

Abstract
The adaptive immune system of placental mammals has evolved to tolerate the fetus. Rejection of the fetus by adaptive immune responses is therefore a rare event, with abortion being caused more frequently by inflammation in the placenta. This review will cover recent aspects of immune privilege and the innate immune system at the feto-maternal interface, citing examples of the role played by microbial infections in fetal demise.
Introduction
Placental mammals have been subjected to two opposing selective pressures during evolution, as survival of the species depends on the ability to eliminate microbial pathogens while at the same time protecting fetuses from immune rejection. In this respect, it is noteworthy that placentation had to evolve in animals that already possessed a major histocompatibility complex (MHC). One could therefore speculate that the innate immune system at the feto-maternal interface underwent less stringent selective pressures to ensure quick and efficient local protection against infection, while the adaptive immune system had to remain under full control to prevent rejection of the semi-allogeneic fetus. Given the high selective pressures at work, pregnancy failures unequivocally related to immune dysregulation are therefore rare events, whether in the human species or laboratory animals. Conversely, there are many examples of abortion or fetal distress due to placental inflammation and/or infection.
A number of excellent reviews have been published recently on adaptive immune responses during pregnancy [1-6]. The local activation of some components of the innate immune system at the feto-maternal interface is attracting a growing interest from the reproductive immunology community. This review will emphasize aspects of the innate immune system that could contribute to reproductive failure.
Immune privilege at the feto-maternal interface
Apoptosis can be triggered by the Th1 cytokine, TNFα, or the Fas ligand (Fas-L). As human syncytiotrophoblasts and cytotrophoblasts in placental villi and chorionic extravillous trophoblasts produce the Fas-L, it has been proposed that trophoblast Fas-L may contribute to placental immune privilege during pregnancy by promoting apoptosis of activated, Fas-bearing maternal lymphocytes at the feto-maternal interface (Fig. 1). This view is supported by studies with isolated human peripheral blood lymphocytes co-cultured with trophoblasts [7], but the data are less clear in animal models. The lpr mutation (defect in the function of Fas) had no effect on the outcome of pregnancy; but gld mice (lacking functional Fas-L) displayed extensive leukocyte infiltrates and cell death at the decidual-placental interface, and delivered small litters [8].
Figure 1.
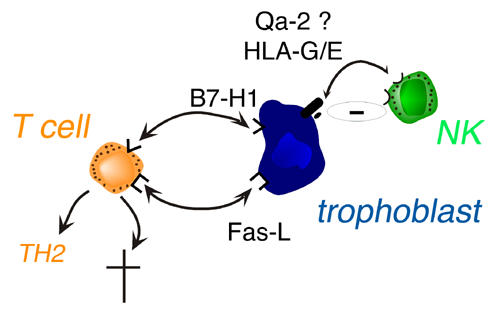
Trophoblast versus maternal T or NK cell interactions. NK: natural killer cell.
Some newly-discovered co-stimulatory molecules of the B7 family, such as B7-H1, can induce T cell apoptosis (Fig. 1). However, they can also deviate immune responses towards a Th2 phenotype, and these molecules are apparently present in the placenta [9]. Thus, the roles played by the Fas-L and the B7 family molecules in immune privilege at the fetal-maternal interface needs to be re-evaluated, especially given the possibility that the B7 molecules may affect local Th2 cytokine production.
It was thought that the main function of HLA-G may be to inhibit the cytolytic activity of maternal NK cells, but this function is being reappraised [10]. HLA-G may also interact with decidual macrophages at the feto-maternal interface, perhaps altering the profile of macrophage cytokine production (Fig. 2). The leader peptides of nascent HLA-G proteins are presented efficiently by HLA-E molecules, thus enhancing cell surface expression of HLA-E, which interacts with surface receptors on NK cells, macrophages and a variety of T cell types. One function of HLA-G, expressed by extravillous trophoblast, may thus be to fine-tune innate immunity by modulating macrophage function and indirectly inhibiting the activity of maternal NK and NK-like cells via HLA-E (Fig. 1) [10]. Recent evidence suggests that soluble HLA-G1 is immunosuppressive, induces apoptosis of activated CD8+ T cells and down-modulates CD4+ T cell proliferation. Moreover, soluble HLA-G1 could also play a role during implantation [11]. Finally, HLA-G may also be expressed in peripheral tissues during viral infections and organ transplantion, where it may protect the tissues during inflammatory responses by favoring Th2-type responses [12].
