

Abstract
Objective:
We investigated mitochondrial DNA (mtDNA) variants in children with a first episode of acquired demyelinating syndromes (PD-ADS) of the CNS and their relationship to disease phenotype, including subsequent diagnosis of multiple sclerosis (MS).
Methods:
This exploratory analysis included the initial 213 children with PD-ADS in the prospective Canadian Pediatric Demyelinating Study and 166 matched healthy sibling controls from the Canadian Autism Genome Project. A total of 31 single nucleotide polymorphisms (SNPs) were analyzed, including haplogroup-defining SNPs and mtDNA variants previously reported to be associated with MS.
Results:
Primary Leber hereditary optic neuropathy (LHON) mutations and other known pathogenic mtDNA mutations were absent in both patients with pediatric acquired demyelinating syndromes and controls. The 13708A haplogroup J–associated variant, previously linked to adult MS, was more frequent among subjects with PD-ADS (13.0%) compared to controls (6.2%; odds ratio [OR] 2.27; 95% confidence interval [CI] 1.06 to 4.83) and haplogroup M was associated with an earlier age at onset of PD-ADS (−1.74 years; 95% CI −3.33 to −0.07). In contrast, the haplogroup cluster UKJT, as well as 3 other SNPs, were each associated with a lower risk of PD-ADS. A total of 33 subjects with PD-ADS were diagnosed with MS during a mean follow-up period of 3.11 ± 1.14 (SD) years. No single SNP was associated with the risk of subsequent diagnosis of MS. However, haplogroup H was associated with an increased risk of MS (OR 2.60; 95% CI 1.21 to 5.55).
Conclusion:
These data suggest an association between mtDNA variants and the risk of PD-ADS and of a subsequent MS diagnosis. Replication of these findings in an independent population of subjects with PD-ADS is required.
Approximately 3%–5% of patients with multiple sclerosis (MS) experience initial MS symptoms as children.1 At the time of the first episode of an acquired demyelinating syndrome (ADS) of the CNS in the pediatric population, referred to as PD-ADS, it is difficult to predict which patients eventually will be diagnosed with MS. This emphasizes the need for biomarkers predictive of MS outcome.
A role for mitochondrial dysfunction and oxidative stress in MS pathogenesis has been suggested.2 Mitochondrial complex I and complex III activity is reduced in the motor cortex of patients with secondary progressive MS compared to controls, and oxidative DNA damage is increased in MS plaques compared to normal-appearing white matter.3 Mitochondrial haplogroup J has been associated with the presentation of optic neuritis4 and haplogroup J or K with heterogeneous phenotypes.5 The association of the K haplogroup with adult MS risk has recently been replicated.6 Other mtDNA variants, such as 13966G or 14798C, have been published in rare case reports of patients with MS.7 A restriction endonuclease splice variant in the 15927/15928 region of the mitochondrial tRNA (thr) gene was reportedly more frequent in patients with MS with severe optic neuropathy compared to controls, although the specific mutation responsible for this association was not identified.8
An increased frequency of MS also has been reported in pedigrees of patients with Leber hereditary optic neuropathy (LHON),9 which is caused by mutations in mtDNA, and optic nerve involvement is common in both conditions. Primary LHON mutations (3460A, 11778A, 14484C10) have been identified in rare subjects with MS-like illnesses, an observation not reproduced in all studies.11 Among patients with MS presenting with severe optic neuropathy, very rarely primary and combinations of secondary LHON mutations have been reported.12 Conversely, case reports exist of white matter changes in patients with primary LHON mutations.13,14 To date, primary or secondary LHON mutations have not been found in children with MS,15,16 or children with MS and severe optic neuropathy.17
These data, although mixed, raise the possibility of a role for mtDNA mutations in MS. However, a comprehensive analysis of mtDNA mutations and haplogroups in PD-ADS has not been carried out. An association of mtDNA genetic variants with PD-ADS or MS would suggest a role for mitochondrial dysfunction in the pathogenesis of these disorders rather than being a consequence of the disease process. Here, we analyzed mtDNA variants in a well-characterized prospective cohort of children followed from onset of an initial demyelinating event.
METHODS
Subjects.
DNA samples were analyzed from the first 213 Canadian children enrolled from onset of an initial demyelinating event between September 2004 and October 2008. All participants were less than 16 years of age (with the exception of one subject presenting at age 16.8 years), and have been followed prospectively as part of the multicenter 8-year Canadian Pediatric Demyelinating Disease study.
ADS is defined as neurologic dysfunction lasting at least 24 hours presenting as optic neuritis, transverse myelitis, acute disseminated encephalomyelitis (ADEM), monofocal-other (monofocal deficits localized to brain regions extrinsic to the optic nerve or spinal cord), or multifocal CNS demyelination.18 All children were examined quarterly in year 1 and annually thereafter, as well as at time of any new neurologic event.
