

Abstract
Background and Objectives
Nitric oxide (NO) is a major endothelium dependent vasomediator and growth inhibitor. NO synthesis is catalyzed by endothelial nitric oxide synthase (eNOS), and NO can also produce peroxynitrite, which activates matrix metalloproteinases (MMPs). The purpose of this study was to determine the gene expression of eNOS and MMP-2 in the lungs of a rat model of pulmonary hypertension after bosentan treatment.
Materials and Methods
Six-week-old male Sprague-Dawley rats were treated as follows: control group, subcutaneous (sc) injection of saline; monocrotaline (MCT) group, sc injection of MCT (60 mg/kg); and bosentan group, sc injection of MCT (60 mg/kg) plus 20 mg/day bosentan orally. The rats were sacrificed after 1, 5, 7, 14 and 28 days.
Results
The right ventricle/(left ventricle+septum) ratio significantly increased in the MCT group compared to the control group on day 14 and 28. The expression of eNOS messenger ribonucleic acid was significantly increased in the MCT group compared to the control group on day 28. MMP-2 gene expression was significantly increased in the MCT-treated rats compared to the control group on day 5 and 28. Following bosentan treatment to reduce pulmonary hypertension, the expression levels of MMP-2 gene were significantly decreased on day 7 and 28. eNOS and tissue inhibitor of MMPs genes were also significantly decreased on day 28 after bosentan treatment.
Conclusion
These results suggest that elevated eNOS expression may be responsible for MMP-2 activation. The causal relationship between eNOS and MMP-2 and their role in pulmonary hypertension require further investigations.
Keywords: Pulmonary hypertension, Monocrotaline, Gene expression, Nitric oxide synthase, Matrix metalloproteinases
Introduction
Pulmonary hypertension is a condition characterized by progressive increase in pulmonary vascular resistance, leading to right ventricular (RV) failure and death. To date, various mechanisms underlying the development of pulmonary hypertension have been explored. One of which is the role of vasoactive mediators produced from the pulmonary vascular endothelium which regulates normal pulmonary vascular tone.
Both nitric oxide (NO) and endothelin (ET)-1 have been identified as major endothelium dependent vasomediator. NO is a potent, endogenous vasodilator which contributes not only to the local regulation of vascular smooth muscle tone, but also to smooth muscle cell (SMC) proliferation.1) NO is produced in pulmonary endothelial cells (ECs) from the amino acid L-arginine, and molecular oxygen by endothelial nitric oxide synthase (eNOS).2) This led to a hypothesis that eNOS expression might play a key role in the regulation of vascular tone. A number of clinical and animal experiments have investigated to test this hypothesis but have shown conflicting results. Whether the expression of eNOS is altered in association with right ventricular hypertrophy (RVH) has not yet been fully elucidated.
Bosentan is an orally active ET receptor antagonist which has a non-selective, dual action against ETA and ETB. It has been shown that bosentan significantly reduces pulmonary hypertension, pulmonary vascular hypertrophy and RVH in rats with hypoxia-induced pulmonary hypertension.3) Similarly, the development of pulmonary hypertension was attenuated by bosentan in rats treated with monocrotaline (MCT).4) In this study, we used MCT to induce pulmonary hypertension and selective RVH in rats. Matrix metalloproteinases (MMPs) are a large family of endopeptidases which has an important role in the degradation of extracellular matrix (ECM). Previous studies have shown increased levels and activation of MMP-2 in pulmonary arteries from experimental pulmonary arterial hypertension models, suggesting that MMPs contribute to the progression of pulmonary arterial hypertension in animals.5),6) The purposes of this study were to investigate the gene expressions of eNOS, MMP and tissue inhibitor of matrix metalloproteinases (TIMP) in the lungs of MCT induced pulmonary hypertension rats, and the effect of bosentan treatment on their gene expressions.
Materials and Methods
Six-week-old male Sprague-Dawley rats, weighing between 200-250 g, were used for this study. All rats were house in climate-controlled conditions with a 12 hours light: 12 hours dark cycle, and had free access to chow and water.
Pulmonary hypertension was induced by subcutaneous (sc) injection of 60 mg/kg MCT (Sigma Chemicals, St. Louis, MO, USA) dissolved in 0.5 N NaCl solution. The rats were grouped as follows: control (n=36), sc injection of normal saline (0.1 mL/kg); MCT (n=36), sc injection of MCT; bosentan (n=36), sc injection of MCT plus 20 mg/day bosentan (Actelion Pharmaceuticals Ltd., Allischwil, Switzerland) by gavage feeding. The rats were sacrificed after 1, 5, 7, 14 and 28 days. Lung tissues were removed and immediately frozen at -70℃ for enzyme analysis, post-fixed in 10% formalin, and processed routinely for paraffin embedding. The study protocols were approved by the Institutional Review Board of the School of Medicine of Ewha Womans University.
