

Abstract
Recent studies suggest that the heart possesses an intrinsic system that is intended to delimit tissue injury, as well as orchestrate homoeostatic responses within the heart. The extant literature suggests that this intrinsic stress response is mediated, at least in part, by a family of pattern recognition receptors that belong to the innate immune system, including CD14, the soluble pattern recognition receptor for lipopolysaccharide, and Toll like receptors-2, 3, 4, 5, 6, 7 and 9. Although this intrinsic stress response system provides a short-term adaptive response to tissue injury, the beneficial effects of this phylogenetically ancient system may be lost if myocardial expression of these molecules either becomes sustained and/or excessive, in which case the salutary effects of activation of these pathways is contravened by the known deleterious effects of inflammatory signaling. Herein we present new information with regard to activation of innate immune gene expression in the failing human heart, as well as review the novel TLR antagonists that are being developed for other indications outside of heart failure.
This review will discuss the interesting possibility that the TLR pathway may represent a new target for the development of novel heart failure therapeutics.
Overview of Innate Immunity
The adult heart responds to tissue injury by synthesizing a variety of proteins that delimit myocardial injury through upregulation of cytoprotective factors, as well as by activating mechanisms that facilitate tissue repair. While, the exact mechanisms that are responsible for orchestrating these stress responses within the heart are not known, there is a growing body of literature which suggests that the innate immune system plays an important role in terms of initiating, integrating, and perpetuating an ongoing the myocardial response to tissue injury. Our understanding of the molecular components that regulate innate immunity and inflammation and that lead to the induction of pro-inflammatory cytokines has increased dramatically with the discovery of a family of phylogenetically ancient receptors termed Toll-like receptors (TLRs) [1]. TLRs serve as pattern recognition receptors (PRRs) that recognize conserved motifs on pathogens, so called pathogen-associated molecular patterns (PAMPs). More recently it has become clear that TLRs also recognize molecular signatures emanating from endogenous host material that is released during cellular injury or death, referred to as damage associated molecular patterns (DAMPs) [2, 3], thereby providing a potential link between tissue injury, activation of inflammatory mediators, and the pathogenesis of heart failure.
Expression and Regulation of Toll Receptors in Animal Models
The heart expresses pattern recognition receptors belonging to the innate immune system, including CD14, the soluble pattern recognition receptor for lipopolysaccharide [4], and Toll like receptors-2, 3, 4, 5, 6, 7 and 9 (TLR-2, TLR-3. TLR-4, TLR-5 and TLR-6, TLR-7, TLR-8, TLR-9 respectively) [5, 6]. TLR 2, 4, 5 and 6 are expressed on the cell surface of murine and rat cell types residing within the heart, including TLR2 and TLR4 expression in cardiac myocytes, whereas TLR 3, 7 and 9 are expressed in intracellular compartments, primarily endosomes and the endoplasmic reticulum, with the ligand binding domains facing the lumen of the vesicle. There are three general categories of TLR ligands: proteins (TLR5), nucleic acids (TLR3,7,9) and lipid-based elements (TLR2, TLR4, TLR6, TLR2/TLR6) [7]. At the time of this writing, very little is known with regard to the regulation and/or spatial localization TLR expression within the heart, although TLR4 appears to be upregulated in the failing human heart [8, 9].
One of the first TLR signaling pathways to be elucidated was the TLR4 signaling pathway (Figure 1). All TLRs (except for TLR3) interact with an adaptor protein termed myeloid differentiation factor 88 (MyD88) via their Toll Interleukin Receptor (TIR) domains. When stimulated, MyD88 recruits IL-1 receptor associated kinase (IRAK) to the receptor complex. IRAK is then activated by phosphorylation on serine/threonine residues and associates with tumor necrosis receptor associated factor 6 (TRAF6), leading to NF-κB activation.[10] Although the adaptor molecule TIR domain-containing adapter protein (TIRAP) was initially thought to contribute to MyD88 independent signaling, studies have shown that TIRAP is required for TLR2 and TLR4 mediated activation of NF-κB. The exact ligands that activate TLR signaling in the heart are not known. In this regard it is interesting to note it that in addition to activation by the classic pathogen associated molecular patterns (e.g. lipolysaccharide), TLR receptors are activated by damaged proteins released by injured and/or dying cells [2, 3]. For example, both heat shock protein 60 and 70 are sufficient to activate TLR signaling in the heart [11, 12], whereas fibronectin can activate TLR signaling in non-myocytes [13]. Once these damage associated molecular patterns are recognized by pattern recognition receptors, they activate the components of the innate signaling pathway, including NF-κB, pro-inflammatory cytokines and nitric oxide [14], that in turn provoke immune cell recruitment and activation.