The diagnosis of MS was conferred if clinical features and examination confirmed a second demyelinating attack, separated 30 days or more from the onset of the first attack and involving new areas of the nervous system.18 MRI evidence of clinically silent lesions was not used for the diagnosis of MS in this study, and the diagnosis of MS was determined as of the data closure date of November 2009.
Extensive laboratory investigations were performed in all participants to exclude other diagnoses, as previously described.19 One patient (who died of an illness clinically characterized as ADEM) was subsequently excluded based on autopsy studies showing mitochondrial Leigh syndrome. None of the children included in this study met the criteria for recurrent ADEM, multiphasic ADEM, or neuromyelitis optica.
Race for the patient and for both biological parents was divided into 4 categories: white, black, Asian, and mixed. Ancestry was divided into European, Asian, Caribbean, South/Central American, Middle Eastern, African, Aboriginal, and mixed. The patients with PD-ADS were ethnically diverse, representative of the general pediatric population in Canada, with 62% of the children of maternal and paternal European descent.
DNA samples from 166 age-, sex-, and ethnicity-matched controls were obtained from the healthy sibling cohort of the Canadian Autism Genome Project.20 The mean age ± SD of subjects with PD-ADS at study entry was 10.2 ± 4.3 years (range 0.6–16.8 years). The mean age ± SD for controls (10.77 ± 5.0 years) was not significantly different from that of the subjects with PD-ADS. Controls ranged in age more broadly from 1 to 27, with all but 19 being age 16 or less. These 19 subjects above the age of 16 were included to optimize matching by race and ancestry.
Standard protocol approvals, registrations, and patient consents.
This study was approved by the individual institutions comprising the Canadian Pediatric Demyelinating Disease Network and the Canadian Autism Genome Project. Written informed consent was obtained from all patients or their parents or legal guardians.
Selection of mtDNA variants.
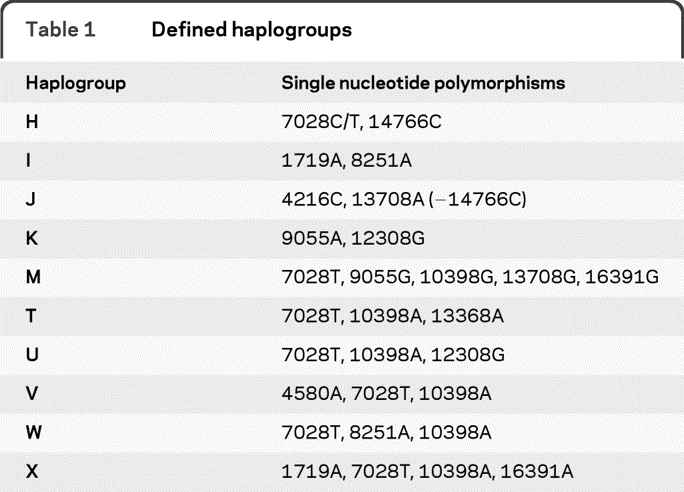
A total of 31 mtDNA variants or mutations were analyzed, including 11 variants selected to define the most common haplogroups21 (table 1). mtDNA mutations previously implicated (but not confirmed) in MS also were analyzed (4298A,22 13966G,23 14798C,23 15927A,24 and 15928A24). The 3 most common primary LHON mutations—3460A, 11778A, and 14484C—as well as secondary LHON mtDNA variants—3394C, 4216C, 4917G, and 13708A—and one variant with an uncertain association with LHON (15257A)10 were also analyzed, as were mutations associated with LHON plus dystonia: 14459A25 and 14596A.26 We additionally screened for the most common mtDNA mutations associated with mitochondrial encephalopathy with ragged red fibers (8344G), mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (3243G), neuropathy, ataxia, and retinitis pigmentosa (8993G),27 and the 4336C28 and 5460A29 variants variably reported in association with neurodegenerative disease. The 10398G variant was included both for haplogroup analyses and due to variable reports of an association with a lower risk of PD,30,31 and an earlier age at onset in spinocerebellar ataxia type 2.32
Table 1.
Defined haplogroups
Genotyping methods.
Genotyping was performed using Sequenom matrix-assisted laser desorption ionization– time of flight mass spectrometry through the Harvard Partners Center for Genetics and Genomics High Throughput Genotyping Facility (Boston, MA). A subset of samples was also analyzed by restriction endonuclease assay or direct sequencing; in each case this confirmed the validity of the Sequenom results. For each sample that tested positive for the 15927A mutation, the mutation was confirmed by restriction endonuclease analysis as previously described.30 Because this restriction endonuclease analysis does not distinguish between the 15927A and 15928A variants, presence of the 15927A variant also was confirmed in each case by direct sequencing. The restriction endonuclease analysis indicated heteroplasmy in each case (a mix of wild-type and mutant mtDNA). TA cloning of the DNA (Invitrogen TOPO TA cloning kit) of a subset of these cases, followed by selection of at least 20 clones for PCR amplification and repeat restriction endonuclease analysis for the 15927A mutation, revealed some clones positive and others negative for the mutation, confirming the heteroplasmy.