Organ weights
The rats were weighed and observed for general appearance during the study period. The animals were sacrificed at the scheduled time, and the hearts and lungs were rapidly removed. The wet weights of the RV, left ventricle and septum (LV+S), and lungs were measured, and the ratio of organ weight to body weight was calculated. The RV to LV+S ratio {RV/(LV+S)} was used as an index of RVH.
Hemodynamic parameters
The animals were placed in the supine position and instrumented with an arterial pressure line (Physiological Pressure Transducer, MLT1199; AD Instruments, Oxfordshire, UK). Hemodynamic parameters were recorded at baseline, and after 5, 7, 14 and 28 days. The catheter was placed in the RV to estimate the mean ventricular pressure. The mean right ventricular pressure was measured. Arterial pressures were calibrated using input from an ambient-pressure reference (APR-1; Data Sciences).
Matrix metalloproteinase-2 and nitric oxide level in serum
At the scheduled times, serum was collected to determine MMP-2 and NO concentrations. Serum MMP-2 level was measured using a mouse immunoassay kit (Quantikine, Mouse MMP-2 Immunoassay kit, R&D system Inc., Minneapolis, MN, USA), and serum NO level was measured using molecular devices (Molecular Devices V-MAX 220 VAC enzyme linked immuno sorbent assay reader, USA) by a sandwich enzyme immunoassay technique. A monoclonal antibody specific for MMP-2 had been pre-coated onto a microplate. Standards and samples were pipetted into the wells and any MMP present were bound by an immobilized antibody. After washing away any unbound substances, an enzyme-linked monoclonal antibody specific for MMP-2 was added to the wells. Following a wash to remove any unbound antibody-enzyme reagent, an enhanced luminal/peroxide substrate solution was added to the wells and light was produced in proportion to the amount of MMP-2 bound in the initial step. The identical method was used to measure NO. A microplate luminometer was used to measure the intensity of the light emitted.
Lung microarray
Total RNA was extracted from the rat lung tissue using the TRI REAGENT (MRC, OH) according to the manufacturer's instructions. Each total RNA sample (10 ug) was labeled with Cyanine (Cy5) conjugated dCTP (Amersharm, Piscataway, NJ) by a reverse transcription reaction using reverse transcriptase, SuperScrip ll (Invitrogen, Carlsbad, California). The labeled cDNA was then concentrated using the ethanol precipitation method. Concentrated Cy5 labeled cDNAs were re-suspended in 10 µL of hybridization solution (GenoCheck, Korea). After the labeled cDNAs were placed on Roche NimbleGen Rat genome 12-plex array (Roche NimbleGen, Inc., WI) and covered by a NimbleGen H12 mixer (Roche NimbleGen, Inc., WI). The slides were hybridized for 12 hours at 42℃ MAUI system (Biomicro systems, Inc. UT). The hybridized slides were washed in 2 X SSC (sodium chloride sodium citrate), 0.1% sodium dodecyl sulfate for 2 minutes, 1 X SSC for 3 minutes, and then 0.2 X SSC for 2 minutes at room temperature. The slides were centrifuged at 3,000 rpm for 20 s to dry.
The arrays were analyzed using an Axon GenePix 4000B scanner with associated software (Molecular Devices Corp., Sunnyvale, CA). Gene expression levels were calculated with NimbeScan Version 2.4 (Roche NimbleGen, Inc., WI). Relative signal intensities for each gene were generated using the Robust Multi-Array Average algorithm. The data were processed based on median polish normalization method using NimbeScan Version 2.4 (Roche NimbleGen, Inc., WI). This normalization method aims to make the distribution of intensities for each array in a set of arrays identical. The normalized, and log transformed intensity values were then analyzed using GeneSpring GX 7.3.1 (Agilent technologies, CA). Fold change filters included the requirement that the genes be present in at least 150% of controls for up-regulated genes, and lower than 66.67% of controls for down-regulated genes. Hierarchical clustering data were clustered groups that behave similarly across experiments using GeneSpring GX 7.3.1 (Agilent technologies, CA). Clustering algorithm was Euclidean distance, average linkage.
Ribonucleic acid extraction and complementary deoxyribonucleic acid synthesis
Total RNA was extracted using trizol reagent™ (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol, and re-suspended in diethyl pyrocarbonate water. The final RNA amount was spectrophotometrically determined at 260/280 nm. Quality was assessed as the absence of smear of 18S and 28S bands analyze by means of Bio analyzer 2100 (Agilent). RNA samples were stored at -70℃ until used. cDNAs were synthesized by 1 ug of total RNA, according to the manufacture's protocol (High Capacity RNA-to-cDNA kit, Applied Biosystems, USA).
