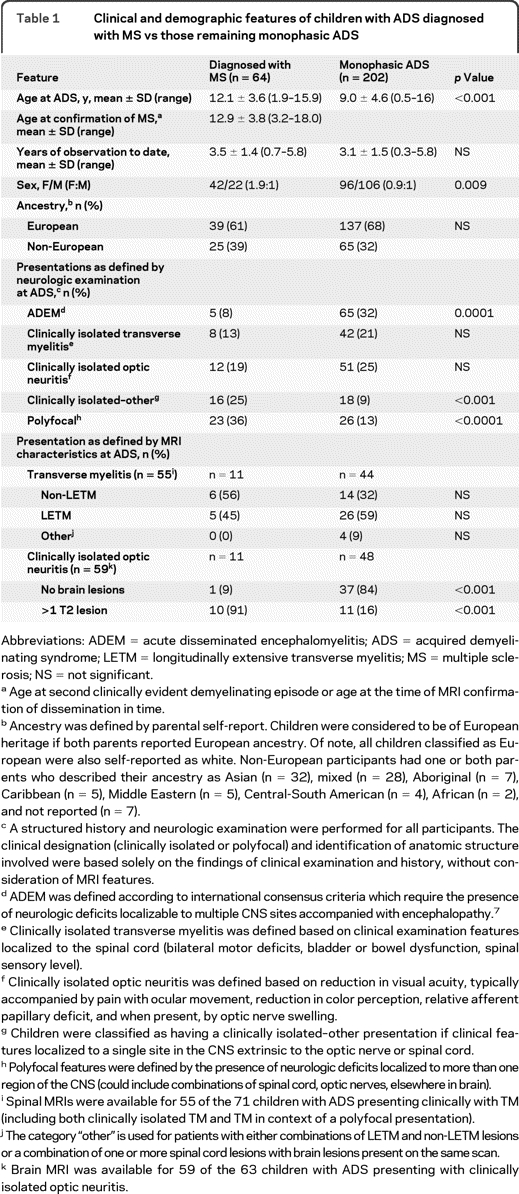
Abbreviations: ADEM = acute disseminated encephalomyelitis; ADS = acquired demyelinating syndrome; LETM = longitudinally extensive transverse myelitis; MS = multiple sclerosis; NS = not significant.
Age at second clinically evident demyelinating episode or age at the time of MRI confirmation of dissemination in time.
Ancestry was defined by parental self-report. Children were considered to be of European heritage if both parents reported European ancestry. Of note, all children classified as European were also self-reported as white. Non-European participants had one or both parents who described their ancestry as Asian (n = 32), mixed (n = 28), Aboriginal (n = 7), Caribbean (n = 5), Middle Eastern (n = 5), Central-South American (n = 4), African (n = 2), and not reported (n = 7).
A structured history and neurologic examination were performed for all participants. The clinical designation (clinically isolated or polyfocal) and identification of anatomic structure involved were based solely on the findings of clinical examination and history, without consideration of MRI features.
ADEM was defined according to international consensus criteria which require the presence of neurologic deficits localizable to multiple CNS sites accompanied with encephalopathy.7
Clinically isolated transverse myelitis was defined based on clinical examination features localized to the spinal cord (bilateral motor deficits, bladder or bowel dysfunction, spinal sensory level).
Clinically isolated optic neuritis was defined based on reduction in visual acuity, typically accompanied by pain with ocular movement, reduction in color perception, relative afferent papillary deficit, and when present, by optic nerve swelling.
Children were classified as having a clinically isolated–other presentation if clinical features localized to a single site in the CNS extrinsic to the optic nerve or spinal cord.
Polyfocal features were defined by the presence of neurologic deficits localized to more than one region of the CNS (could include combinations of spinal cord, optic nerves, elsewhere in brain).
Spinal MRIs were available for 55 of the 71 children with ADS presenting clinically with TM (including both clinically isolated TM and TM in context of a polyfocal presentation).
The category “other” is used for patients with either combinations of LETM and non-LETM lesions or a combination of one or more spinal cord lesions with brain lesions present on the same scan.