Haplogroup analysis.
Haplogroups were defined based on previously grouped mitochondrial SNPs in European populations21 (see table 1).
Data collection.
Clinical information from individual centers was compiled in a central database at the Hospital for Sick Children, Toronto, Canada. Laboratory samples were shipped and stored at the Montreal Neurological Institute, and DNA samples were genotyped through the Harvard Partners Center for Genetics and Genomics High Throughput Genotyping Facility (Boston, MA).
Statistical analyses.
Unpaired Student t test (2-tailed) was performed for comparisons of ages at onset. Fisher exact test (2-tailed) was used for comparisons of mutation or haplogroup frequencies and for comparisons of the frequency of dichotomous clinical features. As an exploratory study, we did not apply a correction factor for multiple comparisons, warranting future confirmatory studies. Subjects with insufficient SNPs to determine a genotype were classified as “missing data”; those with SNPs that did not fit into a prespecified haplogroup were categorized as “unknown.”
RESULTS
Demographics and clinical information.
At time of analysis, 213 children with confirmed PD-ADS were followed for a mean of 3.11 ± 1.14 (SD) years (range 0.76–5.80 years) from presentation. Demographic and clinical data are presented in table 2. To date, 33 of the 213 children (15.5%) have been diagnosed with MS (mean interval between initial and second demyelinating events of 0.82 ± 0.65 years; table e-1 on the Neurology® Web site at www.neurology.org). This is consistent with published data indicating a relatively short mean interval of less than 1 year between a first and second demyelinating event in children.33 The mean age at onset of PD-ADS was older for subjects with PD-ADS subsequently diagnosed with MS (13.09 ± 2.93 years) compared to those not diagnosed with MS during the follow-up period (9.71 ± 4.38 years; p < 0.001). Twelve of the 213 children had a maternal family history of MS and only one of these children has a confirmed MS diagnosis thus far. There was no maternal family history of known mitochondrial disease for any of the 213 children.
Table 2.
Clinical neurologic presentation of 213 children with PD-ADS

Abbreviations: ADEM = acute disseminated encephalomyelitis; ADS = acquired demyelinating syndrome; CI = confidence interval; MS = multiple sclerosis; ON = optic neuritis; OR = odds ratio; PD-ADS = first episode of acquired demyelinating syndrome; TM = transverse myelitis.
Clinical phenotypes based solely on neurologic examination without consideration of MRI findings.
Presenting phenotypes were unclassified for 5 subjects with monophasic ADS.
Significant.
Includes patients with ADEM who had concurrent ON or TM.
None of these patients were diagnosed with neuromyelitis optica at follow-up.
Monofocal: other neurologic dysfunction = clinical findings attributable to a single location in the brain extrinsic to the optic nerve or spinal cord; multifocal: other neurologic dysfunction = clinical findings attributable to greater than one location in the brain or spinal cord, other than ADEM; may include ON or TM as part of symptom constellation.
LHON mutations.
None of the 213 ADS cases or 166 controls had a primary LHON mutation (11778A, 14484C, 3460A). The frequency of the 13708A variant, a SNP associated with the J haplogroup and considered to be a “secondary” LHON mutation due to its apparent influence on the penetrance of a primary LHON mutation,34 was present in 13% of subjects with PD-ADS compared to 6.2% of controls (odds ratio [OR] 2.27; 95% confidence interval [CI] 1.063 to 4.834; p=0.04). The frequency of another secondary LHON mutation, 4917G, was lower in the subjects with PD-ADS (6.2%) compared to controls (11.3%) (OR=0.45; 95% CI 0.22 to 0.92; p=0.03). Neither of these variants was associated with risk of conversion to MS during the study period. A single patient with PD-ADS harbored the 15257A variant; none had the 3394C variant.
15927A and 15928A mtDNA variants.
A prior study found that a restriction endonuclease variant in the mitochondrial tRNA threonine gene was present at a higher frequency in patients with MS compared to controls.24 This restriction endonuclease variant could be caused either by the 15927A variant or by the 15928A variant. There was a trend toward a higher frequency of the 15927A variant in PD-ADS (8/209; 3.8%) compared to controls (2/162; 1.2%) (p=0.09). The mutation was heteroplasmic in each of these 8 cases, representing a potentially important observation as heteroplasmy is a common feature of pathogenic mtDNA mutations. Of the 8 children with the 15927A mutation, 3 (37.5%) were diagnosed with MS during the period of follow-up, compared to 30 out of 180 (16.7%) children diagnosed with MS who do not have the mutation (p=0.10). Among the 8 children positive for the 15927A mtDNA mutation, clinical presentations, ethnicities, and age at onset varied. The mean age at onset of PD-ADS was 7.9 ± 1.8 years for the 8 cases positive for the 15927A mutation compared to 10.3 ± 0.3 years among the negative cases (p=0.16). The frequency of the 15928A variant was lower in subjects with PD-ADS (6.6%) compared to controls (13.3%) (OR 0.47; 95% CI 0.23 to 0.94; p=0.034). The risk of MS was not significantly different among subjects with PD-ADS with this variant compared to those without this variant. None of the analyses were significant when combining all subjects with either the 15927A or the 15928A variants.
