

Abstract
Niemann-Pick disease type C (NPC) is a model for inborn errors of metabolism whose gene product mediates molecular trafficking rather than catabolizing macromolecules, as in classic lipidoses. We report the case of an infant who presented with hepatosplenomegaly without neurological abnormalities. Decreased activity of acid β-glucosidase and elevated serum chitotriosidase and tartrate-resistant acid phosphatase on repeated measurements led to initial diagnosis of Gaucher disease (GD). Failure to respond to enzyme replacement therapy after one year, however, put the diagnosis in question. Cholesterol esterification assays in cultured skin fibroblasts and NPC gene analysis led to the correct diagnosis of NPC. The patient had markedly reduced cholesterol esterification and was a compound heterozygote for a known and a novel mutation in the NPC gene (395delC and 2068insTCCC), which are both predicted to lead to protein truncation. Although the full phenotype of NPC involves hepatosplenomegaly and neurodegenerative disease, the initial presentation in a pediatric patient may be restricted to visceral disease. Of interest, this patient had decreased activity of leukocyte acid β-glucosidase activity and elevated serum chitotriosidase to levels often seen in GD. Although acid β-glucosidase activity in leukocytes was low, it was in the normal range in skin fibroblasts. Therefore, diagnostic delay may occur in NPC due to false positive testing for GD. Diagnosis of NPC requires a high index of suspicion and should be considered in a patient with hepatosplenomegaly even in the absence of neurodevelopmental signs. Prompt diagnosis will become increasingly important as effective therapies are developed for NPC.
Introduction
Niemann-Pick disease type C (NPC) (OMIM catalogue number 257220 Type C1, and 607625 Type C2) is a rare autosomal recessive neurovisceral lysosomal lipid storage disorder caused by mutations in the NPC1 (95%) or NPC2 (5%) genes (Santos et al. 2008; Vanier and Millat 2003). Its incidence has been calculated as 1:150,000 for the Western European population (Patterson et al. 2001). Indistinguishable phenotypes are produced by mutations in two distinct genes, designated NPC1 and NPC2, which play key roles in the intracellular trafficking of lipids (Patterson 2003). NPC1 encodes the NPC1 protein, a transmembrane protein involved in lipid trafficking in lysosomes, but whose precise function remains to be elucidated. Loss-of-function mutations in the NPC1 gene lead to an accumulation of broad range of lipids including sphingomyelin, cholesterol, glycosphingolipids, (GSLs) and, sphingosine (Lloyd-Evans and Platt 2010). Recent studies suggest that transfer of cholesterol between NPC1 and NPC2 is required for exit of lipoprotein-derived cholesterol from lysosomes (Kwon et al. 2009).
There is striking phenotypic variability in NPC, and it may present at any age from fetal life (with hydrops fetalis) up to the seventh decade of life (Patterson 2003). An alteration in cholesterol and glycolipid homeostasis leads to a broad spectrum of symptoms that include hepatosplenomegaly, liver dysfunction, and neurological abnormalities, such as progressive ataxia, cognitive decline, dystonia, cataplexy, vertical supranuclear gaze palsy, seizures, and impairment of swallowing reflexes (Patterson et al. 2001). However, hepatosplenomegaly may be the sole presenting feature (Vanier and Millat 2003).
In children with organomegaly, sphingolipidoses such as Niemann–Pick disease type A/B, NPC, and Gaucher disease (GD) must be considered in the differential diagnosis. In many cases, severe organ enlargement is the leading sign. In others, the splenomegaly can be mild and may be dominated by neurological signs or symptoms. Clinically, the diagnosis of NPC may be challenging, as examination for the characteristic vertical supranuclear gaze palsy is not always performed, organomegaly is often absent, and neuroimaging and standard biochemical screening studies are usually normal (Patterson 2003). Performing a ‘lysosomal panel’ for diagnostic work-up will miss NPC since its diagnosis requires demonstration of the trafficking defect in cultured fibroblasts, supplemented in selected cases by genotyping (Ries et al. 2006).