Brain MRI was available for 59 of the 63 children with ADS presenting with clinically isolated optic neuritis.
Children presenting with ADS who harbored one or both DRB1*15 alleles were more likely to be diagnosed with MS during the period of follow-up (37% [34/93 children]) as compared to children who did not harbor DRB1*15 alleles (17% [30/173]; χ2 = 12.2, p < 0.001; odds ratio [OR] = 2.7). The increased MS risk conferred by DRB1*15 appeared to be greater in children reporting European ancestry: 37% (21/57) of DRB1*15-positive children were diagnosed with MS, vs 15% (18/119) of DRB1*15-negative children (χ2 = 10.5, p = 0.001; OR = 3.3). In contrast, the risk of MS diagnosis in children with ADS of non-European ancestry was not influenced by the presence of DRB1*15 alleles: 36% (13/36) of DRB1*15-positive children were diagnosed with MS, vs 22% (12/54) of DRB1*15-negative children (χ2 = 2.1, p = 0.15). Survival analysis (time to MS diagnosis) also revealed that DRB1*15-positive children had a greater propensity to be diagnosed with MS (log-rank [Mantel-Cox] test: χ2 = 12.6, p = 0.0004) (figure). While sex and age at ADS were associated with a greater likelihood of MS diagnosis, logistic regression accounting for both age and sex revealed that HLA-DRB1*15 status remained an independent predictor of MS diagnosis (OR = 3.2, 95% confidence interval 1.7–6.1).
Figure.

Time to multiple sclerosis diagnosis in children following acquired demyelinating syndrome (ADS) as a function of HLA-DRB1*15 status
For all children presenting with ADS (n = 266), the allele frequency for DRB1*15 was 0.20 (109/532 alleles). The DRB1*15 allele frequencies were higher in children with ADS subsequently diagnosed with MS, compared to children with monophasic ADS (χ2 = 12.0, p = 0.001; OR = 2.2). In the 176 ADS patients of European ancestry (n = 137 monophasic ADS; n = 39 MS), DRB1*15 was the most frequent allele (18%, 64/352 alleles). Comparison across groups revealed higher DRB1*15 allele frequencies in children with ADS subsequently diagnosed with MS, compared to those with monophasic ADS (χ2 = 10.7, p = 0.001; OR = 2.6) or controls (χ2 = 12.5, p < 0.001; OR = 2.7). No difference was seen comparing children with monophasic ADS to controls (χ2 = 0.01, p = 0.90). No differences in allele frequencies were noted between groups for other DRB1 alleles. Of the 39 children with ADS diagnosed with MS, 21 (54%) had at least one DRB1*15 allele, compared to only 36 of 137 (26%) children with monophasic ADS (χ2 = 10.5, p = 0.001). The proportion of controls with at least one DRB1*15 allele was 25%, similar to that of monophasic ADS, but lower than in the children with ADS who were diagnosed with MS (χ2 = 12.9, p < 0.001). While comparison to controls is restricted to Northern Europeans, one cannot exclude potential confounding of associations based on population stratification.
As an exploratory analysis, we examined the association of DRB1*15 alleles with MS diagnosis as a function of different clinical presentations of the 266 children with ADS (table 2). We observed that in clinically isolated TM presentations, the presence of DRB1*15 alleles seemed to increase the risk of a subsequent MS diagnosis, though this trend did not reach statistical significance. However, when MRI features of spinal cord involvement were considered, it was evident that ADS presentations of TM that were not longitudinally extensive (non-LETM) were significantly more likely to be diagnosed with MS if they had at least one DRB1*15 allele. Among children presenting with ON, presence of MRI brain lesions was, as expected, associated with increased risk of a subsequent MS diagnosis, though this risk did not appear to be significantly influenced by DRB1*15 status. As only 5 out of 70 children presenting with ADEM have been diagnosed with MS, meaningful analysis of an HLA-DRB1*15 effect was not possible in this subgroup.
Table 2.
Evaluation of HLA as a function of clinical presentations and MS outcome

Abbreviations: ADEM = acute disseminated encephalomyelitis; ADS = acquired demyelinating syndrome; HLA = human leukocyte antigen; LETM = longitudinally extensive transverse myelitis; MS = multiple sclerosis; NS = not significant.