Other mtDNA variants and mutations.
The haplogroup T–associated 13368A variant, which was genotyped for the purpose of haplogroup analyses, was less frequent among subjects with PD-ADS compared to controls (OR 0.46; 95% CI 0.23 to 0.93; p=0.03). Frequencies of the 4336C, 5460A, 10398G, 13966G, and 14798C variants were not different between groups, and no single SNP was associated with an altered risk of being diagnosed with MS. None of the subjects with PD-ADS or control subjects harbored the 3243G, 4298A, 8334G, 8993G, 14459A, or 14596A mutations.
Haplogroup analysis.
Mitochondrial haplogroups could be defined unambiguously by the genotyped SNPs in 90% of patients with ADS and in 93% of controls (table 3). In the PD-ADS group, haplogroup H was the most common (40.8%), followed by M (15%), U (8.5%), T (6.1%), J (5.6%), X (4.7%), K (3.3%), I (4.2%), W (0.9%), and V (0.5%). No significant differences were observed for the frequencies of any individual haplogroup in subjects with PD-ADS compared to controls. However, the haplogroup cluster UKJT, which has been associated with a reduced risk of PD,35 was present at a lower frequency in the subjects with PD-ADS (23.5%) compared to controls (34.3%; OR 0.587; 95% CI 0.37 to 0.92; p = 0.02). Haplogroup M was present at a higher frequency in subjects with PD-ADS (15.0%) compared to controls (8.4%; p = 0.06).
Table 3.
Frequencies of haplogroups in PD-ADS and controlsa

Abbreviations: CI=confidence interval; OR=odds ratio; PD-ADS=first episode of acquired demyelinating syndrome.
Comparisons of haplogroups between all patients and controls as well as a subgroup comparison of only Caucasian patients with PD-ADS of European descent with Caucasian controls of European descent did not show significant differences in haplogroup frequencies.
Among the 87 children with PD-ADS with haplogroup H, 20 (23%) were diagnosed with MS during follow-up, compared to only 13 (10.3%) of 126 non-H haplogroup children (OR 2.60; 95% CI 1.21 to 5.55; p = 0.02). None of the haplogroups (individually or UKJT cluster) or individual SNP frequencies were associated with a significantly different age at onset of PD-ADS, with the exception of haplogroup M, which was associated with an earlier mean age at onset of 8.7 ± 0.75 years, vs 10.43 ± 0.32 years for non-M haplogroup patients (p = 0.04).
Subgroup analysis of Caucasian subjects of European descent.
As the majority of our patients and controls were Caucasian and of European ancestry (both biological parents), we conducted a subgroup analysis of this relatively homogenous population. The mean age of patients was 10.27 ± 4.5 years vs 10 ± 4 years for controls. Findings were similar to the entire group comparisons. Haplogroup analysis did not reveal any differences in frequencies of haplogroups except for the haplogroup T–associated variant 13368A, which was decreased in the PD-ADS group (6.6%) compared to controls (14.8%) (OR 0.4; 95% CI 0.18 to 0.9; p = 0.03). There was a trend for an increased frequency of 13708A in subjects with PD-ADS (OR 2.24; 95% CI 0.98 to 5.1; p = 0.07). The UKJT cluster frequency was lower in the PD-ADS group (OR 0.6; 95% CI 0.36 to 0.99; p = 0.04). Haplogroup H among this subgroup of Caucasian subjects with PD-ADS was associated with a trend toward higher risk for MS (p = 0.32).
DISCUSSION
Our study is unique in the analysis of mtDNA in an unbiased cohort of prospectively recruited children with PD-ADS, a proportion of whom are representative of the youngest at-risk population for MS. A potentially important finding is that mitochondrial haplogroup H subjects with PD-ADS were significantly more likely to have a subsequent diagnosis of MS, compared to children with PD-ADS with non-H haplogroups. This suggests that haplogroup H, which represented 41% of all patients with PD-ADS in this study, confers a higher risk of an MS diagnosis in these children at risk. However, as an exploratory study without correction for multiple comparisons, this finding should not be considered conclusive until replicated. Haplogroup M had a borderline association with an increased risk of PD-ADS and was associated with an earlier age at onset of ADS, an interesting finding as this haplogroup is more common in children of non-European ethnicities, a group generally considered to be at lower risk for demyelinating events.