Figure 2.
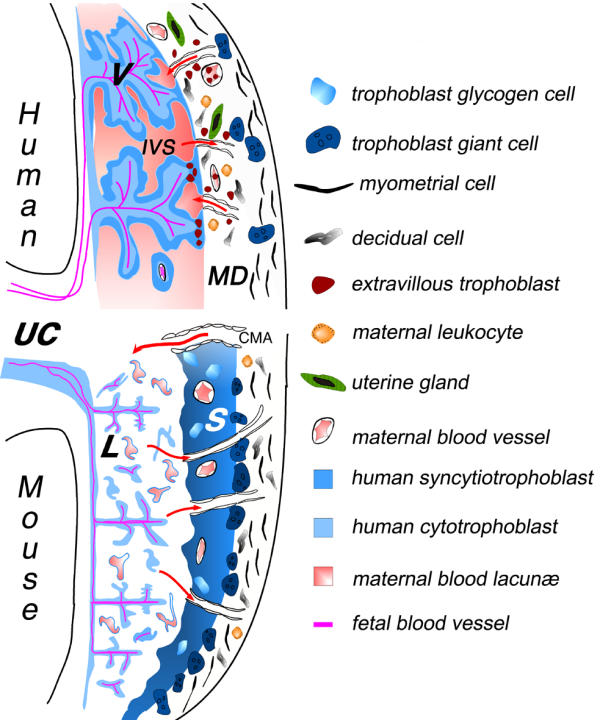
Schematic illustration of the fetal-maternal interface in humans and mice. The placenta, representing the main interface between the mother and fetus, is composed of two parts: the trophoblast of embryonic origin, and the decidua of maternal origin. During implantation, the trophoblasts derived from the early trophectoderm proliferate rapidly and invade, much like tumor cells, the uterine endometrial tissue. The cell wall of maternal blood vessels encountered by trophoblasts is degraded, causing trophoblasts to be bathed by maternal blood. At the same time, the surrounding maternal tissue is modified extensively, leading to the formation of the decidua. In the human placenta, the syncytiotrophoblast cover of the villi is the main site for all maternofetal transfer and secretory functions, and some of the extravillous cytotrophoblast migrate to an endovascular location, where they can form a new vessel lining, in spiral arteries in particular [51]. Although many differences can be distinguished at the histological level, an increasing number of similarities can be found in the cellular and molecular mechanisms involved in implantation and placental function [52,53]. Thus, the fetal-maternal interface comprises two main zones of contact, between the fetal trophoblast layers and the maternal decidua, or maternal blood. Red arrows indicate the blood flow to and from the placenta via maternal arteries or veins, respectively. V: villous trophoblast; IVS: intervillous space; CMA: mouse central maternal artery; S : spongiotrophoblast; L: labyrinthine trophoblast; UC: umbilical cord; MD : maternal decidua.
Components of innate immunity at the feto-maternal interface
Both rodents and higher primates have a hemochorial placenta, in which fetally-derived trophoblast tissue is bathed in maternal blood lacunae. The maternal decidua represents another site of direct contact between fetal trophoblasts and maternal cells (Fig. 2). During pregnancy, the major cell type found at the site of implantation within the maternal decidua are the uterine NK cells. The time-course of their appearance and recent evidence from NK-deficient mouse models suggests that they play an important role in implantation [13-15]. Similar cell types expressing high concentrations of the cytolytic pore-forming protein, perforin, and formerly called "granulated metrial gland" cells, are found in the murine uterus [16,17]. Finally, a type of NK cell sharing some properties with T cells (the NKT cell) is also present and appear to play an important role in some pregnancy loss (see below).
Innate immunity during abortion
To the best of our knowledge, there are no convincing examples showing that the semi-allogeneic fetus is rejected by the maternal adaptive immune system, in the same way as it might reject an allogeneic graft. On the contrary, experimental evidence indicates that fetal alloantigens are recognized by the maternal adaptive immune system, but this recognition induces tolerance of specific maternal T or B cells, as demonstrated in antigen-receptor transgenic models [18-21]. Instead, most of the current evidence suggests that inflammation, complement activation and/or leukocyte infiltration precede abortion. The events leading to abortion share many of the salient features of an innate immune response, such as rapid activation, little or no specificity, and no memory. Furthermore, a growing body of evidence from animal models strongly suggest that abortions are triggered when innate immune responses or their regulators are perturbed (for reviews, see [22,23]).