Figure 1.
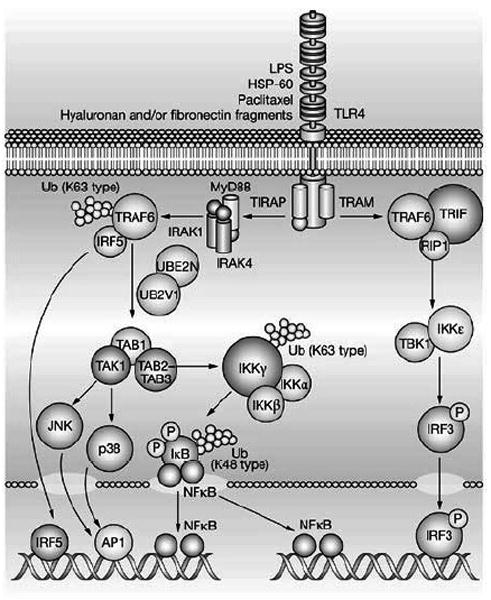
The Toll-like receptor signaling pathway. (Key: AP1, activator protein 1; HSP-60, heat shock protein 60; IκB, inhibitor of nuclear factor κB; IKKα, inhibitor of nuclear factor κ-B kinase α; IKKß, inhibitor of nuclear factor κB kinase-ß; IKKε, inhibitor of nuclear factor κ-B kinase ε; IKKγ, inhibitor of nuclear factor κ-B kinase γ; IRAK1, interleukin 1 receptor-associated kinase 1; IRAK4, interleukin 1 receptor-associated kinase 4; IRF3, interferon regulatory factor 3; IRF5, interferon regulatory factor 5; JNK, c-jun N-terminal kinase; LPS, lipopolysaccharide; MyD88, myeloid differentiation primary response protein; NF-κB, nuclear factor κB; RIP1, receptor-interacting protein 1; TAB1, TAK1 - binding protein 1; TAB2-TAB3, TAK1 -binding proteins 2 and 3; TAK1 (M3K7), transforming growth factor-ß-activated kinase 1; TBK1, serine-threonine-protein kinase; TIRAP, TIR domain-containing adaptor protein; TLR4, Toll-like receptor 4; TRAF6, tumor necrosis factor receptor-associated factor 6; TRAM, TRIF-related adaptor molecule; TRIF, TIR-domain-containing adaptor inducing interferon ß; Ub, ubiquitin; UB2V1, ubiquitin-conjugating enzyme E2 variant 1; UBE2N, ubiquitin-conjugating enzyme E2N (Reproduced with permission from Frantz, S., Ertl, G., & Bauersachs, J. Mechanisms of disease: Toll-like receptors in cardiovascular disease. Nat.Clin.Pract.Cardiovasc.Med. 4, 444-454, 2007).
Functional role of TLR Signaling in Experimental Models
Although the exact role of TLR signaling in the heart is not known, the extant literature supports the point of view that short-term activation of TLR signaling confers cytoprotective responses in the heart, whereas long-term signaling is maladaptive and can lead to cardiac remodeling (see below). For example, activation of TLR4 by LPS in vivo or ex vivo protects the myocardium following myocardial ischemia reperfusion (I/R) injury (reviewed in [15]). Hearts isolated from rats that were pretreated with a low dose of LPS (0.5 mg/kg) 24 hours prior to terminal sacrifice had preserved LV function after I/R injury compared with the saline treated control hearts [16]. The cytoprotective effects of LPS were manifest 12-24 h after the administration of LPS, were sensitive to inhibition with cycloheximide, and required TLR4 signaling [17]. Analogous to ischemic pre-conditioning, the cytoprotective effects of LPS were mediated by NOS2 and Akt [15]. Consistent with these studies, a recent report from our laboratory showed that ischemic preconditioning is mediated via a TLR2-TIRAP dependent signaling pathway that involves protein kinase C [18]. Interestingly, in these latter studies the phosphorylation of GSK-3β was TLR2-TIRAP dependent, whereas Akt phosphorylation was not. Given that GSK-3β has been implicated in opening of the mitochondrial transition pore and cell death in ischemia reperfusion injury, and that inactivation of GSK3β through phosphorylation leads to pre-conditioning by preventing opening of the mitochondrial transition pore [19], the observation GSK-3β phosphorylation was TLR2-TIRAP dependent suggest that one of the mechanisms whereby innate responses are cytoprotective is through inhibition of the formation of mitochondrial permeability transition pores trigger cell death following ischemia reperfusion injury.