For definitions of clinical presentations, see table 1.
For definitions of imaging presentations, see table 1.
DISCUSSION
Unique to our work is the evaluation of HLA alleles in a nonselected prospectively followed population of children with ADS, representing a population at risk for the diagnosis of MS. We show that presence and frequencies of DRB1*15 alleles in these children confers increased risk for a subsequent diagnosis of pediatric-onset MS. This was not the case for monophasic children with ADS, indicating that DRB1*15 does not represent a risk factor for development of acquired demyelination in general. The association between DRB1*15 and diagnosis of MS in our ADS population was influenced by the increased frequency of DRB1*15 in children with European ancestry, while DRB1*15 conveyed no increased MS risk in children of non-European ancestry.
In a single prior study examining HLA alleles in children with established MS,5 44.6% of Russian pediatric patients with MS were found to have at least one HLA-DRB1*15 allele, similar to the frequency in our children with ADS who subsequently developed the diagnosis of pediatric MS (all children with MS: 34/64 [53%]; European children with MS: 21/39 [54%]).
Conceptually, presentations with ADS may represent several diverse biologies with differential MS risk, as evidenced by the particular influence of HLA-DRB1*15 status on MS outcome in children presenting with TM and non-LETM lesions. We did not observe an influence of DRB1*15 on MS outcome in children presenting with ON, as has been reported in adults.15 Exploration of the association of HLA alleles and MS outcome in children presenting initially with ADEM features will require a longer observation period, as the time from initial presentation to confirmed MS relapse in younger children (who are more likely to have an ADEM event) can exceed 5 years.2
ACKNOWLEDGMENT
The authors thank the children and their families; the Canadian Pediatric Demyelinating Disease Network, which includes pediatric neurologists and their coordinators at 23 sites across Canada (coinvestigators); and the research staff at the Experimental Therapeutics Program and the McConnell Brain Imaging Centre at the Montreal Neurological Institute.
- ADEM
- acute disseminated encephalomyelitis
- ADS
- acquired demyelinating syndrome
- CCPGSMS
- Canadian Collaborative Project on the Genetic Susceptibility to Multiple Sclerosis
- HLA
- human leukocyte antigen
- LETM
- longitudinally extensive transverse myelitis
- MHC
- major histocompatibility complex
- MS
- multiple sclerosis
- ON
- optic neuritis
- OR
- odds ratio
- TM
- transverse myelitis.
COINVESTIGATORS
Coinvestigators are listed in alphabetical order by site–investigator: Mark Awuku, MD (Windsor Regional Hospital, Site Investigator); Louise Roberts, RN (Windsor Regional Hospital, Site Coordinator); J. Burke Baird, MD (Sudbury Regional Hospital, Site Investigator); Nancy Cacciotti, RN (Sudbury Regional Hospital, Site Coordinator); Brenda Banwell, MD (The Hospital for Sick Children, Site Investigator); Melissa McGowan, MHK, Julia O'Mahony, BSc, Emily Ursell, BSc, Courtney Fairbrother, BA, Julia Kennedy, MSc, Jennifer Hamilton, Samantha Irwin, MSc, Sandra Magalhaes, MSc (The Hospital for Sick Children, Site Coordinators); Virender Bhan, MBBS (Dalhousie University, Site Investigator); Trudy Campbell, NP, Lucy Sagar, BSc, MEd (Dalhousie University, Site Coordinators); Frances Booth, MD, Namrata Shah, MD, Ruth Ann Marrie, MD, PhD (Winnipeg Health Sciences Centre, Site Investigators); Joan Kupchak, RN (Winnipeg Health Sciences Centre, Site Coordinator); David Buckley, MD (Janeway Children's Health and Rehabilitation Centre, Site Investigator); Dianne McGrath, RN, Sharon Penney, RN (Janeway Children's Health and Rehabilitation Centre, Site Coordinators); Mary Connolly, MD (BC Children's Hospital, Site Investigator); Shelia Kent, RN (BC Children's Hospital, Site Coordinator); Pamela