Complement activation, a component of the inflammatory response, leads to necrosis, and uncontrolled complement activation in the placenta results in fetal loss [23]. Clinically, these responses could be triggered by local necrotic lesions (due to stress or ischemia) or infections of the placenta. Thus, anti-phospholipid syndrome (APS) results in recurrent fetal loss occurring in the presence of anti-phospholipid (aPL) antibodies. Some of these antibodies may target phosphatidylserine (PS) exposed on the surface of trophoblasts, as shown by both animal and in vitro models [24]. But complement activation is indispensable for aPL antibody-mediated fetal loss, as the fetuses are resistant to aPL-induced damage in mice deficient in complement C3 [25]. Likewise, fetal loss is provoked by a deficiency in the murine complement regulator, Crry, which results in complement deposition and placenta inflammation, and fetuses are rescued from lethality when Crry-deficient mice are bred to C3-deficient mice. Along similar lines, semi-allogeneic fetuses are resorbed after treatment with an indoleamine 2,-3-dioxygenase (IDO) inhibitor, which triggers extensive inflammation, complement deposition (even in the absence of antibodies) and hemorrhagic necrosis at the feto-maternal interface [26].
The enzyme IDO, which degrades tryptophan, is expressed in syncytiotrophoblasts and macrophages during gestation. In vitro, macrophages suppress T cell activity due to degradation of tryptophan by IDO, and the suppression is reversed by treatment with an IDO inhibitor. Likewise, the tryptophan concentration is significantly lower in pregnant women than in nonpregnant controls. IDO activity appears to protect the fetus by suppressing T-cell dependent inflammatory responses to fetal alloantigens, since syngeneic fetuses are not rejected after treatment with IDO inhibitor [26]. Thus, trophoblasts and antigen-presenting cells in the placenta may protect the fetus via tryptophan catabolism, thus preventing a unique type of inflammation that involves T cell-dependent, antibody-independent complement activation. However, alternative mechanisms have been proposed to account for the results obtained with the IDO inhibitor, including indirect effects of IDO inhibition on vasoconstriction and macrophage activation [27], and death of T cells due to release of by-products of the IDO reaction, such as kynurerine, 3-hydroxykynurenine and 3-hydroxyanthranilic acid [28-30].
Xu et al. have elegantly demonstrated the important role of Crry in the outcome of murine pregnancy [31]. Triggering of resorptions by administration of IDO inhibitor to pregnant mice was reported to depend on T cell recognition of fetal alloantigens in the presence of increased concentrations of tryptophan [26]. These resorptions were subsequently shown to result from local complement activation and inflammation. More recently, a fascinating case of serendipity led to the discovery that a deficiency in the N-terminal domain of TBP (TATA binding protein, used for promoter recognition during transcription initiation by RNA polymerases) results in a placental malfunction that triggers inflammation, hemorrhages and clotting, such that over 90 % of fetuses die in midgestation [32]. Curiously, the local placental damage was avoided by rearing the mutant fetuses in RAG-deficient mothers. Hence, although the precise cellular and molecular pathways remain to be elucidated, it is remarkable that the destruction of the feto-placental units by local innate mechanisms is prevented in the absence of a functional adaptive maternal immune system.
Stimulation of innate immune responses and abortion by microbial infections
Infections of the genital tract of pregnant mothers by Chlamydia trachomatis, Neisseria gonorrhoeae, and Trichomonas vaginalis cause a large but undetermined fraction of miscarriages, and about 40 % of premature births [33-36]. Pronounced hemorrhaging and necrosis has been described during infection of the placenta by some pathogens, under conditions where direct fetal damage is not observed [37,38]. It is conceivable that stimulation of TNFα secretion by necrotic cells in the placenta may be one of the triggers for embryo loss. Elevated levels of TNFα in the environment of the embryo has been associated with early embryo loss. Stress, which can trigger abortions, also increases the levels of TNFα, and leukocytes producing TNFα are present in the uterus and placenta. In spontaneously aborting mice, an increased expression of TNFα is observed mainly in uterine and trophoblast cells, and the same cells display enhanced expression of the TNFα receptor [39].