Although the above studies suggest a beneficial role for TLR signaling in the heart, it should be recognized that the signaling pathways evolved in organisms with relatively short life spans (weeks to months), and thus never intended to provide long-term adaptive responses to the host organism. And indeed “loss of function studies” in experimental heart failure models suggest that sustained activation of TLRs is maladaptive and can contribute to adverse cardiac remodeling (Table 1). For example, mice with a missense mutation of TLR4 or targeted disruption of TLR4 [20-22], TLR2 [23], or MyD88 [24] have reduced infarct sizes when compared to wild-type controls. Correspondingly, mice pre-treated with a TLR4 antagonist (Eritoran) [25] had smaller infarct sizes when compared to vehicle treated animals. Mortality and LV remodeling are reduced in mice with targeted disruption of TLR4 or TLR2 [26, 27]. Although the mechanism(s) for the deleterious effects of TLR signaling following I/R injury and/or myocardial infarction have not been elucidated, a study in an experimental systemic sepsis model suggested that TLR4 receptors on bone marrow-derived hematopoietic cells were required for the neutrophil recruitment to the myocardium with resultant adverse cardiac remodeling [28]. Whether the mechanisms involved in an experimental model of sepsis will also obtain in experimental models of cardiac injury remains to be determined.
Table 1.
TLR signaling Modulation of Myocardial Ischemia Reperfusion Injury and Cardiac Remodeling
| Mice | Infarct Models | Effects in Knockout Mice |
|---|---|---|
| TLR2 signaling | ||
| TLR2-/- | I/R (30’ I/60R’)[23] | smaller infarct sizes, reduced neutrophil recruitment, reduced ROS and cytokines |
| TLR2-/- | Permanent coronary ligation[26] | Improved survival rate, attenuated remodeling, but same infarct sizes at 4 wk |
| TLR4 signaling | ||
| C57 BL/10 ScCr * C3H/HeJ** | I/R (60’ I/24 h R)[22] | Smaller infarct sizes, reduced MPO activity and complement 3 deposition |
| C3H/HeJ** | I/R (60’ I/120’ R)[20] | Smaller infarct sizes, decreased cardiac expression of TNF, MCP-1, and ILs |
| C3H/HeJ** | I/R (60’ I/24 h R)[21] | Smaller infarct sizes, but no gain in LV function |
| WT with eritoran | I/R (30’ I/120’ R)[25] | Smaller infarct sizes, reduced pJNK, reduced cytokine expression |
| C3H/HeJ** | Permanent coronary ligation[40] | Reduced LV remodeling, improved systolic function, reduced cytokine expression |
| C57 BL/10 ScCr* | Permanent coronary ligation[27] | Improved LV function on day 6 after infarction, improved survival rate, reduced LV remodeling and apoptosis at 4 wk. |
| MyD88−/− | I/R (30’ I/24 h R)[24] | Smaller infarct sizes, improved LV function, and attenuated cytokine expression and neutrophil recruitment |
C57 BL/10 ScCr mice contain a null mutation in the Lps gene and
C3H/HeJ mice contain a missense point mutation (Pro → His) in the Lps gene, rendering these mice hyporesponsive to lipoplysaccharide.