GD type 1 is the most common hereditary lysosomal storage disorder (LSD), characterized by hepatosplenomegaly, cytopenia and skeletal disease. It is caused by a deficiency of the lysosomal enzyme glucocerebrosidase, which catalyzes the hydrolysis of glucocerebroside and glucosylsphingosine (Grabowski 2008). The gold standard for diagnosis is the demonstration of low lysosomal glucocerebrosidase (GCase) activity in blood leukocytes and/or skin fibroblasts. Genotyping for common mutations within the GBA1 (glucocerebrosidase) gene may be helpful, however a negative finding does not rule out GD. GD can now be effectively treated with enzyme replacement therapy (ERT) with imiglucerase, a recombinant, mannose-terminated β-glucocerebrosidase. Biomarkers, such as chitotriosidase, have proven to be beneficial in supporting the diagnosis and monitoring treatment efficacy (Aerts et al. 2006).
Case report
Patient A was born at 36 weeks gestation to non-consanguineous parents of Irish-German ancestry. She had an uneventful neonatal course, but was found at her 8-week Well-Child visit to have hepatosplenomegaly. She was noted to have a firm hepatomegaly 5 cm below the costal margin and splenomegaly 4–5 cm below the costal margin. She had no neurological findings, ophthalmologic exam was normal, and she had no developmental delays. Her serum transaminases were slightly elevated, but her blood counts were normal. Abdominal ultrasound demonstrated hepatosplenomegaly with homogeneous echotexture, and this was confirmed by CT scan. CMV and toxoplasma titers revealed evidence of perinatal CMV infection. Other viral serologies were negative.
The patient was followed with serial ultrasounds, which showed persistent splenomegaly. She was also noted to be hyperlipidemic: serum HDL, LDL, and TG were 33, 445, and 233 mg/dl respectively at 4 months of age. She developed mild thrombocytopenia (platelets 120×109/L). Further work-up at 10 months of age led to the diagnosis of GD. Assay of leukocyte acid β-glucosidase activity was low at 4 nm/h/mg (normal 12.5–16.9). Leukocyte acid β-glucosidase activity was repeated; the result was again low at 4.5 nm/h/mg. The glucosidase activity was assayed using 4-methylumbelliferyl-B-D-glucopyranoside as the substrate (Beutler and Kuhl 1970) at the Mayo diagnostic laboratories (http://www.mayomedicallaboratories.com/test-catalog/Clinical+and+Interpretive/8788).
In addition, the patient had elevated biomarkers of GD activity: chitotriosidase was increased at 1,516 nmoles/hr/ml (normal 8–65), and tartrate-resistant acid phosphatase (TRAP) was also elevated at 36 IU/L (normal 4.8–8.2). Mutation analysis for the four most common Gaucher mutations was negative. However, with a consistent clinical picture, she was started on ERT on a regimen of imiglucerase 60 U/kg by IV infusion every 2 weeks. After 2 months, chitotriosidase fell to 968. TRAP remained stable. She continued to make developmental progress and had no evidence of regression or changes in her behavior or cognitive abilities. She was followed by pediatric neurology for periodic neurologic assessment (Table 1).
Table 1.
Diagnostic Work-up
| Age (months) |
|||||||
|---|---|---|---|---|---|---|---|
| 3 | 8 | 10 | 12a | 14 | 16 | 20 | |
| Hemoglobin (g/dL) | 12 | 13 | 10.8 | ||||
| Platelets (×109 cells/L) | 244 | 127 | 96 | ||||
| Chitotriosidase (nmol/hr/ml) | 1,558 | 1,516 | 968 | 1,137 | |||
| TRAP | 31 | 36 | 31 | 32 | |||
| Liver size (cm) | 10 | ULN | WNL | Mildly enlarged | |||
| Spleen size (cm) | 10.0×9.5 | 7.6 | 10.9 | 12.3×11.4 | |||
ULN upper limit of normal, WNL within normal limits
Imiglucerase started hereafter (see text)
After 8 months of ERT, she continued to thrive but there was no improvement in her thrombocytopenia (platelet count 96×109/L), or in the extent of hepatosplenomegaly. Serum chitotriosidase appeared initially to decline, but subsequently rose to 1,137 nmoles/hr/ml. After 8 months of treatment with imiglucerase, this lack of response to ERT is at variance with the expected response to ERT in GD (Weinreb et al. 2008). Sequencing of the coding regions and splice sites of the GBA1 gene was undertaken. We did not find any mutations in GBA1. In light of the lack of response to ERT and absence of GBA1 gene mutations, further evaluation was undertaken for other lysosomal storage diseases in particular NPC.