In mice, during infection with bacteria such as Listeria monocytogenes or Chlamydia trachomatis, maternal neutrophils are recruited to the fetomaternal interface and act as the main immune effector cells against the bacteria. The macrophage growth factor, colony stimulating factor (CSF)-1, is produced in large quantities by the uterine epithelium during pregnancy, and induces trophoblasts to synthesize neutrophil chemoattractants, cytokine-induced neutrophil chemoattractant (KC) (CXCL1), and macrophage inflammatory protein (MIP)-2 (CXCL2) [40] (Fig. 3). In the absence of CSF-1, neutrophils are not recruited and bacterial infection is unrestrained, leading to fetal demise. In parallel, the macrophage migration inhibitory factor (MIF), which is expressed in the human endometrium in early pregnancy, inhibits NK cell-mediated cytolysis and could thus contribute to immune privilege at the feto-maternal interface. Conversely, MIF also stimulates macrophage phagocytosis and secretion of TNFα and IL-1β, which could lead to recruitment and maintenance of a pool of activated phagocytes in the endometrium [41].
Figure 3.
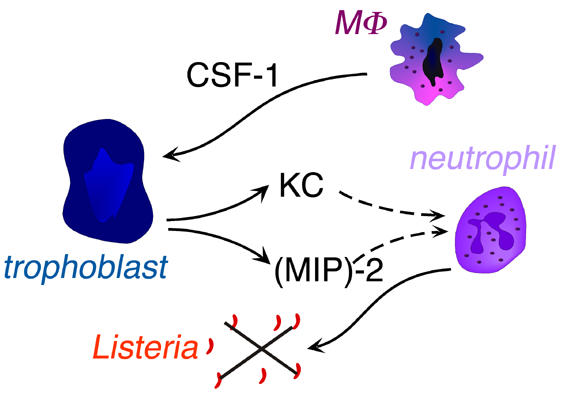
Cross-talk between fetal trophoblast and maternal macrophages and neutrophils during placental infection by Listeria. MΦ: macrophage; CSF-1: colony stimulating factor 1; KC: cytokine-induced neutrophil chemoattractant; (MIP)-2: macrophage inflammatory protein-2.
The Toll-like receptors TLR2 and TLR4 play different roles in pathogen recognition. TLR4 is stimulated by LPS, the most proinflammatory cell wall component of Gram-negative bacteria, while TLR2 has a broader role as a pattern recognition receptor for a variety of microbes and Gram-positive ligands [42]. Consistent with the pattern recognition preferences of TLR, mutations in the TLR4 receptor predispose humans to develop septic shock with Gram-negative bacteria [43]. TLR2 and TLR4 are expressed in the placenta, mainly in the villous and intermediate trophoblasts (Fig. 2), and incubation of placenta cultures with LPS induces IL-6 and IL-8 cytokine production [44]. At the same time, urogenital infection caused by Gram-negative bacteria is a known risk factor for premature births, and TLR4 mutations are associated with an increased risk of premature birth [43]. Thus, TLR mutations are detrimental to the outcome of pregnancy when expressed in the maternal immune system.
Shifting the immune response toward the Th2 pattern (IL-4, IL-5, IL-6) may benefit the fetus, whereas development of pro-inflammatory Th1 cells (secreting IL-2, IFNγ, TNFα) may be harmful. This view is supported by studies on different mouse strains infected by Leishmania major during pregnancy, which have shown that the Th1 anti-infectious response (spontaneous in C57B1/6 mice) is associated with some increases in fetal resorption and implantation failure rates [45]. In the BALB/c strain characterized by a Th2 (non-protective) response to infection, fetal resorption and implantation failure rates were unaffected. Conversely, in C57B1/6 mice, pregnancy was associated with decreased resistance to infection, decreased IFN-γ production, and increased levels of Th2 cytokines.