While a higher proportion of the patients with PD-ADS had the mtDNA 15927A tRNA threonine mutation compared to pediatric healthy controls, this did not meet statistical significance and thus the association of this mutation with PD-ADS remains uncertain. The potential pathogenicity of the 15927A mutation is nonetheless of interest, and supported by its identification in subjects of several different haplogroups, as well as the presence of heteroplasmy in all 8 patients. This mutation is quite rare in adult (predominantly Caucasian) subjects, being found in none of 213 controls and only 1 of 271 subjects with PD.30 Of the 8 children with PD-ADS with the 15927A mutation, 3 (37.5%) were diagnosed with MS during the follow-up period, compared to 15.5% among subjects with PD-ADS diagnosed with MS who lack this mutation (p = 0.1). Further studies in a larger population of patients with PD-ADS are needed to determine whether or not these trends represent a true influence of the 15927A variant on the risk of PD-ADS or MS.
None of the PD-ADS cohort harbored primary LHON mutations. Given the very low frequency (approximately 0.5%) of primary LHON mutations in a previously reported adult MS population,9 the current study may have been underpowered to find rare primary LHON mutations. The 13708A complex I gene variant was present at an increased frequency in our PD-ADS group, similar to a previous study of adult patients with MS showing a higher frequency of secondary LHON mutations compared to their control population.36 In the current study, this variant was not associated with a specific clinical subtype or severity of ADS presentation, or with a higher likelihood of a diagnosis of MS.
A limitation of this study is that the age- and ethnicity-matched Canadian controls who were available for analysis consisted of healthy siblings of children with autism spectrum disorder rather than children without a family history of neurologic disorders. A previous study identified that 7.2% of patients with autism had mitochondrial respiratory chain abnormalities (all had severe autism), although no mtDNA mutations were identified.37 Case reports have described children with clinical features of autism and other neurologic deficits in association with mtDNA mutations (e.g., G8363A38 and A4323G39), and children with proven mitochondrial electron chain defects can manifest with autism and neurologic deficits.40 However, there are no reports demonstrating mtDNA mutations in healthy family members of affected children with autism. Still, we acknowledge that our control population may not be entirely representative of the general population.
A complex interaction between genes and environment is thought to underlie the pathobiology of MS, and possibly also influence susceptibility to monophasic ADS. Our results do not support the hypothesis of a major role for primary LHON mutations in children presenting with ADS, but raise the possibility that specific mitochondrial variants or haplogroups may influence the age at onset of PD-ADS and subsequent MS risk. Further study in larger PD-ADS populations is required to confirm such observations.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Julia Kennedy, Sandra Magalhaes, and Katherine Sansom for their assistance.
- ADEM
- acute disseminated encephalomyelitis
- ADS
- acquired demyelinating syndrome
- CI
- confidence interval
- LHON
- Leber hereditary optic neuropathy
- MS
- multiple sclerosis
- mtDNA
- mitochondrial DNA
- OR
- odds ratio
- PD-ADS
- first episode of acquired demyelinating syndrome
- SNP
- single nucleotide polymorphism
COINVESTIGATORS
Coinvestigators are listed in alphabetical order by site–investigator: Mark Awuku, MD (Windsor Regional Hospital, Site Investigator); Louise Roberts, RN (Windsor Regional Hospital, Site Coordinator); J. Burke Baird, MD (Sudbury Regional Hospital, Site Investigator); Nancy Cacciotti, RN (Sudbury Regional Hospital, Site Coordinator); Brenda Banwell, MD (The Hospital for Sick Children, Site Investigator); Melissa McGowan, MHK, Julia O'Mahony, BSc, Emily Ursell, BSc, Courtney Fairbrother, BA, Julia Kennedy, MSc, Jennifer Hamilton, Samantha Irwin, MSc, Sandra Magalhaes, MSc (The Hospital for Sick Children, Site Coordinators); Virender Bhan, MBBS (Dalhousie University, Site Investigator); Trudy Campbell, NP, Lucy Sagar, BSc, MEd (Dalhousie University, Site Coordinators); Frances Booth, MD, Namrata Shah, MD, Ruth Ann Marrie, MD, PhD (Winnipeg Health Sciences Centre, Site Investigators); Joan Kupchak, RN (Winnipeg Health Sciences Centre, Site Coordinator); David