(Key: MPO, myeloperoxidase; MCP-1, monocyte chemoattractant protein-1; MyD88, myeloid differentiation primary-response gene 88; pJNK, phosphorylated JNK; TLR, Toll-like receptor; ROS, reactive oxygen species)
(Modified from Chao W, Am. J. Heart Circ Physiol 296: H1-H12, 2009)
Functional role of TLR Signaling in Human Heart Failure
As noted above the experimental literature suggests that sustained activation of TLR signaling following cardiac injury is maladaptive and can lead to a heart failure phenotype, very little is known regarding the innate immune system in the failing human heart. Two studies have shown that expression of TLR4 mRNA and TLR4 protein is increased in the hearts of patients with advanced heart failure [8, 9]. However, until recently it has been unclear whether or innate immune genes are expressed differently in human heart failure. To this end, we examined the expression profiles of genes that were involved in innate immune signaling using the Cardiogenomics Consortium data base (http://www.cardiogenomics.med.harvard.edu]), which was obtained from explanted hearts from patients with ischemic cardiomyopathy (ICM), idiopathic dilated cardiomyopathy (DCM), and viral cardiomyopathy (VCM), as well as non-failing (NF) hearts. Expression data for innate immune signaling genes were analyzed by Principal Component Analysis (PCA), a mathematical modeling procedure that transforms a number of possibly correlated variables into a smaller number of uncorrelated variables that are termed principal components[29]. The 3-dimension PCA plot illustrated in Figure 2 shows two important findings with respect to innate immune gene expression in human heart failure. First, the numerical values for the PCA plots for NF hearts were clustered differently than the numerical values for the PCA plots for the ICM, DCM, VCM hearts, which tended to cluster together (best observed in Figure 2A). The second salient finding is that the PCA profiles were different in ICM patients when compared to DCM patients. This raises the intriguing possibility that that the innate immune system is activated differentially in response to the nature of the pathological tissue injury pattern. Inspection of Figure 2A shows that the PCA plots for VCM patients were similar to that observed in DCM. This particular observation is of interest, insofar as occult and/or persistent viral myocarditis has been suggested as a potential etiology for idiopathic dilated cardiomyopathy [30]. An unsupervised hierarchical clustering analysis of DCM, ICM, VCM and non-failing human hearts confirmed that there were (1) distinct gene expression profiles for innate immune genes in failing and non-failing hearts, and (2) that there were distinct gene expression profiles for innate immune genes in ICM and DCM hearts [31]. Indeed there were 37, 37, and 27 transcripts, respectively, whose expression levels were significantly different in DCM, ICM, and VCM when compared to non-failing hearts. Interestingly there were only 14 transcripts whose changes were similar in all forms of cardiomyopathy, of which there were 3 genes whose expression levels increased and 11 immune genes whose expression decreased in all forms of cardiomyopathy [31]. Although any inferences with respect to a cause and effect relationship vis-à-vis differential expression of innate immune genes in the failing human heart and the pathogenesis of human heart failure must be regarded as provisional until detailed studies of protein levels of TLRs are analyzed in failing human hearts, the observation that NF-κB activation and pro-inflammatory cytokines gene and protein expression are increased in the failing human heart [32-34] raises the interesting possibility that enhanced innate immune signaling may contribute to the pathogenesis of human heart failure by provoking the expression of pro-inflammatory mediators that are sufficient to contribute to a heart failure phenotype (reviewed in [35]).
Figure 2.
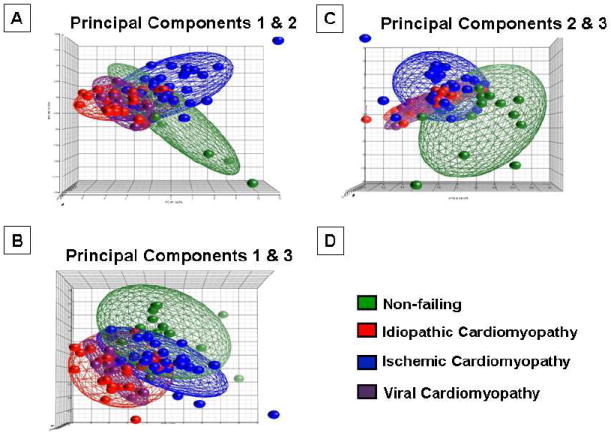
Principal component analysis of changes in innate immune gene expression in failing and non-failing human hearts. Innate immune genes were subjected to a principal component analysis (PCA), and the first, second and third principal components were displayed in a 3-D graphic format. (From Mann DL, Topkara VK, Evans S, Barger PM. Innate immunity in the adult Mammalian heart: for whom the cell tolls. Trans Am Clin Climatol Assoc 2010;121:34-50).
Translation Potential of TLR Signaling in Human Heart Failure
There has been significant interest in developing TLR antagonists as novel therapeutics in diseases such as sepsis, systemic lupus erythematosis and rheumatoid arthritis, wherein the immune system and inflammatory mediators are inappropriately overactive. Currently, there are a variety of novel antagonists that are being developed for TLR 2, 4, 7, and 9 (reviewed in [7]). Given the focus of the present review on innate immunity in heart failure, we will focus on TLR2 and TL4 antagonism (see Table 2), for which there is the most direct evidence of TLR involvement in heart failure.
Table 2.