The maternal decidua also has an expanded population of Vα 14 NKT lymphocytes, which as in other tissues can be stimulated with α-galactosylceramide (α-GalCer) [46]. NKT cells can lyse infected or stressed cells with a pore-forming protein (perforin) and secrete inflammatory cytokines. NKT cells can also be stimulated by glycosyl-phosphatidylinositols derived from protozoan parasites known to provoke abortion, such as Leishmania mexicana, Plasmodium falciparum, and Trypanosoma brucei. Interestingly, stimulation of pregnant mice with α-GalCer induces abortion through a mechanism requiring NKT cell-mediated perforin-dependent killing and IFNγ and TNFα secretion, and histological inspection showed that embryonic trophoblasts die selectively following α-GalCer treatment (Fig. 4) [47]. NKT cells recognize glycolipids presented by the MHC-related molecule, CD1d, which is present on human trophoblasts [48-50]. However, direct interaction between NKT cells and CD1d in the placenta has not been demonstrated, and it is not known whether the microbial pathogens may still induce abortion in CD1d-deficient mice.
Figure 4.
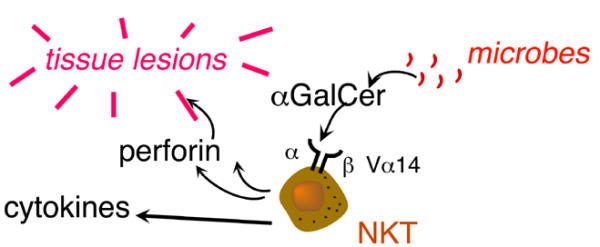
Decidual NKT cell activation causes tissue lesions within the placenta. α-GalCer : α-galactosyl ceramide.
Conclusion
Recent research on the mechanisms leading to abortion or premature birth have focused increasingly on the role played by the innate immune system. A Th2 cytokine profile favors successful pregnancy, while production of Th1 inflammatory cytokines and complement activation result in higher rates of abortion. Approximately 10 % of all births are preterm, and the incidence of preterm birth is increasing and remains the main cause of perinatal morbidity and mortality. Given that preterm births account for 70 % of perinatal mortality and nearly half of long-term neurologic morbidity [33], clinical intervention that could decrease the risk of inflammation during pregnancy would also contribute to the well-being of the children who are born. Although one could conceivably envision the use of therapeutic agents that interfere with complement activation or modulate TLR-dependent inflammation, the main challenge will be to develop a strategy that attenuates inflammatory responses, without jeopardizing the mother's ability to ward off microbial infections.
Acknowledgments
Acknowledgements
This work was supported by the Centre National pour la Recherche Scientifique, the Ministere de l'Education Nationale, de la Recherche et de la Technologie, the National Institutes of Health (grant AI05462), Université Paris 7, and the Fondation pour la Recherche Médicale.
Contributor Information
Colette Kanellopoulos-Langevin, Email: kanellopoulos@ijm.jussieu.fr.
Stéphane M Caucheteux, Email: caucheteux@ijm.jussieu.fr.
Philippe Verbeke, Email: verbeke@ijm.jussieu.fr.
David M Ojcius, Email: ojcius@ijm.jussieu.fr.
References
- Vacchio MS, Jiang SP. The fetus and the maternal immune system: pregnancy as a model to study peripheral T-cell tolerance. Crit Rev Immunol. 1999;19:461–480. [PubMed] [Google Scholar]
- Gaunt G, Ramin K. Immunological tolerance of the human fetus. Am J Perinatol. 2001;18:299–312. doi: 10.1055/s-2001-17861. [DOI] [PubMed] [Google Scholar]
- Meeusen EN, Bischof RJ, Lee CS. Comparative T-cell responses during pregnancy in large animals and humans. Am J Reprod Immunol. 2001;46:169–179. doi: 10.1111/j.8755-8920.2001.460208.x. [DOI] [PubMed] [Google Scholar]
- Szekeres-Bartho J, Barakonyi A, Miko E, Polgar B, Palkovics T. The role of gamma/delta T cells in the feto-maternal relationship. Sem Immunol. 2001;13:229–233. doi: 10.1006/smim.2000.0318. [DOI] [PubMed] [Google Scholar]