Cooper, MD (Rouge Valley-Centenary HC, Site Investigator); Loris Aro, RN (Rouge Valley-Centenary HC, Site Coordinator); Marie-Emmanuelle Dilenge, MD (Montreal Children's Hospital, Site Investigator); Heather Davies, MSc (Montreal Children's Hospital, Site Coordinator); Asif Doja, MD, Daniela Pohl, MD, PhD, Sharon Whiting, MD (Children's Hospital of Eastern Ontario, Site Investigators); Chantal Horth, Sheila Ledoux (Children's Hospital of Eastern Ontario, Site Coordinators); Francois Grand'Maison, MD (Hôpital Charles LeMoyne, Site Investigator); Julie Lafrenière, RN, BScN (Hôpital Charles LeMoyne, Site Coordinator); Simon Levin, MD (Children's Hospital of Western Ontario, Site Investigator); Mala Ramu, BA (Children's Hospital of Western Ontario, Site Coordinator); Anne Lortie, MD (Sainte-Justine Hospital, Site Investigator); Sophie Morin, RN, Fabiola Breault, RN, Stephanie Pellerin, RN (Sainte-Justine Hospital, Site Coordinators); E. Athen MacDonald, MD (Kingston General Hospital, Site Investigator); Vee McBride, RN, BScN (Kingston General Hospital, Site Coordinator); Jean Mah, MD, MSc (Alberta Children's Hospital, Site Investigator); Caitlin Wright, BSc, Natarie Liu, BSc, Catherine Riddell (Alberta Children's Hospital, Site Coordinators); Brandon Meaney, MD, Dave Callen, PhD, MD (McMaster Children's Hospital, Site Investigators); Leah Morgenstern, RN, Laurie Wyllie, RN, Heather Neuman, RN (McMaster Children's Hospital, Site Coordinators); David Meek, MD (Saint John Regional Hospital, Site Investigator); Alison Crowell, RN, BScN (Saint John Regional Hospital, Site Coordinator); Noel Lowry, MD (Royal University Hospital, Site Investigator); Doris Newmeyer, RN (Royal University Hospital, Site Coordinator); Guillaume Sébire, MD (Université de Sherbrooke, Site Investigator); Christian Houde, RN (Université de Sherbrooke, Site Coordinator); Kati Wambera, MA, MD, Colleen Adams, MD (Victoria General Hospital, Site Investigators); Laurie Robson, RN (Victoria General Hospital, Site Coordinator); Ellen Wood, MD (IWK Health Centre, Site Investigator); Elaine Woolridge, RN, Edythe Smith, RN (IWK Health Centre, Site Coordinators); Jerome Yager, MD (Children's Stollery Hospital, Site Investigator); Marjorie Berg, RN, Hope Chick, RN (Children's Stollery Hospital, Site Coordinators); Conrad Yim, MD, PhD (Trillium Health Centre, Site Investigator); Leanne Montgomery, RN (Trillium Health Centre, Site Coordinator).
DISCLOSURE
Dr. Disanto, S. Magalhaes, Dr. Handel, and K.M. Morrison report no disclosures. Dr. Sadovnick has received funding for travel and speaker honoraria from Bayer Canada, Teva Pharmaceutical Industries Ltd., EMD Serono, Inc., and Biogen Idec; and has received research support from the MS Society of Canada Scientific Research Foundation. Dr. Ebers serves on the editorial boards of the International Multiple Sclerosis Journal and Multiple Sclerosis; has received a speaker honorarium from Roche; served as a consultant to UCB; and receives research support from Bayer Schering Pharma, the Multiple Sclerosis Society of the United Kingdom, and the Multiple Sclerosis Society of Canada Scientific Research Foundation. Dr. Banwell serves on a scientific advisory board for Biogen Idec; serves on the editorial boards of Neurology® and Multiple Sclerosis; and receives research support from the Multiple Sclerosis Society of Canada, the Multiple Sclerosis Scientific Research Foundation, and the Canadian Institutes of Health Research. Dr. Bar-Or serves on scientific advisory boards for DioGenix, Inc., Ono Pharmaceutical Co. Ltd., and Roche; serves on the editorial board of Neurology®; has received speaker honoraria from Biogen Idec, Bayhill Therapeutics, Bayer Schering Pharma (Berlex), Eli Lilly and Company, Genentech, Inc., GlaxoSmith Kline, Merck Serono, Novartis, Wyeth, and Teva Pharmaceutical Industries Ltd.; and receives research support from, Biogen Idec, Genentech, Inc., and Teva Pharmaceutical Industries Ltd.