Development Status of TLR2 and TLR4 antagonists
| Compound | Indications | Target | Drug Class | Clinical Phase |
|---|---|---|---|---|
| OPN-305 | Inflammation, autoimmunity, ischemia/reperfusion | TLR2 antagonist | Antibody | Orphan status for prevention of ischemia reperfusion injury |
| OPN-401 | IBD, rheumatoid arthritis | TLR2/TLR4 antagonist | viral-derived peptide | Preclinical |
| AP177 | NS | TLR2 antagonist | DNA aptamer | Preclinical |
| Eritoran (E5564) | Sepsis | TLR4 antagonist | Synthetic lipodisaccharide | Phase III |
| Lidid-IVa | NS | TLR4 antagonist | Lipid A partial mimetic | Preclinical |
| TAK-242 | Sepsis | TLR4 antagonist | Small molecule inhibitor | Suspended in phase III |
| 1A6 | Colitis | TLR4 antagonist | Antibody | Preclinical |
| CPG-52364 | SLE | PolyTLR antagonist | Quinazoline derivative | Phase I |
| Ibudilast (AV411) | Pain management, withdrawal | TLR4 antagonist | Small-molecule phosphodiesterase inhibitor | Phase II |
Modified from Hennessy et al., Nat. Rev Drug Discov. 9, 293-307; 2010
TLR2
OPN-305, a TLR2-specific monoclonal anti-body that inhibits TLR2-mediated pro-inflammatory cytokine production, was granted orphan status for the prevention of the ischemia and reperfusion injury associated with organ transplantation. The first human trials as a potential treatment of inflammatory diseases are expected to begin in 2010. Given the role of TLR2 in mediating ischemia reperfusion injury, this molecule is attractive for testing in phase I clinical trials in patients with ischemic cardiomyopathy. AP177, which is a DNA aptamer that was identified by a SELEX (systematic evolution of ligands by exponential enrichment) screen [36], binds to TLR and competitively antagonizes TLR2 ligand binding, thereby inhibiting NF-κB activity and pro-inflammatory cytokine production [7].
TLR4
There are a number of strategies that have been undertaken to inhibit TLR4 activation [7]. Eritoran (E5564), which reduces the binding of lipid-A (the biologically active part of the lipolysaccharide molecule), reduced mortality by 6.4% compared with the placebo group in a phase II sepsis trial, and is currently undergoing evaluation in Phase III sepsis trials (NCT00334828). Given that the pharmacodynamic profile of Eritoran requires administration as a continuous infusion or by repeated intravenous injections, this TLR4 antagonist may not be practical for treating chronic heart failure. However, it may be useful during myocardial inflammatory states or in the setting of an acute coronary syndromes that lead to the development of heart failure. Alternative approaches have been to develop variations of lipid-A that bind TLR4, but have reduced agonist activity (e.g., CRX-527, lipid ▪ IVa). TAK-242 also targets TLR4- dependent signaling, although the precise target is not known. Development of this compound was discontinued during a Phase III sepsis clinical trial because the drug’s profile did not meet the criteria required to support continued development, not because of drug safety issues (NCT00633477). Ibudilast (AV411) is another TLR4 antagonist, that suppresses pro-inflammatory cytokines such as TNF and IL-6, and may induce the anti-inflammatory cytokine IL-10, is undergoing phase II trials for opioid dependence (NCT00723177). OPN-401, is a viral protein-derived peptide that inhibits TLR4-dependent signaling is also in preclinical development.
Direction of Future Research
The foregoing review suggests that activation of TLR signaling in the heart confers short-term benefits in the heart when activated acutely, but that the beneficial effects of TLR signaling are lost in the chronic setting, wherein ongoing tissue damage can lead to sustained TLR signaling that is sufficient to provoke a heart failure phenotype. Thus, analogous to the renin angiotensin system and the adrenergic nervous system, activation of the innate immune system can provoke disparate responses, depending on the context, as well as duration of activation of this phylogenetically conserved signaling system. Although the data linking increased TLR signaling to heart failure is provisional at the time of this writing, the extant literature suggests that increased TLR signaling, perhaps secondary to DAMPs, may contribute to adverse cardiac remodeling secondary to increased NF-κB activation and increased expression of pro-inflammatory cytokines. Whereas the initial clinical heart failure trials that employed targeted anti-inflammatory approaches yielded disappointing