- Mellor AL, Munn DH. Immunology at the maternal-fetal interface: lessons for T cell tolerance and suppression. Annu Rev Immunol. 2000;18:367–391. doi: 10.1146/annurev.immunol.18.1.367. [DOI] [PubMed] [Google Scholar]
- Rukavina D, Podack ER. Abundant perforin expression at the maternal-fetal interface: guarding the semiallogeneic transplant? Immunol Today. 2000;21:160–163. doi: 10.1016/S0167-5699(00)01603-0. [DOI] [PubMed] [Google Scholar]
- Guller S, LaChapelle L. The role of placental Fas ligand in maintaining immune privilege at maternal-fetal interfaces. Sem Reprod Endocrinol. 1999;17:39–44. doi: 10.1055/s-2007-1016210. [DOI] [PubMed] [Google Scholar]
- Hunt JS, Vassmer D, Ferguson TA, Miller L. Fas ligand is positioned in mouse uterus and placenta to prevent trafficking of activated leukocytes between the mother and the conceptus. J Immunol. 1997;158:4122–4128. [PubMed] [Google Scholar]
- Petroff MG, Chen L, Phillips TA, Hunt JS. B7 family molecules: novel immunomodulators at the maternal-fetal interface. Placenta. 2002;23:S95–S101. doi: 10.1053/plac.2002.0813. [DOI] [PubMed] [Google Scholar]
- Bainbridge D, Ellis S, Le Bouteiller P, Sargent I. HLA-G remains a mystery. Trends Immunol. 2001;22:548–552. doi: 10.1016/S1471-4906(01)02031-2. [DOI] [PubMed] [Google Scholar]
- Le Bouteiller P, Legrand-Abravanel F, Solier C. Soluble HLA-G1 at the materno-foetal interface – a review. Placenta. 2003;24:S10–S15. doi: 10.1053/plac.2002.0931. [DOI] [PubMed] [Google Scholar]
- Carosella ED, Moreau P, Aractingi S, Rouas-Freiss N. HLA-G: a shield against inflammatory aggression. Trends Immunol. 2001;22:553–555. doi: 10.1016/S1471-4906(01)02007-5. [DOI] [PubMed] [Google Scholar]
- Ashkar AA, Croy BA. Functions of uterine natural killer cells are mediated by interferon gamma production during murine pregnancy. Sem Immunol. 2001;13:235–241. doi: 10.1006/smim.2000.0319. [DOI] [PubMed] [Google Scholar]
- Miller JS. The biology of natural killer cells in cancer, infection, and pregnancy. Exp Hematol. 2001;29:1157–1168. doi: 10.1016/S0301-472X(01)00696-8. [DOI] [PubMed] [Google Scholar]
- Zhang JH, He H, Borzychowski AM, Takeda K, Akira S, Croy BA. Analysis of cytokine regulators inducing interferon production by mouse uterine natural killer cells. Biol Reprod. 2003;69:404–411. doi: 10.1095/biolreprod.103.015529. [DOI] [PubMed] [Google Scholar]
- Zheng LM, Liu CC, Ojcius DM, Young JD. Expression of lymphocyte perforin in the mouse uterus during pregnancy. J Cell Sci. 1991;99:317–323. doi: 10.1242/jcs.99.2.317. [DOI] [PubMed] [Google Scholar]
- Croy BA, Chantakru S, Esadeg S, Ashkar AA, Wei Q. Decidual natural killer cells: key regulators of placental development (a review) J Reprod Immunol. 2002;57:151–168. doi: 10.1016/S0165-0378(02)00005-0. [DOI] [PubMed] [Google Scholar]
- Ait-Azzouzene D, Gendron MC, Houdayer M, Langkopf A, Burki K, Nemazee D, Kanellopoulos-Langevin C. Maternal B lymphocytes specific for paternal histocompatibility antigens are partially deleted during pregnancy. J Immunol. 1998;161:2677–2683. [PubMed] [Google Scholar]
- Jiang SP, Vacchio MS. Multiple mechanisms of peripheral T cell tolerance to the fetal "allograft". J Immunol. 1998;160:3086–3090. [PubMed] [Google Scholar]
- Ait-Azzouzene D, Caucheteux S, Tchang F, Wantyghem J, Moutier R, Langkopf A, Gendron MC, Kanellopoulos-Langevin C. Transgenic major histocompatibility complex class I antigen expressed in mouse trophoblast affects maternal immature B cells. Biol Reprod. 2001;65:337–344. doi: 10.1095/biolreprod65.2.337. [DOI] [PubMed] [Google Scholar]