First, a bone marrow biopsy was performed, which revealed scattered single histiocytes compatible with a storage disease; however, the cytological characteristics were not typical of GD (Figs. 1 and 2) A skin biopsy was obtained from the patient to culture fibroblasts. Acid β-glucosidase activity was found to be 71.5 nm/h/mg (range 8–12 nm/h/mg, GD <2 nm/h/mg). Filipin staining in cultured skin fibroblasts for excess free cholesterol was strongly positive. Cholesterol esterification was undetectable (<5% normal), confirming the diagnosis of Niemann-Pick disease, type C (NPC). Subsequently, NPC1 gene analysis revealed the patient to be compound heterozygote for 395delC and 2068insTCCC mutations in exons 4 and 13, respectively. The 395delC was first discovered by Sun et al. (2001). The 2068insTCCC mutation has not been previously reported in the literature. Both mutations are predicted to result in protein truncation.
Fig. 1.
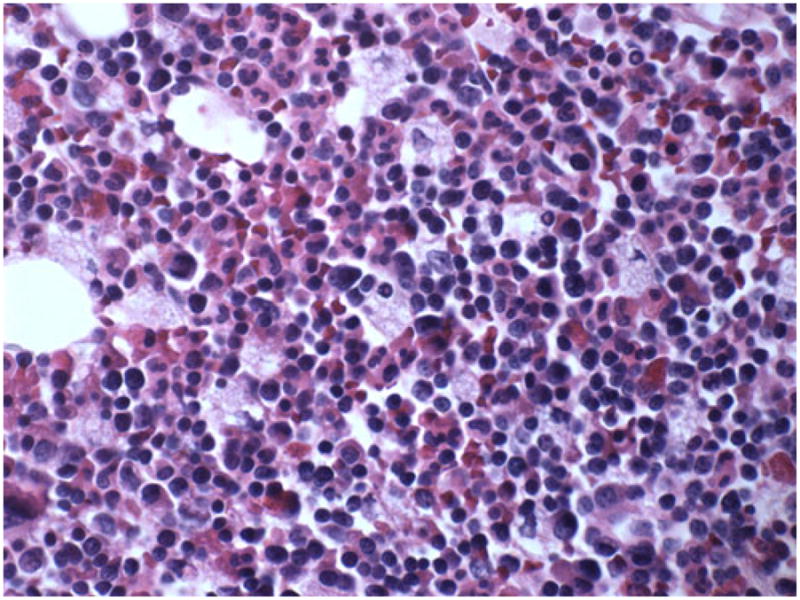
Macroscopic photograph of the bone marrow in our patient with NPC. Niemann-Pick cells can be seen with small eccentric nuclei and foamy cytoplasm. (Hematoxylin-Eosin; G ×40)
Fig. 2.