Gene expression analysis by real time reverse transcription-polymerase chain reaction
Real-time quantitative polymerase chain reaction (PCR) was performed in triplicate in 384-well plates. A 384-well high-throughput analysis was performed using the ABI Prism 7900 Sequence Detection System (Applied Biosystems, Calfornia, USA), and white colored 384-well plates (ABgene, Hamburg, Germany) for intensification of the fluorescent signals by a factor of three. The system operates using a thermal cycler and a laser that is directed via fiber optics to each well. Fluorescence emission from each sample was collected by a charge-coupled device-camera, and quantitative data were analyzed using the Sequence Detection System software (SDS version 2.0, Applied Biosystems, California, USA). Reaction mixtures contained 10 pmol/uL of each primer and 2X SYBR Green PCR Master Mix (Applied Biosystems, California, USA), which included the HotStarTaqt DNA-Polymerase in an optimized buffer, the dNTP mix (with dUTP additive), the SYBRs Green I fluorescent dye, and ROX dye as a passive reference. Each of the 384-well real-time quantitative PCR plate included serial dilutions (1, 1/2 and 1/4) of cDNA, which were used to generate relative standard curves for genes.
The resulting first-strand of cDNA was normalized by the glyceraldehyde 3-phosphate dehydrogenase gene. The normalized cDNA was used for the PCR procedure as a template. The specific primers for rat inducible NO synthase (NOS2) was 5'-GGG CCA CCT TTA TGT TTG TG-3' (forward) and 5'-CCT CAA CCT GCT CCT CAC TC-3' (reverse). The spe-cific primers for rat endothelial NO synthase (eNOS, NOS3) were 5'-CTG CGG TGA TGT CAC TAT GG-3' (forward) and 5'-AAA TGT CCT CGT GGT AGC GT-3' (reverse). The specific primers for rat MMP-2 was 5'-AAG AGG CCT GGT TAC CCT GT-3' (forward) and 5'-AAG TAG CAC CTG GGA GGG AT-3'(reverse). The specific primers for rat TIMP were 5'-GAC CTA TAG TGC TGG CTG TG-3' (forward) and 5'-GATC GCT CTG GTA GCC CTT CT-3' (reverse) (Table 1). All primers were amplified using the same conditions. Thermal cycling conditions 50℃ for 2 minutes and 95℃ for 10 minutes, followed by 40 cycles of 95℃ for 30 s and 60℃ for 30 s, 72℃ for 30 s. In order to exclude the presence of unspecific products, a melting curve analysis of products was performed routinely after finishing amplification by a high-resolution data collection during an incremental temperature increase from 60℃ to 95℃ at a ramp rate of 0.21℃/s. We then converted real-time PCR cycle numbers to gene amounts (ng) on the basis of the equation. Real-time PCR analysis was performed on an Applied Biosystems Prism 7900 Sequence Detection System (Applied Biosystems, California, USA) (Fig. 1).
Table 1.
Gene sequence of primer of RT-PCR

NOS2: inducible nitric oxide synthase, NOS3: endothelial nitric oxide synthase, MMP-2: matrix metalloproteinase 2, TIMP: tissue inhibitor of matrix metalloproteinases, RT-PCR: reverse transcription-polymerase chain reaction
Fig. 1.
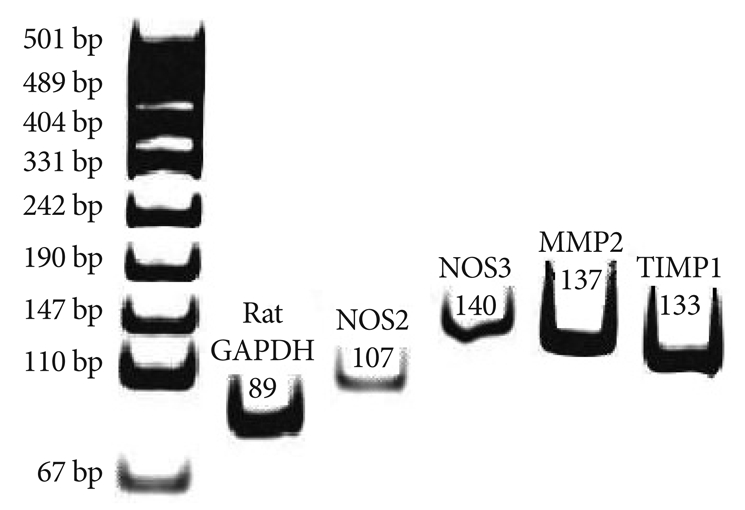
Typical example of RT-PCR products are shown for the level of inducible nitric oxide synthase endothelial nitric oxide synthase, matrix metalloproteinase 2 and tissue inhibitor of matrix metalloproteinases messenger ribonucleic acid in lung tissue. The RT-PCR products from the transcripts of NOS2, NOS3, MMP2, TIMP and GAPDH were 107 bp, 140 bp, 137 bp, 133 bp and 89 bp. NOS 2: inducible nitric oxide synthase, NOS3: endothelial nitric oxide synthase, MMP2: matrix metalloproteinase 2,TIMP1: tissue inhibitor of matrix metalloproteinases, RT-PCR: reverse transcription-polymerase chain reaction.
Statistical analysis
Results are expressed as the mean±standard deviation. An unpaired two-tailed t-test and Mann-Whitney test were used, and a p<0.05 was considered statistically significant.