- Tafuri A, Alferink J, Moller P, Hammerling GJ, Arnold B. T cell awareness of paternal alloantigens during pregnancy. Science. 1995;270:630–633. doi: 10.1126/science.270.5236.630. [DOI] [PubMed] [Google Scholar]
- Moffett-King A. Natural killer cells and pregnancy. Nature Reviews Immunol. 2002;2:656–663. doi: 10.1038/nri886. [DOI] [PubMed] [Google Scholar]
- Caucheteux SM, Kanellopoulos-Langevin C, Ojcius DM. At the innate frontiers between mother and fetus: linking abortion with complement activation. Immunity. 2003;18:169–172. doi: 10.1016/s1074-7613(03)00028-1. [DOI] [PubMed] [Google Scholar]
- McIntyre JA. Antiphospholipid antibodies in implantation failures. Am J Reprod Immunol. 2003;49:221–229. doi: 10.1034/j.1600-0897.2003.01197.x. [DOI] [PubMed] [Google Scholar]
- Holers VM, Girardi G, Mo L, Guthridge JM, Molina H, Pierangeli SS, Espinola R, Xiaowei LE, Mao D, Vialpando CG, et al. Complement C3 activation is required for antiphospholipid antibody-induced fetal loss. J Exp Med. 2002;195:211–220. doi: 10.1084/jem.200116116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor AL, Munn DH. Tryptophan catabolism prevents maternal T cells from activating lethal anti-fetal immune responses. J Reprod Immunol. 2001;52:5–13. doi: 10.1016/S0165-0378(01)00118-8. [DOI] [PubMed] [Google Scholar]
- Bonney EA, Matzinger P. Much IDO about pregnancy. Nature Med. 1998;4:1128–1129. doi: 10.1038/2624. [DOI] [PubMed] [Google Scholar]
- Terness P, Bauer TM, Rose L, Dufter C, Watzlik A, Simon H, Opelz G. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med. 2002;196:447–457. doi: 10.1084/jem.20020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. 2002;196:459–468. doi: 10.1084/jem.20020121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, Fioretti MC, Puccetti P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002;9:1069–1077. doi: 10.1038/sj.cdd.4401073. [DOI] [PubMed] [Google Scholar]
- Xu C, Mao D, Holers VM, Palanca B, Cheng AM, Molina H. A critical role for murine complement regulator crry in fetomaternal tolerance. Science. 2000;287:498–501. doi: 10.1126/science.287.5452.498. [DOI] [PubMed] [Google Scholar]
- Hobbs NK, Bondareva AA, Barnett S, Capecchi MR, Schmidt EE. Removing the vertebrate-specific TBP N terminus disrupts placental β 2m-dependent interactions with the maternal immune system. Cell. 2002;110:43–54. doi: 10.1016/s0092-8674(02)00806-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg RL, Hauth JC, Andrews WW. Intrauterine infection and preterm delivery. New Engl J Med. 2000;342:1500–1507. doi: 10.1056/NEJM200005183422007. [DOI] [PubMed] [Google Scholar]
- Locksmith G, Duff P. Infection, antibiotics, and preterm delivery. Semin Perinatol. 2001;25:295–309. doi: 10.1053/sper.2001.27163. [DOI] [PubMed] [Google Scholar]
- Gibbs RS, Eschenbach DA. Use of antibiotics to prevent preterm birth. Am J Obstet Gynecol. 1997;177:375–380. doi: 10.1016/s0002-9378(97)70201-1. [DOI] [PubMed] [Google Scholar]
- Hillier SL, Nugent RP, Eschebach DA, Krohn MA, Gibbs RS, Martin DH, Cotch MF, Edelman R, Pastorek JG, Rao AV, et al. Association between bacterial vaginosis and preterm delivery of a low birth-weight infant. New Engl J Med. 1995;333:1737–1742. doi: 10.1056/NEJM199512283332604. [DOI] [PubMed] [Google Scholar]
- Pal S, Peterson EM, de la Maza LM. A murine model for the study of Chlamydia trachomatis genital infections during pregnancy. Infect Immun. 1999;67:2607–2610. doi: 10.1128/iai.67.5.2607-2610.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner W, Bachmann S. Histological and immunocytochemical characterization of Coxiella burnetii-associated lesions in the murine uterus and placenta. Infect Immun. 1992;60:5232–5241. doi: 10.1128/iai.60.12.5232-5241.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argiles JM, Carbo N, Lopez-Soriano FJ. TNF and pregnancy: the paradigm of a complex interaction. Cytokine & Growth Factor Reviews. 1997;8:181–188. doi: 10.1016/S1359-6101(97)00012-9. [DOI] [PubMed] [Google Scholar]