Buckley, MD (Janeway Children's Health and Rehabilitation Centre, Site Investigator); Dianne McGrath, RN, Sharon Penney, RN (Janeway Children's Health and Rehabilitation Centre, Site Coordinators); Mary Connolly, MD (BC Children's Hospital, Site Investigator); Shelia Kent, RN (BC Children's Hospital, Site Coordinator); Pamela Cooper, MD (Rouge Valley-Centenary HC, Site Investigator); Loris Aro, RN (Rouge Valley-Centenary HC, Site Coordinator); Marie-Emmanuelle Dilenge, MD (Montreal Children's Hospital, Site Investigator); Heather Davies, MSc (Montreal Children's Hospital, Site Coordinator); Asif Doja, MD, Daniela Pohl, MD, PhD, Sharon Whiting, MD (Children's Hospital of Eastern Ontario, Site Investigators); Chantal Horth, Sheila Ledoux (Children's Hospital of Eastern Ontario, Site Coordinators); Francois Grand'Maison, MD (Hôpital Charles LeMoyne, Site Investigator); Julie Lafrenière, RN, BScN (Hôpital Charles LeMoyne, Site Coordinator); Simon Levin, MD (Children's Hospital of Western Ontario, Site Investigator); Mala Ramu, BA (Children's Hospital of Western Ontario, Site Coordinator); Anne Lortie, MD (Sainte-Justine Hospital, Site Investigator); Sophie Morin, RN, Fabiola Breault, RN, Stephanie Pellerin, RN (Sainte-Justine Hospital, Site Coordinators); E. Athen MacDonald, MD (Kingston General Hospital, Site Investigator); Vee McBride, RN, BScN (Kingston General Hospital, Site Coordinator); Jean Mah, MD, MSc (Alberta Children's Hospital, Site Investigator); Caitlin Wright, BSc, Natarie Liu, BSc, Catherine Riddell (Alberta Children's Hospital, Site Coordinators); Brandon Meaney, MD, Dave Callen, PhD, MD (McMaster Children's Hospital, Site Investigators); Leah Morgenstern, RN, Laurie Wyllie, RN, Heather Neuman, RN (McMaster Children's Hospital, Site Coordinators); David Meek, MD (Saint John Regional Hospital, Site Investigator); Alison Crowell, RN, BScN (Saint John Regional Hospital, Site Coordinator); Noel Lowry, MD (Royal University Hospital, Site Investigator); Doris Newmeyer, RN (Royal University Hospital, Site Coordinator); Guillaume Sébire, MD (Université de Sherbrooke, Site Investigator); Christian Houde, RN (Université de Sherbrooke, Site Coordinator); Kati Wambera, MA, MD, Colleen Adams, MD (Victoria General Hospital, Site Investigators); Laurie Robson, RN (Victoria General Hospital, Site Coordinator); Ellen Wood, MD (IWK Health Centre, Site Investigator); Elaine Woolridge, RN, Edythe Smith, RN (IWK Health Centre, Site Coordinators); Jerome Yager, MD (Children's Stollery Hospital, Site Investigator); Marjorie Berg, RN, Hope Chick, RN (Children's Stollery Hospital, Site Coordinators); Conrad Yim, MD, PhD (Trillium Health Centre, Site Investigator); Leanne Montgomery, RN (Trillium Health Centre, Site Coordinator).
DISCLOSURE
Dr. Venkateswaran, Dr. Zheng, M. Sacchetti, and D. Gagne report no disclosures. Dr. Arnold serves on scientific advisory boards for Biogen Idec, Genentech, Inc., and Teva Pharmaceutical Industries Ltd.; has received speaker honoraria from Biogen Idec; holds a patent re: Method of evaluating the efficacy of drug on brain nerve cells; serves as a consultant for Biogen Idec, Elan Corporation, GlaxoSmithKline, and Roche; and has received research support from Biogen Idec and the Canadian Institutes of Health Research. Dr. Sadovnick has received funding for travel and speaker honoraria from Bayer Canada, Teva Pharmaceutical Industries Ltd., EMD Serono, Inc., and Biogen Idec; and has received research support from the MS Society of Canada Scientific Research Foundation. Dr. Scherer reports no disclosures. Dr. Banwell serves on a scientific advisory board for Biogen Idec; serves on the editorial boards of Neurology® and Multiple Sclerosis; and receives research support from the Multiple Sclerosis Society of Canada, the Multiple Sclerosis Scientific Research Foundation, and the Canadian Institutes of Health Research. Dr. Bar-Or serves on scientific advisory boards for DioGenix, Inc., Ono Pharmaceutical Co. Ltd., and Roche; serves on the editorial board of Neurology®; has received speaker honoraria from Biogen Idec, Bayhill Therapeutics, Bayer Schering Pharma (Berlex), Eli Lilly and Company, Genentech, Inc., GlaxoSmith Kline, Merck Serono, Novartis, Wyeth, and Teva Pharmaceutical Industries Ltd.; and receives research support from Biogen Idec, Genentech, Inc., and Teva Pharmaceutical Industries Ltd. Dr. Simon has served as a consultant for Gerson Lehrman Group (GLG), UCB, and Link Medicine; and receives research support from the NIH/NINDS, the Michael J. Fox Foundation, and the National Parkinson Foundation.