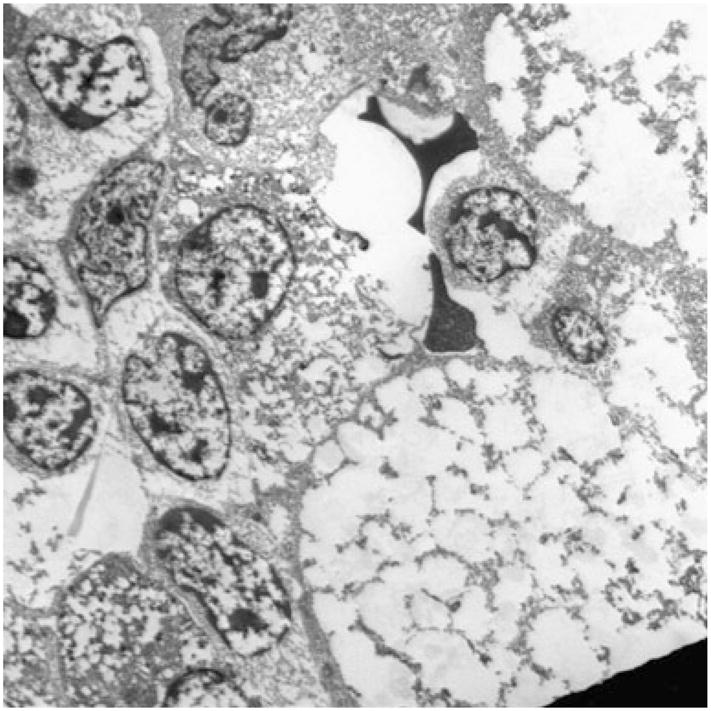
Electron microscopy photograph of the bone marrow in our patient. Ultrastructure of foam cells, the characteristic macrophages in NPC, can be seen with their distinctive foamy cytoplasm
The patient was not eligible to participate in the clinical trial of N butyl-dexoxynojirimicin (miglustat tm) due to her young age. Compassionate use of miglustat was denied. At the request of the parents, imiglucerase was continued in the hopes of delaying progression, since there have been experimental data that glucocerebrosidase (GCase) is also reduced in NPC, and that the glycolipid that accumulates in GD (glucosylceramide) also accumulates in NPC. However, the patient developed mild developmental delay, progressing, by 20 months of age, to obvious neurologic deficits. Eventually, 2 years later, at age 3.5 years, the patient commenced therapy with miglustat after it was approved for use in Europe for NPC. Unfortunately, she has continued to develop progressive neurologic deterioration during the last 3.5 years despite miglustat substrate reduction therapy. Miglustat has not yet been approved in the US for use in NPC although clinical trials are in progress.
Discussion
We describe a patient with NPC who was initially diagnosed with GD due to the low activity of GCase in peripheral blood leucocytes, elevated chitotriosidase, elevated TRAP and compatible clinical presentation of hepatosplenomegaly and thrombocytopenia. Failure to respond to ERT as expected led to questioning the diagnosis of GD (Weinreb et al. 2008). Further investigations led to the ultimate diagnosis of NPC. The patient was found to be a compound heterozygote for one known and a novel mutation in NPC gene that are predicted to result in protein truncation: 395delC and 2068insTCCC. There was almost 2 years of diagnostic delay, underscoring the importance of maintaining a high index of suspicion when evaluating children with hepatosplenomegaly, even when enzyme and biomarker studies might be compatible with GD.
NPC was originally viewed simply as a disorder of cholesterol metabolism, but has now been shown to also involve the accumulation of a broad range of lipids: sphingomyelin, glycosphingolipids and sphingosine (Vanier 1983, 1999; Lloyd-Evans and Platt 2010). The link between cholesterol metabolism and sphingolipid trafficking is the focus of ongoing research, and the initiating cause of the pathology seen in NPC remains a topic of debate (Lloyd-Evans et al. 2008; Vanier and Millat 2003; Lloyd-Evans and Platt 2010).
The biochemical hallmark of NPC is the intracellular accumulation of multiple lipids. In peripheral tissues, unesterified cholesterol predominates, but in the CNS, GSLs are proportionately more significant and likely play key roles in the pathogenesis (Patterson 2003). Glucosylceramide, lactosylceramide, sphingosine, bis-monoacyl glycerol phosphate, and GM1 and GM2 gangliosides are present in excess, particularly in the gray matter of the brain (Vanier 1999). Classically, the accumulation of GSLs was believed to be secondary to cholesterol accumulation in NPC1-deficient cells (Puri et al. 1999, 2003). Recently, it has been suggested that sphingosine is the primary accumulating lipid in NPC (Lloyd-Evans et al. 2008; Lloyd-Evans and Platt 2010).
GSLs, especially glucocerebroside, will accumulate within cells deficient in NPC1 protein (Salvioli et al. 2004; Zervas et al. 2001). In NPC, the accumulation of glucocerebroside within the endosomal-lysosomal pathway occurs despite lack of GBA1 mutation. The precursor form of GCase is found at normal levels in NPC fibroblasts; however, there is a decreased level of the mature protein. This pattern suggests an accelerated degradation of mature GCase rather than a decrease in its production (Salvioli et al. 2004). This appears in contrast to GD1, where mutation in the GBA1 gene causes a decrease in the production of GCase.