Results
Body and organ weight
The mean body weight decreased significantly in the MCT group and the bosentan group, compared to the control group on day 1, 5, 7, 14 and 28 (Table 2).
Table 2.
Changes in body weight, RV weight, LV and septum weight and RV/LV+septum, lung/body weight

*vs. control group p<0.05, †vs. control group p<0.01, ‡vs. MCT group p<0.05. MCT: monocrotaline, LV: left ventricle, RV: right ventricle, S: septum
The RV/(LV+septum) ratio significantly increased in the MCT group compared to the control group on day 7 (21.5±2.1% vs. 26.1±3.2%), day 14 (22.6±4.8% vs. 33.2±2.7%), day 28 (19.3±0.8% vs. 46.3±12.1%). The RV/(LV+septum) ratio increased in the bosentan group compared to the control group on day 7 (21.5±2.1% vs. 27.9±1.8%), day 14 (22.6±4.8% vs. 29.2±3.0%). RV/(LV+septum) ratio was also increased in the bosentan group on day 28 compared to the control group {37.0±6.4% (bosentan) vs. 19.3±0.8% (control)}, but was significantly reduced compared to the MCT group {37.0±6.4% (bosentan) vs. 46.3±12.1% (MCT)} (Table 2).
The lung to body weight ratio showed significant increase after MCT administration when compared to the control group on day 14 {0.80±0.08% (MCT) vs. 0.47±0.04% (control)}, day 28 {0.79±0.33% (MCT) vs. 0.44±0.01% (control)}. The lung to body weight ratio was also increased in the bosentan group on day 28 compared to the control group {0.67±0.08% (bosentan) vs. 0.44±0.01% (control)}, but was significantly reduced compared to the MCT group {0.67±0.08% (bosentan) vs. 0.79±0.33% (MCT)} (Table 2).
Mean right ventricular pressure
The mean RV pressure was significantly increased in MCT group compared to the control group on day 14 (22.0±3.7 mmHg vs. 12.3±3.8 mmHg), day 28 (29.5±3.5 mmHg vs. 12.0±2.8 mmHg) (p<0.05) (Table 3). After bosentan treatment, mean RV pressures significantly decreased from 22.0±3.7 mmHg to 16.3±2.5 mmHg on day 14 and from 29.5±3.5 mmHg to 15.0±3.0 mmHg on day 28 (Table 3).
Table 3.
Right ventricular pressure in each group (mmHg)

*vs. control group p<0.05, †vs. MCT group p<0.05. MCT: monocrotaline
Nitric oxide level in serum
Serum NO concentration increased in the MCT and in the bosentan groups on day 1 and 14 but the difference was not statistically significant. There were no significance in the changes of serum NO level on day 28 between the MCT and bosentan groups (Table 4).
Table 4.
Changes in serum nitric oxide level in each group (µmol/L)

p>0.05. MCT: monocrotaline
Matrix metalloproteinase-2 level in serum
Serum MMP-2 level was significantly increased in the MCT group compared to the control group on day 28 {254.21±36.27 ng/mL (MCT) vs. 198.37±10.57 ng/mL (control)} (Table 5). But there were no significant changes in MMP-2 level after bosentan treatment.
Table 5.
Changes in serum MMP-2 level in each group (ng/mL)

*vs. control group p<0.05. MMP: matrix metalloproteinase, MCT: monocrotaline
Microarray of the lung tissue
Genes which expression was more than twice beyond the control group were selected. NOS2, NOS3, MMP-2 and TIMP genes in MCT group were more than twice beyond the control group.
Expression of nitric oxide synthase 2 messenger ribonucleic acid in rat lung tissue
The expression of NOS2 messenger ribonucleic acid (mRNA) was not different between the two groups. Bosentan did not modify the pulmonary expression of the NOS2 gene (Fig. 2).
Fig. 2.

Gene expression of inducible nitric oxide synthase after bosentan treatment.
Expression of nitric oxide synthase 3 messenger ribonucleic acid in rat lung tissue
The expression of NOS3 mRNA was significantly increased in the MCT group, compared to the control group on day 28 (3.51±1.18 vs. 1±0.37).
The expression levels of NOS3 genes were significantly decreased after bosentan treatment on day 28 (2.16±1.37) (Fig. 3).
Fig. 3.

Gene expression of endothelial nitric oxide synthase after bosentan treatment. *p<0.05 significantly different from control.
Expression of matrix metalloproteinas-2 messenger ribonucleic acid in rat lung tissue
MMP-2 gene expressions were significantly increased in MCT-treated rats compared to the control group on day 5 (2.29±0.77 vs. 1±0.09) and day 28 (3.5±1.18 vs. 1±0.37).
The gene expressions of MMP-2 were significantly decreased after bosentan treatment on day 7 (1.14±0.31 vs. 0.52±0.17) and day 28 (3.51±1.18 vs. 1.55±1.14) (Fig. 4).
Fig. 4.

Gene expression of matrix metalloproteinase 2 after bosentan treatment. *p<0.05 significantly different from control.