results [35, 37, 38], targeting the TLR signaling pathway in heart failure may offer a more rationale therapeutic approach, insofar as the TLR signaling pathway modulates a much broader portfolio of inflammatory mediators, and acts as a important upstream nodal mechanism for activating inflammatory signaling in response to tissue injury. Moreover, targeting specific TLR pathways may allow for tailoring anti-inflammatory strategies to specific subsets of heart failure patients. Indeed a recent consensus statement from the Translation Research Committee of the Heart Failure Association of the European Society of Cardiology suggested that there may not be a common inflammatory pathway that characterizes all of the different forms of heart failure, and that going forward it would be important to design specific anti-inflammatory approaches for different types and stages of heart failure, as well as to determine the specific inflammatory pathways that are activated in different forms of heart failure [39]. Based on the extant literature, targeting TLR signaling in the setting of chronic ischemic cardiomyopathy, wherein TLR signaling has been implicated in plaque rupture in atherosclerotic coronary arteries, as well as adverse cardiac remodeling, would appear to be attractive. As with all therapeutic approaches in heart failure, the only way to really answer the question of whether these types of anti-inflammatory strategies will have any added value in heart failure is through well designed clinical trials. However, it bears emphasis that at present our knowledge of the role of TLR signaling is not sufficient to support the evaluation of this therapeutic target in phase I clinical trials. Additional studies in clinical heart failure samples that demonstrate increased protein levels and/or activation of the signal transduction pathways that are downstream from TLR signaling will be required to complement the studies on human heart failure gene expression. Further, it will also be important to determine whether blocking TLR2, TLR4 or TLR2 and TLR4 with antibodies or small molecule inhibitors is more effective with respect to preventing the development of a heart failure phenotype in small animal models, insofar as Eritoran is the only molecule that has been tested in experimental heart failure models. Lastly, it remains to be determined whether antagonizing TLR signaling in heart failure will lead to worsening heart failure because of the potential loss of the beneficial effects of TLR signaling in the heart. Despite these cautionary notes, drug development in this area will benefit from the explosive growth in knowledge that has occurred over the past decade with respect to the biology of innate immune responses in the heart, which in turn will facilitate choosing the appropriate biomarkers and surrogate end-points that would support enrolling patients in well-designed phase I –II clinical trials to test the efficacy of this therapeutic target in heart failure.
Acknowledgments
This research was supported by research funds from the N.I.H. (RO1 HL58081, HL-73017-0, HL089543-01 and T32HL007081).
Footnotes
DISCLOSURE STATEMENT None of the authors have an actual or potential conflict of interest with regard to this publication.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Janeway CA, Jr, Medzhitov R. Introduction: the role of innate immunity in the adaptive immune response. Semin Immunol. 1998;10:349–50. doi: 10.1006/smim.1998.0142. [DOI] [PubMed] [Google Scholar]
- 2.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 3.Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr Opin Immunol. 2001;13:114–9. doi: 10.1016/s0952-7915(00)00191-6. [DOI] [PubMed] [Google Scholar]
- 4.Cowan DB, Poutias DN, del Nido PJ, McGowan FX., Jr CD14-independent activation of cardiomyocyte signal transduction by bacterial endotoxin. Am J Physiol Heart Circ Physiol. 2000;279:H619–H629. doi: 10.1152/ajpheart.2000.279.2.H619. [DOI] [PubMed] [Google Scholar]
- 5.Boyd JH, Mathur S, Wang Y, Bateman RM, Walley KR. Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-kappaB dependent inflammatory response. Cardiovasc Res. 2006;72:384–93. doi: 10.1016/j.cardiores.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 6.Frantz S, Kelly RA, Bourcier T. Role of TLR-2 in the activation of nuclear factor-kappa B by oxidative stress in cardiac myocytes. J Biol Chem. 2001;276:5197–203. doi: 10.1074/jbc.M009160200. [DOI] [PubMed] [Google Scholar]