Because low acid β-glucosidase activity can be seen in both GD and NPC, at least in blood leukocytes, as illustrated by our case report, a second, ancillary diagnostic marker has the potential to decrease the number of inaccurate diagnoses for NPC. In this patient, we observed increased chitotriosidase as well as TRAP that was initially attributed to GD. Chitotriosidase is a human chitinase with markedly elevated activity in a variety of LSDs but most dramatically in GD (Guo et al. 1995). A positive chitotriosidase is non-specific for GD as it may also be elevated in NPC, though the mechanism is not understood (Wajner et al. 2004). CT activities above 200 nmol/h per ml are predictive for GD, sphingomyelinase deficiency (Niemann-Pick disease type A or B) or NPC. Activities above 4,000 nmol/h per ml are predictive for GD (Ries et al. 2006). Therefore, although plasma chitotriosidase can be useful in screening patients for NPC, this marker enzyme is neither sensitive nor specific for NPC as it can vary significantly in affected patients (Ries et al. 2006; NP-C Guidelines Working Group 2009). Hence, the chitotriosidase value in this patient is indicative of a LSD, although the value is non-specific for NPC. Interestingly, we also found an almost 4-fold upper limit of normal elevation of TRAP. This has not been described previously in NPC. TRAP is known to be produced by osteoclasts and GlcCer-laden macrophages in GD. It would be of interest to examine TRAP in a larger number of NPC patients and determine whether it is associated with any aspect of the phenotype.
The standard of care for GD is macrophage-directed ERT in which the defective GCase is supplemented with recombinant glucocerebrosidase, administered by intravenous infusions usually every 2 weeks. Imiglucerase is modified to expose mannose residues that can be recognized by macrophages, a procedure that dramatically improves targeting to and internalization by macrophages, the main cell type affected in GD (Kacher et al. 2008). As expected, our patient did not respond to ERT in either visceral or neurologic compartments. Presumably, ERT reduced the level of glucosylceramide in splenic and hepatic macrophages. Nevertheless, this was insufficient to reverse visceral disease because accumulation of glucosylceramide is not the major abnormality in NPC; moreover, GlcCer accumulation in NPC occurs mostly in the brain but not in peripheral tissues (Vanier 1999).
N-butyldeoxynojirimycin (NB-DNJ) or miglustat, an inhibitor of glucocerebroside synthase, has been licensed for use in substrate reduction therapy (SRT) for GD. In NPC cells treated with NB-DNJ, GSLs and cholesterol fail to accumulate to pathological levels (Zervas et al. 2001). Inhibition of GSL synthesis with NB-DNJ has been shown to delay the onset of disease, reverse manifestations, and prolong survival in NPC in experimental models. Recently, a clinical trial showed miglustat improved or stabilized several clinically relevant indicators of NPC and may slow disease progression (Patterson et al. 2007). Aside from GD, miglustat continues to be used only on an investigational basis in other glycosphingolipidoses including NPC (Patterson et al. 2007), although it was recently approved for use in NPC in Europe (EMEA 2009).
As opposed to classic lipid storage diseases, NPC is a model for inborn errors of metabolism whose gene product mediates molecular trafficking rather than catabolizing macromolecules. The diagnosis of NPC requires a high index of suspicion, particularly given the nonspecific nature of routine screening tests as described above. NPC should be considered in the differential diagnosis of hepatosplenomegaly even in the absence of neurodevelopmental signs. An important lesson to be learnt from this patient is that when a patient with presumed GD does not exhibit absolutely classic features and/or the expected response to ERT, alternative diagnosis should be considered, especially NPC. A timely accurate diagnosis can lead to appropriate counseling of families and early consideration of miglustat therapy and other emerging therapies which may have some efficacy.
Acknowledgments
We thank Dr. M. Paterson for his helpful advice in the management of this patient.
Contract grant sponsor NIH T32 post-doctoral training program in investigative hematology supported S.M.L. NIDDK K24DK066306 mid-career clinical investigator award supported P.K.M. P.K.M. receives research support from Genzyme Corporation for participation in the International Gaucher Registry (ICGG).
Footnotes
Competing interest: None declared.
Contributor Information
Sarah M. Lo, Department of Pediatrics, Yale University School of Medicine, New Haven, CT, USA
Joseph McNamara, Department of Pediatrics, Yale University School of Medicine, New Haven, CT, USA.