- Guleria I, Pollard JW. The trophoblast is a component of the innate immune system during pregnancy. Nature Med. 2000;6:589–593. doi: 10.1038/75074. [DOI] [PubMed] [Google Scholar]
- Arcuri F, Ricci C, Ietta F, Cintorino M, Tripodi SA, Cetin I, Garzia E, Schatz F, Klemi P, Santopietro R, et al. Macrophage migration inhibitory factor in the human endometrium: expression and localization during the menstrual cycle and early pregnancy. Biol Reprod. 2001;64:1200–1205. doi: 10.1095/biolreprod64.4.1200. [DOI] [PubMed] [Google Scholar]
- Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nature Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- Lorenz E, Mira JP, Frees KL, Schwartz DA. Relevance of mutations in the TLR4 receptor in patients with Gram-negative septic shock. Arch Intern Med. 2002;162:1028–1032. doi: 10.1001/archinte.162.9.1028. [DOI] [PubMed] [Google Scholar]
- Holmlund U, Cebers G, Dahlfors AR, Sandstedt B, Bremme K, Ekstrom ES, Scheynius A. Expression and regulation of the pattern recognition receptors Toll-like receptor-2 and Toll-like receptor-4 in the human placenta. Immunology. 2002;107:145–151. doi: 10.1046/j.1365-2567.2002.01491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan L, Guilbert LJ, Wegmann TG, Belosevic M, Mosmann TR. T helper 1 response against Leishmania major in pregnant C57BL/6 mice increases implantation failure and fetal resorptions. Correlation with increased IFN-gamma and TNF and reduced IL-10 production by placental cells. J Immunol. 1996;156:653–662. [PubMed] [Google Scholar]
- Ojcius DM, Delarbre C, Kourilsky P, Gachelin G. MHC and MHC-related proteins as pleiotropic signal molecules. FASEB J. 2002;16:202–206. doi: 10.1096/fj.01-0758com. [DOI] [PubMed] [Google Scholar]
- Ito K, Karasawa M, Kawana T, Akasaka T, Koseki H, Akutsu Y, Kondo E, Sekiya S, Sekikawa K, Harada M, et al. Involvement of decidual Vα 14 NKT cells in abortion. Proc Natl Acad Sci USA. 2000;97:740–744. doi: 10.1073/pnas.97.2.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyson JE, Rybalov B, Koopman LA, Exley M, Balk SP, Racke FK, Schatz F, Masch R, Wilson SB, Strominger JL. CD1d and invariant NKT cells at the human maternal-fetal interface. Proc Natl Acad Sci USA. 2002;99:13741–13746. doi: 10.1073/pnas.162491699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki M, Sasho T, Moriya H, Kanno M, Harada M, Kamada N, Shimizu E, Nakayama T, Taniguchi M. Extrathymic development of Vα 11 T cells in placenta during pregnancy and their possible physiological role. J Immunol. 2001;166:7244–7249. doi: 10.4049/jimmunol.166.12.7244. [DOI] [PubMed] [Google Scholar]
- Dang Y, Heyborne KD. Cutting edge: regulation of uterine NKT cells by a fetal class I molecule other than CD1. J Immunol. 2001;166:3641–3644. doi: 10.4049/jimmunol.166.6.3641. [DOI] [PubMed] [Google Scholar]
- Kaufmann P, Scheffen I. Placental development. In: Polin R, Fox W, editor. In Neonatal and Fetal Physiology. Second. Orlando, FL: W.B. Saunders; 1998. pp. 59–70. [Google Scholar]
- Cross JC, Werb Z, Fisher SJ. Implantation and the placenta: key pieces of the development puzzle. Science. 1994;266:1508–1518. doi: 10.1126/science.7985020. [DOI] [PubMed] [Google Scholar]
- Georgiades P, Ferguson-Smith AC, Burton GJ. Comparative developmental anatomy of the murine and human definitive placentae. Placenta. 2002;23:3–19. doi: 10.1053/plac.2001.0738. [DOI] [PubMed] [Google Scholar]