Expression of tissue inhibitor of matrix metalloproteinases-1 messenger ribonucleic acid in rat lung tissue
The gene expression of TIMP-1 was significantly increased in MCT-treated rats compared to the control group on day 28 (7.09±2.28 vs. 1±0.67).
The gene expression of TIMP-1 was significantly decreased after bosentan treatment on day 28 (3.83±2.74) (Fig. 5).
Fig. 5.

Gene expression of tissue inhibitor of matrix metalloproteinases -1 after bosentan treatment. *p<0.05 significantly different from control.
Discussion
ECs are recognized as major regulators of vascular function, and endothelial dysfunction implies a multifaceted imbalance in EC production of vasoconstrictors versus vasodilators, activators versus inhibitors of SMC growth and migration, prothrombotic versus antithrombotic mediators, and pro-inflammatory versus anti-inflammatory signals.7)
We confirmed MCT-induced pulmonary hypertension by pathologic findings and ET-1 gene expression changes in our previous report.8) The predominant changes in pulmonary vasculature included increased number of intra-acinar muscular arteries from 1 day after MCT administration, and the development of medial thickening in the pulmonary arterioles in the MCT group from 1 week. A progressive increase in the ratio of RV to LV and septum was noted from 2 weeks in our previous study8) and the present study. These pathologic findings were accompanied by significant rise in pulmonary blood pressure and apparent RVH after MCT injection.4),9),10)
In the present study and previous study,11) ET-1 and MMP gene increased as early as 5 days in the course of MCT treatment, suggesting their contribution to progression of cardiopulmonary pathology in rats with MCT-induced pulmonary hypertension. Up-regulation of eNOS gene was noted significantly on day 28, suggesting that it is involved in the compensation mechanism.
Administration of bosentan reduced lung vascular hypertrophy and RVH. The gene expression of MMP significantly decreased on day 7 and day 28 after bosentan treatment. eNOS and TIMP genes significantly decreased on day 28 after bosentan treatment. NO is a potent endogenous vasodilator synthesized by the action of eNOS in lung and other tissues. It is well known that impaired production of NO can be one of the causes leading to pulmonary hypertension,12) but the role eNOS expression plays on pulmonary hypertension is yet to be determined.
Reduced expression of eNOS was observed in the lungs of patients with pulmonary hypertension compared to lungs of normal healthy subjects.13) On the other hand, others have demonstrated up-regulation of eNOS protein expression in pulmonary hypertensive subjects.1),14)
Pulmonary hypertension is induced by MCT experimentally in rats.15) MCT is a pyrrolizine alkaloid which selectively injures the vascular endothelium of the lung and induces pulmonary vasculitis.16) Muscularization and hypertrophy of pulmonary artery media lead to increased vascular resistance and pulmonary arterial pressure.17) MCT-induced pulmonary hypertension is associated with the development of RVH within weeks, depending on the dose of MCT and the age of the rats.17) In our study, rats were treated with sc injection of 60 mg/kg MCT to induce RVH.
In our study, eNOS mRNA significantly increased in MCT-induced pulmonary hypertension rats on day 28, consistent with other studies.18),19) Tyler et al.18) reported increased levels of eNOS mRNA in MCT-treated rats although eNOS protein levels decreased. Up-regulation of arterial eNOS in lungs of MCT treated rats was also demonstrated by Resta et al.19) although some showed that MCT did not alter the expression of eNOS mRNA in rat lungs.20)
The regulation of eNOS expression in other forms of pulmonary hypertension has been investigated in various animal models. eNOS mRNA expression and eNOS protein increased in the lungs of rats with increased pulmonary flow,21) which developed pulmonary artery medial hypertrophy prior to development of pulmonary hypertension.22) Similar results were shown by Black et al.23) who demonstrated early up-regulation of eNOS gene expression and activity in lambs with increased pulmonary blood flow and pulmonary hypertension. They suggested increased pulmonary blood flow resulted in increased shear stress which up-regulated eNOS expression, which in turn increased the production of NO to minimize vascular resistance in the early stages of pulmonary hypertension. However, other models of pulmonary hypertension have also shown up-regulation of eNOS independent of shear stress. eNOS expression was also up-regulated in hypoxia-induced pulmonary hypertension models.24) Le Cras et al.25) suggested that pulmonary blood oxygenation may play a factor in the up-regulation of eNOS in chronic hypoxia. Possible mechanisms proposed for the up-regulation of eNOS after chronic hypoxia are as follows: a result of polycythemia, hypertrophy and hyperplasia of ECs associated with pulmonary vascular remodeling, increased rates of transcription or increased eNOS mRNA stability.26) The hypothesis that hypoxia, rather than hemodynamic factors, contributes to the up-regulation of eNOS was supported by the study performed by Le Cras et al.25) where eNOS expression remained up-regulated in a setting of decreased blood flow due to left lung pulmonary artery stenosis. However, in a study of posthypoxic rats in recovery state from chronic hypoxia, eNOS expression maintained up-regulated. This suggests hypoxia may be the initiating factor for up-regulation of eNOS, but not the only factor that controls eNOS expression. From the increased expression of eNOS in MCT-induced pulmonary hypertension shown in our study and by the others,18),19) we suggest that regulation of eNOS expression is controlled by other factors than hypoxia. However, possible factors leading to the up-regulation of eNOS expression in MCT-induced pulmonary hypertension remains unclear.