Margherita R. Seashore, Department of Pediatrics, Yale University School of Medicine, New Haven, CT, USA, Department of Genetics, Yale University School of Medicine, New Haven, CT, USA
Pramod K. Mistry, Email: pramod.mistry@yale.edu, Department of Pediatrics, Yale University School of Medicine, New Haven, CT, USA, Department of Medicine, Yale University School of Medicine, New Haven, CT, USA, Pediatric Gastroenterology and Hepatology, Yale University School of Medicine, 333 Cedar Street, P.O. Box 208064, New Haven, CT 06520-8064, USA
References
- Aerts JMFG, Hollak CEM, Boot RG, Groener JEM, Maas M. Substrate reduction therapy of glycosphingolipid disorders. J Inherit Metab Dis. 2006;29:449–456. doi: 10.1007/s10545-006-0272-5. [DOI] [PubMed] [Google Scholar]
- Beutler E, Kuhl W. Detection of the defect of Gaucher’s disease and its carrier state in peripheral-blood leucocytes. Lancet. 1970;1 (7647):612–613. doi: 10.1016/s0140-6736(70)91646-6. [DOI] [PubMed] [Google Scholar]
- European Medicines Association (EMEA) Miglustat summary of product characteristics. 2009. [Last accessed 15th May 2009]. Available at: < http://www.emea.europa.eu/humandocs/PDFs/EPAR/zavesca/H-435-PI-en.pdf>.
- Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet. 2008;372(9645):1263–1271. doi: 10.1016/S0140-6736(08)61522-6. [DOI] [PubMed] [Google Scholar]
- Guo Y, He W, Boer AM, et al. Elevated plasma chitotriosidase activity in various lysosomal storage disorders. J Inherit Metab Dis. 1995;18:717–722. doi: 10.1007/BF02436762. [DOI] [PubMed] [Google Scholar]
- Kacher Y, Brumshtein B, Boldin-Adamsky S, Toker L, Shainskaya A, Silman I, Sussman JL, Futerman AH. Acid beta-glucosidase: insights from structural analysis and relevance to Gaucher disease therapy. Biol Chem. 2008;389(11):1361–1369. doi: 10.1515/BC.2008.163. Review. [DOI] [PubMed] [Google Scholar]
- Kwon HJ, Abi-Mosleh L, Wang ML, Deisenhofer J, Goldstein JL, Brown MS, Infante RE. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 2009;137(7):1213–1224. doi: 10.1016/j.cell.2009.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Evans E, Platt FM. Lipids on trial: the search for the offending metabolite in Niemann-Pick type C disease. Traffic. 2010 doi: 10.1111/j.1600-0854.2010.01032.x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, Churchill GC, Schuchman EH, Galione A, Platt FM. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med. 2008;14 (11):1247–1255. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- NP-C Guidelines Working Group. Wraith JE, Baumgartner MR, Bembi B, Covanis A, Levade T, Mengel E, Pineda M, Sedel F, Topçu M, Vanier MT, Widner H, Wijburg FA, Patterson MC. Recommendations on the diagnosis and management of Niemann-Pick disease type C. Mol Genet Metab. 2009;98(1–2):152–165. doi: 10.1016/j.ymgme.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Patterson MC. A riddle wrapped in a mystery: understanding Niemann-Pick disease, type C. Neurologist. 2003;6:301–310. doi: 10.1097/01.nrl.0000094627.78754.5b. [DOI] [PubMed] [Google Scholar]
- Patterson MC, Vanier MT, Suzuki K, et al. Niemann-Pick disease, type C: a lipid trafficking disorder. In: Beaudet AL, Sly WS, Valle D, et al., editors. The metabolic and molecular bases of inherited disease. 8. Vol. 3. McGraw-Hill; New York: 2001. pp. 3611–3633. [Google Scholar]
- Patterson MC, Vecchio D, Prady H, Abel L, Wraith JE. Miglustat for treatment of Niemann–Pick C disease: a randomized controlled study. Lancet Neurol. 2007;6:765–772. doi: 10.1016/S1474-4422(07)70194-1. S1474-4422(07) 70194-1. [DOI] [PubMed] [Google Scholar]