- 7.Hennessy EJ, Parker AE, O’Neill LA. Targeting Toll-like receptors: emerging therapeutics? Nat Rev Drug Discov. 2010;9:293–307. doi: 10.1038/nrd3203. [DOI] [PubMed] [Google Scholar]
- 8.Frantz S, Kobzik L, Kim YD, Fukazawa R, Medzhitov R, Lee RT, et al. Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. J Clin Invest. 1999;104:271–80. doi: 10.1172/JCI6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Birks EJ, Felkin LE, Banner NR, Khaghani A, Barton PJ, Yacoub MH. Increased toll-like receptor 4 in the myocardium of patients requiring left ventricular assist devices. J Heart Lung Transplant. 2004;23:228–35. doi: 10.1016/S1053-2498(03)00106-2. [DOI] [PubMed] [Google Scholar]
- 10.Frantz S, Ertl G, Bauersachs J. Mechanisms of disease: Toll-like receptors in cardiovascular disease. Nat Clin Pract Cardiovasc Med. 2007;4:444–54. doi: 10.1038/ncpcardio0938. [DOI] [PubMed] [Google Scholar]
- 11.Satoh M, Shimoda Y, Akatsu T, Ishikawa Y, Minami Y, Nakamura M. Elevated circulating levels of heat shock protein 70 are related to systemic inflammatory reaction through monocyte Toll signal in patients with heart failure after acute myocardial infarction. Eur J Heart Fail. 2006;8:810–5. doi: 10.1016/j.ejheart.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 12.Kim SC, Stice JP, Chen L, Jung JS, Gupta S, Wang Y, et al. Extracellular heat shock protein 60, cardiac myocytes, and apoptosis. Circ Res. 2009;105:1186–95. doi: 10.1161/CIRCRESAHA.109.209643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, et al. The EDA domain of fibronectin activates toll-like receptor 4. J Biol Chem. 2001;276:10229–33. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- 14.Mann DL. Tumor Necrosis Factor and Viral Myocarditis: The Fine Line Between Innate and Inappropriate Immune Responses in the Heart. Circulation. 2001;103:626–9. doi: 10.1161/01.cir.103.5.626. [DOI] [PubMed] [Google Scholar]
- 15.Chao W. Toll-like receptor signaling: a critical modulator of cell survival and ischemic injury in the heart. Am J Physiol Heart Circ Physiol. 2009;296:H1–12. doi: 10.1152/ajpheart.00995.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown JM, Grosso MA, Terada LS, Whitman GJR, Banerjee A, White CW, et al. Endotoxin pretreatment increases endogenous myocardial catalase activity and decreases ischemia-reperfusion injury of isolated rat hearts. Proc Natl Acad Sci U S A. 1989;86:2516–20. doi: 10.1073/pnas.86.7.2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu X, Zhao H, Graveline AR, Buys ES, Schmidt U, Bloch KD, et al. MyD88 and NOS2 are essential for toll-like receptor 4-mediated survival effect in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2006;291:H1900–H1909. doi: 10.1152/ajpheart.00112.2006. [DOI] [PubMed] [Google Scholar]
- 18.Dong JW, Vallejo JG, Tzeng HP, Thomas JA, Mann DL. Innate immunity mediates myocardial preconditioning through Toll-like receptor 2 and TIRAP-dependent signaling pathways. Am J Physiol Heart Circ Physiol. 2010;298:H1079–H1087. doi: 10.1152/ajpheart.00306.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gross ER, Hsu AK, Gross GJ. Opioid-induced cardioprotection occurs via glycogen synthase kinase beta inhibition during reperfusion in intact rat hearts. Circ Res. 2004;94:960–6. doi: 10.1161/01.RES.0000122392.33172.09. [DOI] [PubMed] [Google Scholar]
- 20.Chong AJ, Shimamoto A, Hampton CR, Takayama H, Spring DJ, Rothnie CL, et al. Toll-like receptor 4 mediates ischemia/reperfusion injury of the heart. J Thorac Cardiovasc Surg. 2004;128:170–9. doi: 10.1016/j.jtcvs.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 21.Kim SC, Ghanem A, Stapel H, Tiemann K, Knuefermann P, Hoeft A, et al. Toll-like receptor 4 deficiency: smaller infarcts, but no gain in function. BMC Physiol. 2007;7:5. doi: 10.1186/1472-6793-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oyama J, Blais C, Jr, Liu X, Pu M, Kobzik L, Kelly RA, et al. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004;109:784–9. doi: 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- 23.Favre J, Musette P, Douin-Echinard V, Laude K, Henry JP, Arnal JF, et al. Toll-like receptors 2-deficient mice are protected against postischemic coronary endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2007;27:1064–71. doi: 10.1161/ATVBAHA.107.140723. [DOI] [PubMed] [Google Scholar]