REFERENCES
- 1. Ghezzi A, Deplano V, Faroni J, et al. Multiple sclerosis in childhood: clinical features of 149 cases. Mult Scler 1997;3:43–46 [DOI] [PubMed] [Google Scholar]
- 2. Kalman B, Laitinen K, Komoly S. The involvement of mitochondria in the pathogenesis of multiple sclerosis. J Neuroimmunol 2007;188:1–12 [DOI] [PubMed] [Google Scholar]
- 3. Lu F, Selak M, O'Connor J, et al. Oxidative damage to mitochondrial DNA and activity of mitochondrial enzymes in chronic active lesions of multiple sclerosis. J Neurol Sci 2000;2000:2. [DOI] [PubMed] [Google Scholar]
- 4. Reynier P, Penisson-Besnier I, Moreau C, et al. mtDNA haplogroup J: a contributing factor of optic neuritis. Eur J Hum Genet 1999;7:404–406 [DOI] [PubMed] [Google Scholar]
- 5. Kalman B, Li S, Chatterjee D, et al. Large scale screening of the mitochondrial DNA reveals no pathogenic mutations but a haplotype associated with multiple sclerosis in Caucasians. Acta Neurol Scand 1999;99:16–25 [DOI] [PubMed] [Google Scholar]
- 6. Vyshkina T, Sylvester A, Sadiq S, et al. Association of common mitochondrial DNA variants with multiple sclerosis and systemic lupus erythematosus. Clin Immunol 2008;129:31–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kalman B, Lublin F, Alder H. Characterization of the mitochondrial DNA in patients with multiple sclerosis. Journal of Neurological Science 1996;140:75–84 [DOI] [PubMed] [Google Scholar]
- 8. Mayr-Wohlfart U, Paulus C, Henneberg A, Rodel G. Mitochondrial DNA mutations in multiple sclerosis patients with severe optic involvement. Acta Neurol Scand 1996;94:167–171 [DOI] [PubMed] [Google Scholar]
- 9. Vanopdenbosch L, Dubois B, D'Hooghe MB, Meire F, Carton H. Mitochondrial mutations of Leber's hereditary optic neuropathy: a risk factor for multiple sclerosis. J Neurol 2000;247:535–543 [DOI] [PubMed] [Google Scholar]
- 10. Mackey D, Oostra R, Rosenberg T, et al. Primary pathogenic mtDNA mutations in multigeneration pedigrees with Leber hereditary optic neuropathy. Am J Hum Genet 1996;59:481–485 [PMC free article] [PubMed] [Google Scholar]
- 11. Kalman B, Alder H. Is the mitochondrial DNA involved in determining susceptibility to multiple sclerosis? Acta Neurol Scand 1998;98:232–237 [DOI] [PubMed] [Google Scholar]
- 12. Buhmann C, Gbadamosi J, Heesen C. Visual recovery in a man with the rare combination of mtDNA 11778 LHON mutation and a MS-like disease after mitoxantrone therapy. Acta Neurol Scand 2002;106:236–239 [DOI] [PubMed] [Google Scholar]
- 13. Lev D, Yanoov-Sharav M, Watemberg N, Leshinsky-Silver E, Lerman-Sagie T. White matter abnormalities in Leber's hereditary optic neuropathy due to the 3460 mitochondrial DNA mutation. Eur J Paediatr Neurol 2002;6:121–123 [DOI] [PubMed] [Google Scholar]
- 14. Kovács G, Höftberger R, Majtényi K, et al. Neuropathology of white matter disease in Leber's hereditary optic neuropathy. Brain 2005;128:35–41 [DOI] [PubMed] [Google Scholar]
- 15. Wilichowski E, Ohlenbusch A, Hanefeld F. Characterization of the mitochondrial genome in childhood multiple sclerosis. II. Multiple sclerosis without optic neuritis and LHON-associated genes. Neuropediatrics 1998;29:307–312 [DOI] [PubMed] [Google Scholar]
- 16. Hanefeld F, Ernst B, Wilichowski E, Christen H. Leber's hereditary optic neuropathy mitochondrial DNA mutations in childhood multiple sclerosis. Neuropediatrics 1994;25:331. [DOI] [PubMed] [Google Scholar]
- 17. Ohlenbusch A, Wilichowski E, Hanefeld F. Characterization of the mitochondrial genome in childhood multiple sclerosis. I. Optic neuritis and LHON mutations. Neuropediatrics 1998;29:175–179 [DOI] [PubMed] [Google Scholar]
- 18. Krupp LB, Banwell B, Tenembaum S. Consensus definitions proposed for pediatric multiple sclerosis and related disorders. Neurology 2007;68:S7–S12 [DOI] [PubMed] [Google Scholar]
- 19. Banwell B, Kennedy J, Sadovnick D, et al. Incidence of acquired demyelination of the CNS in Canadian children. Neurology 2009;72:232–239 [DOI] [PubMed] [Google Scholar]