- Puri V, Watanabe R, Dominguez M, Sun X, Wheatley CL, Marks DL, Pagano RE. Cholesterol modulates membrane traffic along the endocytic pathway in sphingolipid-storage diseases. Nat Cell Biol. 1999;1:386–388. doi: 10.1038/14084. [DOI] [PubMed] [Google Scholar]
- Puri V, Jefferson JR, Singh RD, Wheatley CL, Marks DL, Pagano RE. Sphingolipid storage induces accumulation of intracellular cholesterol by stimulating SREBP-1 cleavage. J Biol Chem. 2003;278 (23):20961–20970. doi: 10.1074/jbc.M300304200. [DOI] [PubMed] [Google Scholar]
- Ries M, Schaefer E, Lührs T, Mani L, Kuhn J, Vanier MT, Krummenauer F, Gal A, Beck M, Mengel E. Critical assessment of chitotriosidase analysis in the rational laboratory diagnosis of children with Gaucher disease and Niemann-Pick disease type A/B and C. J Inherit Metab Dis. 2006;29:647–652. doi: 10.1007/s10545-006-0363-3. [DOI] [PubMed] [Google Scholar]
- Salvioli R, Scarpa S, Ciaffoni F, Tatti M, Ramoni C, Vanier MT, Vaccaro AM. Glucosylceramidase mass and subcellular localization are modulated by cholesterol in Niemann-Pick disease type C. J Biol Chem. 2004;279(17):17674–17680. doi: 10.1074/jbc.M313517200. [DOI] [PubMed] [Google Scholar]
- Santos ML, Raskin S, Telles DS, et al. Treatment of a child diagnosed with Niemann-Pick disease type C with miglustat: A case report in Brazil. J Inherit Metab Dis. 2008 doi: 10.1007/s10545-008-0923-9. Epub ahead of print Oct 21. [DOI] [PubMed] [Google Scholar]
- Sun X, Marks DL, Park WD, Wheatley CL, Puri V, O’Brien JF, Kraft DL, Lundquist PA, Patterson MC, Pagano RE, Snow K. Niemann-Pick C variant detection by altered sphingolipid trafficking and correlation with mutations within a specific domain of NPC1. Am J Hum Genet. 2001;68(6):1361–1372. doi: 10.1086/320599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Li H, Liu JP. Niemann-Pick Disease type C: from molecule to clinic. Clin Exp Pharmacol Physiol. 2009;37(1):132–140. doi: 10.1111/j.1440-1681.2009.05235.x. [DOI] [PubMed] [Google Scholar]
- Vanier MT. Biochemical studies in Niemann-Pick disease. I. Major sphingolipids of liver and spleen. Biochim Biophys Acta. 1983;750:178–184. doi: 10.1016/0005-2760(83)90218-7. [DOI] [PubMed] [Google Scholar]
- Vanier MT. Lipid changes in Niemann-Pick disease type C brain: personal experience and review of the literature. Neurochem Res. 1999;24:481–489. doi: 10.1023/a:1022575511354. [DOI] [PubMed] [Google Scholar]
- Vanier MT, Millat G. Niemann-Pick disease type C. Clin Genet. 2003;64:269–281. doi: 10.1034/j.1399-0004.2003.00147.x. [DOI] [PubMed] [Google Scholar]
- Wajner A, Michelin K, Burin MG, Pires RF, Pereira MLS, Giugliani R, Coelho JC. Biochemical characterization of chitotriosidase enzyme: comparison between normal individuals and patients with Gaucher and with Niemann-Pick diseases. Clin Biochem. 2004;37:893–897. doi: 10.1016/j.clinbiochem.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Weinreb N, Taylor J, Cox T, Yee J, vom Dahl S. A benchmark analysis of the achievement of therapeutic goals for type 1 Gaucher disease patients treated with imiglucerase. Am J Hematol. 2008;83(12):890–895. doi: 10.1002/ajh.21280. [DOI] [PubMed] [Google Scholar]
- Zervas M, Somers KL, Thrall MA, Walkley SU. Critical role of glycosphingolipids in Niemann-Pick disease type C. Curr Biol. 2001;11:1283–1287. doi: 10.1016/s0960-9822(01)00396-7. [DOI] [PubMed] [Google Scholar]