Despite increased levels of eNOS mRNA after 28 days of MCT treatment, there was no significant change in serum NO level in the current study. As in our study, increased eNOS expression did not lead to increased NO production in other studies of pulmonary hypertension.27) Several mechanisms have been proposed for the discordance between NO production and increased eNOS expression in vitro and in vivo.28) They include impaired arginine uptake, decreased heat shock protein-90 levels, and decreased O2 tension. In addition, a limitation of this study is that it remain unclear whether the discordance between eNOS expression and serum NO level was due to reduced translation, or a blunted response at the post-translational level because eNOS protein expression or activity was not measured.
In a recent report by Mukhopadhyay et al.29) the intracellular vesicular trafficking theory was proposed as the mechanism of eNOS up-regulation in MCT-induced pulmonary hypertension. They demonstrated that MCT produced a Golgi blockade in pulmonary artery ECs, leading to sequestration of eNOS away from its functional caveolar location. This provided a novel insight in the mechanisms of the often reported reduction in pulmonary NO levels in experimental pulmonary hypertension, despite sustained eNOS protein levels.
In our study, the mean body weight was significantly decreased in MCT treated rats compared to the control group, which was a sign of heart failure comparable to cachexia in patients with chronic heart failure. The lung to body weight ratio increased in the MCT group compared to the control group, which indicated the presence of an extensive proliferative pulmonary response in the MCT treated rats. These findings were consistent with that shown by Marleen et al.16) The RV/(LV+septum) ratio, which can be thought of, as an index of RVH significantly increased in the MCT group from day 14. Thus, MCT treatment resulted in RVH and pulmonary arterial hypertension in our study as expected.
Bosentan is efficacious in experimental and clinical pulmonary hypertension.4) It is well-known that bosentan improves hemodynamics, increases exercise capacity, and decreases the rate of clinical worsening in patients with World Health Organization Class III or IV pulmonary arterial hypertension.
One of the important findings in our study was that bosentan treatment decreased the RV/(LV+septum) ratio of MCT treated rats on day 28. This means that bosentan treatment attenuated RVH in MCT-induced pulmonary hypertension model. These findings are consistent with previous data reported by other researchers, in which improved survival4) and decreased RVH were demonstrated in MCT-treated rats after bosentan administration.3),4) In other studies, bosentan was shown to improve endothelium-dependent relaxation in various animals4),30) and furthermore, to increase exercise capacity and improve hemodynamics in patients with pulmonary hypertension.3)
In this study, the gene expression of MMP-2 significantly decreased after bosentan treatment on day 7 and 28. The expression levels of NOS3 and TIMP-1 genes significantly decreased after bosentan treatment on day 28.
The inhibitory effects of bosentan on pulmonary hypertension influenced the gene expressions of NOS3, MMP and TIMP. Typical pulmonary artery lesions in idiopathic pulmonary hypertension include adventitial thickening, medial hypertrophy, SMC proliferation and ECM accumulation.6)
MMPs are matrix-degrading enzymes involved in ECM turnover, and in SMC and EC migration and proliferation.6) It has been shown that over-activity of MMPs in the lung is likely to contribute to pulmonary disease progression.31) There is also evidence to suggest that MMP expression is up-regulated in pulmonary arterial hypertension.19) Increased levels of MMP-2 were also reported in the plasma of patients with heart failure, ischemic and idiopathic DCM, implicating that activation of MMPs may play an important role in ventricular remodeling.17) In the present study, serum MMP-2 level was significantly increased in the MCT-treated rats that developed RVH after 28 days of MCT injection. This finding is consistent with a previous report by Umar et al.17) who showed activation of immunoreactive MMP-2 and MMP-9 in the remodeled RV myocardium of rats with pulmonary hypertension induced RV failure. Thus, our results support previous findings by other researchers5),6) that MMPs contribute to pulmonary arterial hypertension progression in animals.
NO and reactive oxygen species produce peroxynitrite, known to activate MMPs.17) Umar et al.17) demonstrated the stimulating effect of NO on the expression of MMP-2, and suggested that neuronal NOS activation may be responsible for increased immunoreactivity of MMP-2. We proposed that eNOS expression be responsible for the activation of MMPs, as increased eNOS expression after 28 days of MCT treatment coincided with the timing of significant elevation of serum MMP level in our study. Further investigation is necessary to assess this hypothesis and elucidate the underlying mechanisms.