- 20. Szatmari P, Paterson AD, Zwaigenbaum L, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet 2007;39:319–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simon DK, Pankratz N, Kissell DK, et al. Maternal inheritance and mitochondrial DNA variants in familial Parkinson's disease. BMC Med Genet 2010;11:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Taylor RW, Chinnery PF, Bates MJ, et al. A novel mitochondrial DNA point mutation in the tRNA(Ile) gene: studies in a patient presenting with chronic progressive external ophthalmoplegia and multiple sclerosis. Biochem Biophys Res Commun 1998;243:47–51 [DOI] [PubMed] [Google Scholar]
- 23. Kalman B, Lublin FD, Alder H. Characterization of the mitochondrial DNA in patients with multiple sclerosis. J Neurol Sci 1996;140:75–84 [DOI] [PubMed] [Google Scholar]
- 24. Mayr-Wohlfart U, Paulus C, Henneberg A, Rodel G. Mitochondrial DNA mutations in multiple sclerosis patients with severe optic involvement. Acta Neurol Scand 1996;94:167–171 [DOI] [PubMed] [Google Scholar]
- 25. Jun A, Brown M, Wallace D. A mitochondrial DNA mutation at nucleotide pair 14459 of the NADH dehydrogenase subunit 6 gene associated with maternally inherited Leber hereditary optic neuropathy and dystonia. Proc Natl Acad Sci USA 1994;91:6206–6210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. De Vries D, Went L, Bruyn G, et al. Genetic and biochemical impairment of mitochondrial complex I activity in a family with Leber hereditary optic neuropathy and hereditary spastic dystonia. Am J Hum Genet 1996;58:703–711 [PMC free article] [PubMed] [Google Scholar]
- 27. DiMauro S, Schon EA. Mitochondrial DNA mutations in human disease. Am J Med Genet 2001;106:18–26 [DOI] [PubMed] [Google Scholar]
- 28. Howell N, Elson JL, Chinnery PF, Turnbull DM. mtDNA mutations and common neurodegenerative disorders. Trends Genet 2005;21:583–586 [DOI] [PubMed] [Google Scholar]
- 29. Petruzzella V, Chen X, Schon EA. Is a point mutation in the mitochondrial ND2 gene associated with Alzheimer's disease. Biochem Biophys Res Commun 1992;186:491–497 [DOI] [PubMed] [Google Scholar]
- 30. Simon DK, Mayeux R, Marder K, Kowall NW, Beal MF, Johns DR. Mitochondrial DNA mutations in complex I and tRNA genes in Parkinson's disease. Neurology 2000;54:703–709 [DOI] [PubMed] [Google Scholar]
- 31. Van Der Walt JM, Nicodemus KK, Martin ER, et al. Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. Am J Hum Genet 2003;72:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Simon DK, Zheng K, Velazquez L, et al. Mitochondrial complex I gene variant associated with early age at onset in spinocerebellar ataxia type 2. Arch Neurol 2007;64:1042–1044 [DOI] [PubMed] [Google Scholar]
- 33. Ghezzi A, Pozzilli C, Liguori M, et al. Prospective study of multiple sclerosis with early onset. Mult Scler 2002;8:115–118 [DOI] [PubMed] [Google Scholar]
- 34. Simon DK, Johns DR. Mitochondrial disorders: clinical and genetic features. Annu Rev Med 1999;50:111–127 [DOI] [PubMed] [Google Scholar]
- 35. Pyle A, Foltynie T, Tiangyou W, et al. Mitochondrial DNA haplogroup cluster UKJT reduces the risk of PD. Ann Neurol 2005;57:564–567 [DOI] [PubMed] [Google Scholar]
- 36. Kalman B, Lublin FD, Alder H. Mitochondrial DNA mutations in multiple sclerosis. Mult Scler 1995;1:32–36 [DOI] [PubMed] [Google Scholar]
- 37. Oliveira G, Diogo L, Grazina M, et al. Mitochondrial dysfunction in autism spectrum disorders: a population-based study. Dev Med Child Neurol 2005;47:185–189 [DOI] [PubMed] [Google Scholar]
- 38. Graf WD, Marin-Garcia J, Gao HG, et al. Autism associated with the mitochondrial DNA G8363A transfer RNA(Lys) mutation. J Child Neurol 2000;15:357–361 [DOI] [PubMed] [Google Scholar]
- 39. Pons R, Andreu AL, Checcarelli N, et al. Mitochondrial DNA abnormalities and autistic spectrum disorders. J Pediatr 2004;144:81–85 [DOI] [PubMed] [Google Scholar]
- 40. Weissman JR, Kelley RI, Bauman ML, et al. Mitochondrial disease in autism spectrum disorder patients: a cohort analysis. PLoS One 2008;3:e3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.