REFERENCES
- 1. Banwell B, Ghezzi A, Bar-Or A, Mikaeloff Y, Tardieu M. Multiple sclerosis in children: clinical diagnosis, therapeutic strategies, and future directions. Lancet Neurol 2007;6:887–902 [DOI] [PubMed] [Google Scholar]
- 2. Mikaeloff Y, Suissa S, Vallee L, et al. First episode of acute CNS inflammatory demyelination in childhood: prognostic factors for multiple sclerosis and disability. J Pediatr 2004;144:246–252 [DOI] [PubMed] [Google Scholar]
- 3. Lincoln MR, Montpetit A, Cader MZ, et al. A predominant role for the HLA class II region in the association of the MHC region with multiple sclerosis. Nat Genet 2005;37:1108–1112 [DOI] [PubMed] [Google Scholar]
- 4. Fogdell A, Hillert J, Sachs C, Olerup O. The multiple sclerosis- and narcolepsy-associated HLA class II haplotype includes the DRB5*0101 allele. Tissue Antigens 1995;46:333–336 [DOI] [PubMed] [Google Scholar]
- 5. Boiko AN, Gusev EI, Sudomoina MA, et al. Association and linkage of juvenile MS with HLA-DR2(15) in Russians. Neurology 2002;58:658–660 [DOI] [PubMed] [Google Scholar]
- 6. Idrissova ZR, Boldyreva MN, Dekonenko EP, et al. Acute disseminated encephalomyelitis in children: clinical features and HLA-DR linkage. Eur J Neurol 2003;10:537–546 [DOI] [PubMed] [Google Scholar]
- 7. Krupp LB, Banwell B, Tenembaum S. Consensus definitions proposed for pediatric multiple sclerosis and related disorders. Neurology 2007;68(suppl 2):S7–S12 [DOI] [PubMed] [Google Scholar]
- 8. Banwell B, Kennedy J, Sadovnick D, et al. Incidence of acquired demyelination of the CNS in Canadian children. Neurology 2009;72:232–239 [DOI] [PubMed] [Google Scholar]
- 9. Poser CM. The Diagnosis of Multiple Sclerosis. New York: Thieme-Stratton;1984 [Google Scholar]
- 10. Polman CH, Reingold SC, Edan G, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria.” Ann Neurol 2005;58:840–846 [DOI] [PubMed] [Google Scholar]
- 11. Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485–1489 [DOI] [PubMed] [Google Scholar]
- 12. Sadovnick AD, Risch NJ, Ebers GC. Canadian collaborative project on genetic susceptibility to MS, phase 2: rationale and method: Canadian Collaborative Study Group. Can J Neurol Sci 1998;25:216–221 [DOI] [PubMed] [Google Scholar]
- 13. Olerup O, Zetterquist H. HLA-DR typing by PCR amplification with sequence-specific primers (PCR-SSP) in 2 hours: an alternative to serological DR typing in clinical practice including donor-recipient matching in cadaveric transplantation. Tissue Antigens 1992;39:225–235 [DOI] [PubMed] [Google Scholar]
- 14. Banwell B, Krupp L, Kennedy J, et al. Clinical features and viral serologies in pediatric multiple sclerosis: results of multinational study. Lancet Neurol 2007;6:773–781 [DOI] [PubMed] [Google Scholar]
- 15. Nilsson P, Larsson EM, Maly-Sundgren P, Perfekt R, Sandberg-Wollheim M. Predicting the outcome of optic neuritis: evaluation of risk factors after 30 years of follow-up. J Neurol 2005;252:396–402 [DOI] [PubMed] [Google Scholar]