- 24.Feng Y, Zhao H, Xu X, Buys ES, Raher MJ, Bopassa JC, et al. Innate immune adaptor MyD88 mediates neutrophil recruitment and myocardial injury after ischemia-reperfusion in mice. Am J Physiol Heart Circ Physiol. 2008;295:H1311–H1318. doi: 10.1152/ajpheart.00119.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimamoto A, Chong AJ, Yada M, Shomura S, Takayama H, Fleisig AJ, et al. Inhibition of Toll-like receptor 4 with eritoran attenuates myocardial ischemia-reperfusion injury. Circulation. 2006;114:I270–I274. doi: 10.1161/CIRCULATIONAHA.105.000901. [DOI] [PubMed] [Google Scholar]
- 26.Shishido T, Nozaki N, Yamaguchi S, Shibata Y, Nitobe J, Miyamoto T, et al. Toll-like receptor-2 modulates ventricular remodeling after myocardial infarction. Circulation. 2003;108:2905–10. doi: 10.1161/01.CIR.0000101921.93016.1C. [DOI] [PubMed] [Google Scholar]
- 27.Riad A, Jager S, Sobirey M, Escher F, Yaulema-Riss A, Westermann D, et al. Toll-like receptor-4 modulates survival by induction of left ventricular remodeling after myocardial infarction in mice. J Immunol. 2008;180:6954–61. doi: 10.4049/jimmunol.180.10.6954. [DOI] [PubMed] [Google Scholar]
- 28.Tavener SA, Long EM, Robbins SM, McRae KM, Van Remmen H, Kubes P. Immune cell Toll-like receptor 4 is required for cardiac myocyte impairment during endotoxemia. Circ Res. 2004;95:700–7. doi: 10.1161/01.RES.0000144175.70140.8c. [DOI] [PubMed] [Google Scholar]
- 29.Ringner M. What is principal component analysis? Nat Biotechnol. 2008;26:303–4. doi: 10.1038/nbt0308-303. [DOI] [PubMed] [Google Scholar]
- 30.Parrillo JE, Cunnion RE, Epstein SE, Parker ME, Suffredini AF, Brenner M, et al. A prospective randomized controlled trial of prednisone for dilated cardiomyopathy. N Engl J Med. 1989;321:1061–8. doi: 10.1056/NEJM198910193211601. [DOI] [PubMed] [Google Scholar]
- 31.Mann DL, Topkara VK, Evans S, Barger PM. Innate immunity in the adult Mammalian heart: for whom the cell tolls. Trans Am Clin Climatol Assoc. 2010;121:34–50. [PMC free article] [PubMed] [Google Scholar]
- 32.Frantz S, Fraccarollo D, Wagner H, Behr TM, Jung P, Angermann CE, et al. Sustained activation of nuclear factor kappa B and activator protein 1 in chronic heart failure. Cardiovasc Res. 2003;57:749–56. doi: 10.1016/s0008-6363(02)00723-x. [DOI] [PubMed] [Google Scholar]
- 33.Grabellus F, Levkau B, Sokoll A, Welp H, Schmid C, Deng MC, et al. Reversible activation of nuclear factor-kappaB in human end-stage heart failure after left ventricular mechanical support. Cardiovasc Res. 2002;53:124–30. doi: 10.1016/s0008-6363(01)00433-3. [DOI] [PubMed] [Google Scholar]
- 34.Torre-Amione G, Kapadia S, Lee J, Bies RD, Lebovitz R, Mann DL. Expression and functional significance of tumor necrosis factor receptors in human myocardium. Circulation. 1995;92:1487–93. doi: 10.1161/01.cir.92.6.1487. [DOI] [PubMed] [Google Scholar]
- 35.Mann DL. Inflammatory Mediators and the Failing Heart: Past, Present, and the Foreseeable Future. Circ Res. 2002;91:988–98. doi: 10.1161/01.res.0000043825.01705.1b. [DOI] [PubMed] [Google Scholar]
- 36.Chang YC, Kao WC, Wang WY, Wang WY, Yang RB, Peck K. Identification and characterization of oligonucleotides that inhibit Toll-like receptor 2-associated immune responses. FASEB J. 2009;23:3078–88. doi: 10.1096/fj.09-129312. [DOI] [PubMed] [Google Scholar]
- 37.Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT. Randomized, Double-Blind, Placebo-Controlled, Pilot Trial of Infliximab, a Chimeric Monoclonal Antibody to Tumor Necrosis Factor-alpha, in Patients With Moderate-to-Severe Heart Failure: Results of the Anti-TNF Therapy Against Congestive Heart failure (ATTACH) Trial. Circulation. 2003;107:3133–40. doi: 10.1161/01.CIR.0000077913.60364.D2. [DOI] [PubMed] [Google Scholar]
- 38.Mann DL, McMurray JJV, Packer M, Swedberg K, Borer JS, Colucci WS, et al. Targeted anti-cytokine therapy in patients with chronic heart failure: results of the Randomized EtaNcercept Worldwide evALuation (RENEWAL) Circulation. 2004;109:1594–602. doi: 10.1161/01.CIR.0000124490.27666.B2. [DOI] [PubMed] [Google Scholar]
- 39.Heymans S, Hirsch E, Anker SD, Aukrust P, Balligand JL, Cohen-Tervaert JW, et al. Inflammation as a therapeutic target in heart failure? A scientific statement from the Translational Research Committee of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. 2009;11:119–29. doi: 10.1093/eurjhf/hfn043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Timmers L, Sluijter JP, Van Keulen JK, Hoefer IE, Nederhoff MG, Goumans MJ, et al. Toll-Like Receptor 4 Mediates Maladaptive Left Ventricular Remodeling and Impairs Cardiac Function Following Myocardial Infarction. Circ Res. 2007;102:257–64. doi: 10.1161/CIRCRESAHA.107.158220. [DOI] [PubMed] [Google Scholar]