In conclusion, MCT treated rats developed RV hypertrophy and pulmonary arterial hypertension. The eNOS , MMP-2 and TIMP-1 genes were up-regulated in MCT-treated rats, which suggests cardiac hypertrophy increases synthesis of ECM component. Treatment with bosentan attenuated RVH and the gene expressions of MMP-2, TIMP-1 and eNOS in the MCT treated rats. More invasive studies are needed to investigate the changes of eNOS and MMP gene expressions in a chronic MCT-induced pulmonary hypertension model.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education Science and Technology (KRF-2009-0064784).
Footnotes
The authors have no financial conflicts of interest.
References
- 1.Murata T, Sato K, Hori M, Ozaki H, Karaki H. Decreased endothelial nitric-oxide synthase (eNOS) activity resulting from abnormal interaction between eNOS and its regulatory proteins in hypoxia-induced pulmonary hypertension. J Biol Chem. 2002;277:44085–44092. doi: 10.1074/jbc.M205934200. [DOI] [PubMed] [Google Scholar]
- 2.Everett AD, Le Cras TD, Xue C, Johns RA. eNOS expression is not altered in pulmonary vascular remodeling due to increased pulmonary blood flow. Am J Physiol. 1998;274:L1058–L1065. doi: 10.1152/ajplung.1998.274.6.L1058. [DOI] [PubMed] [Google Scholar]
- 3.Channick RN, Simonneau G, Sitbon O, et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet. 2001;358:1119–1123. doi: 10.1016/S0140-6736(01)06250-X. [DOI] [PubMed] [Google Scholar]
- 4.Clozel M, Hess P, Rey M, Iglarz M, Binkert C, Qiu C. Bosentan, sildenafil, and their combination in the monocrotaline model of pulmonary hypertension in rats. Exp Biol Med (Maywood) 2006;231:967–973. [PubMed] [Google Scholar]
- 5.Frisdal E, Gest V, Vieillard-Baron A, et al. Gelatinase expression in pulmonary arteries during experimental pulmonary hypertension. Eur Respir J. 2001;18:838–845. doi: 10.1183/09031936.01.00084601. [DOI] [PubMed] [Google Scholar]
- 6.Lepetit H, Eddahibi S, Fadel E, et al. Smooth muscle cell matrix metalloproteinases in idiopathic pulmonary arterial hypertension. Eur Respir J. 2005;25:834–842. doi: 10.1183/09031936.05.00072504. [DOI] [PubMed] [Google Scholar]
- 7.Morrell NW, Adnot S, Archer SL, et al. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54(1 Suppl):S20–S31. doi: 10.1016/j.jacc.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lim KA, Shim JY, Cho SH, Kim KC, Han JJ, Hong YM. Effect of endothelin receptor blockade on monocrotaline-induced pulmonary hypertension in rats. Korean J Pediatr. 2009;52:689–695. [Google Scholar]
- 9.Miyauchi T, Yorikane R, Sakai S, et al. Contribution of endogenous endothelin-1 to the progression of cardiopulmonary alterations in rats with monocrotaline-induced pulmonary hypertension. Circ Res. 1993;73:887–897. doi: 10.1161/01.res.73.5.887. [DOI] [PubMed] [Google Scholar]
- 10.Itoh T, Nagaya N, Fujii T, et al. A combination of oral sildenafil and beraprost ameliorates pulmonary hypertension in rats. Am J Respir Crit Care Med. 2004;169:34–38. doi: 10.1164/rccm.200303-346OC. [DOI] [PubMed] [Google Scholar]
- 11.Lim KA, Kim KC, Cho MS, Lee BR, Kim HS, Hong YM. Gene expression of endothelin-1 and endothelin receptor A on monocrotaline-induced pulmonary hypertension in rats after bosentan treatment. Korean Circ J. 2010;40:459–464. doi: 10.4070/kcj.2010.40.9.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hampl V, Herget J. Role of nitric oxide in the pathogenesis of chronic pulmonary hypertension. Physiol Rev. 2000;80:1337–1372. doi: 10.1152/physrev.2000.80.4.1337. [DOI] [PubMed] [Google Scholar]
- 13.Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995;333:214–221. doi: 10.1056/NEJM199507273330403. [DOI] [PubMed] [Google Scholar]
- 14.Komai H, Naito Y, Aimi Y, Kimura H. Nitric oxide synthase expression in lungs of pulmonary hypertensive patients with heart disease. Cardiovasc Pathol. 2001;10:29–32. doi: 10.1016/s1054-8807(00)00063-6. [DOI] [PubMed] [Google Scholar]
- 15.Kim MY, Kim YK, Jung YW, Kim WT, Kwon TH, Lee DS. The protective effect of simvastatin on monocrotaline-induced pulmonary hypertension in rats. Korean Circ J. 2008;38:313–319. [Google Scholar]
- 16.Hessel MH, Steendijk P, den Adel B, Schutte CI, van der Laarse A. Characterization of right ventricular function after monocrotaline-induced pulmonary hypertension in the intact rat. Am J Physiol Heart Circ Physiol. 2006;291:H2424–H2430. doi: 10.1152/ajpheart.00369.2006. [DOI] [PubMed] [Google Scholar]
- 17.Umar S, Hessel M, Steendijk P, et al. Activation of signaling molecules and matrix metalloproteinases in right ventricular myocardium of rats with pulmonary hypertension. Pathol Res Pract. 2007;203:863–872. doi: 10.1016/j.prp.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 18.Tyler RC, Muramatsu M, Abman SH, et al. Variable expression of endothelial NO synthase in three forms of rat pulmonary hypertension. Am J Physiol. 1999;276:L297–L303. doi: 10.1152/ajplung.1999.276.2.L297. [DOI] [PubMed] [Google Scholar]
- 19.Resta TC, Gonzales RJ, Dail WG, Sanders TC, Walker BR. Selective upregulation of arterial endothelial nitric oxide synthase in pulmonary hypertension. Am J Physiol. 1997;272:H806–H813. doi: 10.1152/ajpheart.1997.272.2.H806. [DOI] [PubMed] [Google Scholar]
- 20.Mathew R, Yuan N, Rosenfeld L, Gewitz MH, Kumar A. Effects of monocrotaline on endothelial nitric oxide synthase expression and sulfhydryl levels in rat lungs. Heart Dis. 2002;4:152–158. doi: 10.1097/00132580-200205000-00004. [DOI] [PubMed] [Google Scholar]
- 21.Chou TF, Wu MS, Chien CT, Yu CC, Chen CF. Enhanced expression of nitric oxide synthase in the early stage after increased pulmonary blood flow in rats. Eur J Cardiothorac Surg. 2002;21:331–336. doi: 10.1016/s1010-7940(01)01129-0. [DOI] [PubMed] [Google Scholar]
- 22.Dai ZK, Tan MS, Chai CY, Chen IJ, Jeng AY, Wu JR. Effects of increased pulmonary flow on the expression of endothelial nitric oxide synthase and endothelin-1 in the rat. Clin Sci. 2002;103(Suppl 48):289S–293S. doi: 10.1042/CS103S289S. [DOI] [PubMed] [Google Scholar]
- 23.Black SM, Fineman JR, Steinhorn RH, Bristow J, Soifer SJ. Increased endothelial NOS in lambs with increased pulmonary blood flow and pulmonary hypertension. Am J Physiol. 1998;275:H1643–H1651. doi: 10.1152/ajpheart.1998.275.5.H1643. [DOI] [PubMed] [Google Scholar]
- 24.Xue C, Johns RA. Upregulation of nitric oxide synthase correlates temporally with onset of pulmonary vascular remodeling in the hypoxic rat. Hypertension. 1996;28:743–753. doi: 10.1161/01.hyp.28.5.743. [DOI] [PubMed] [Google Scholar]
- 25.Le Cras TD, Xue C, Rengasamy A, Johns RA. Chronic hypoxia upregulates endothelial and inducible NO synthase gene and protein expression in rat lung. Am J Physiol. 1996;270:L164–L170. doi: 10.1152/ajplung.1996.270.1.L164. [DOI] [PubMed] [Google Scholar]
- 26.Resta TC, Chicoine LG, Omdahl JL, Walker BR. Maintained upregulation of pulmonary eNOS gene and protein expression during recovery from chronic hypoxia. Am J Physiol. 1999;276:H699–H708. doi: 10.1152/ajpheart.1999.276.2.H699. [DOI] [PubMed] [Google Scholar]
- 27.Le Cras TD, McMurtry IF. Nitric oxide production in the hypoxic lung. Am J Physiol Lung Cell Mol Physiol. 2001;280:L575–L582. doi: 10.1152/ajplung.2001.280.4.L575. [DOI] [PubMed] [Google Scholar]
- 28.Fagan KA, Morrissey B, Fouty BW, et al. Upregulation of nitric oxide synthase in mice with severe hypoxia-induced pulmonary hypertension. Respir Res. 2001;2:306–313. doi: 10.1186/rr74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mukhopadhyay S, Xu F, Sehgal PB. Aberrant cytoplasmic sequestration of eNOS in endothelial cells after monocrotaline, hypoxia, and senescence: live-cell caveolar and cytoplasmic NO imaging. Am J Physiol Heart Circ Physiol. 2007;292:H1373–H1389. doi: 10.1152/ajpheart.00990.2006. [DOI] [PubMed] [Google Scholar]
- 30.Wang QD, Li XS, Pernow J. The nonpeptide endothelin receptor antagonist bosentan enhances myocardial recovery and endothelial function during reperfusion of the ischemic rat heart. J Cardiovasc Pharmacol. 1995;26(Suppl 3):S445–S447. [PubMed] [Google Scholar]
- 31.Winkler MK, Foldes JK, Bunn RC, Fowlkes JL. Implications for matrix metalloproteinases as modulators of pediatric lung disease. Am J Physiol Lung Cell Mol Physiol. 2003;284:L557–L565. doi: 10.1152/ajplung.00195.2002. [DOI] [PubMed] [Google Scholar]