

Abstract
Cytochrome c oxidase (CcO) is the terminal enzyme of the respiratory chain. This redox-driven proton pump catalyzes the four-electron reduction of molecular oxygen to water, one of the most fundamental processes in biology. Elucidation of the intermediate structures in the catalytic cycle is crucial for understanding both the mechanism of oxygen reduction and its coupling to proton pumping. Using CcO from Paracoccus denitrificans, we demonstrate that the artificial F state, classically generated by reaction with an excess of hydrogen peroxide, can be converted into a new P state (in contradiction to the conventional direction of the catalytic cycle) by addition of ammonia at pH 9. We suggest that ammonia coordinates directly to CuB in the binuclear active center in this P state and discuss the chemical structures of both oxoferryl intermediates F and P. Our results are compatible with a superoxide bound to CuB in the F state.
Keywords: artificial intermediates, catalase activity, electron paramagnetic resonance spectroscopy, optical difference spectroscopy
Cytochrome c oxidase (CcO) is a redox-driven proton pump. It catalyzes the four-electron reduction of molecular oxygen to water and couples this exergonic reaction to the generation of an electrochemical proton gradient across the membrane into which it is embedded (for recent reviews, see refs. 1–3). Electrons are delivered to the bimetallic CuA center by cytochrome c and transferred, via heme a, into the binuclear heme a3-CuB center where the reduction of oxygen takes place. The oxidized O state is successively reduced by two electrons to the R state. Molecular oxygen binds to the doubly reduced binuclear heme a3-CuB center resulting in the oxygen adduct A. Oxygen is fully reduced in one step by four electrons from 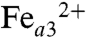 ,
,  , and (probably) tyrosine 280 (Paracoccus denitrificans numbering) to prevent the formation of reactive oxygen species (P state). Receipt of a third electron leads to the formation of the F state and the fourth electron closes the cycle by regenerating the O state (for a schematic representation of the catalytic cycle, see Fig. 1A).
, and (probably) tyrosine 280 (Paracoccus denitrificans numbering) to prevent the formation of reactive oxygen species (P state). Receipt of a third electron leads to the formation of the F state and the fourth electron closes the cycle by regenerating the O state (for a schematic representation of the catalytic cycle, see Fig. 1A).
Fig. 1.

Schematic catalytic cycle and artificial intermediates of CcO. (A) The fully oxidized O state is successively reduced to the E and then to the R state. The R state binds molecular oxygen resulting in the transient A state. Oxygen receives four electrons simultaneously from the protein resulting in complete reduction (P state). Receiving the third electron leads to building of the F state. The fourth electron completes the cycle by regenerating the O state. (B) The P state can be formed artificially by treating O state CcO with either one to five equivalents of hydrogen peroxide at pH 9 or with carbon monoxide in the presence of molecular oxygen at pH > 7. The F state can be reached by treatment with an excess (we used 500 equivalents) of H2O2. This F state can be transformed, as shown in this study, by treatment with ammonia at pH 9 to the previously undescribed PN state.
P and F intermediates were first described in reversed electron transfer studies using coupled mitochondria (4, 5) and observed later in the forward reaction of fully reduced CcO with oxygen (6, 7). These intermediates were characterized by difference UV/visible optical absorption spectroscopy and defined by their optical properties. P exhibits a distinct peak at 607 nm with a shoulder around 570 nm and F a maximum near 580 nm; the Soret region of both species is red-shifted compared to the oxidized enzyme. The redox states of P and F differ by 2 and 3 electrons, respectively, from the O state (8).
The chemical structures of these intermediates, especially the ligands of CuB, are difficult to identify because the active center is EPR spectroscopy silent due to a strong antiferromagnetic coupling (9). Heme a3 can still be visualized by its intense optical properties, but this does not apply to CuB.
There are different methods to generate artificial intermediates of CcO (summarized in Fig. 1B), which are related to but not necessarily the same as the physiological intermediates. In contrast to the physiological ones, these intermediates are relatively stable and can be characterized and investigated by various spectroscopic methods. A P state, termed PH, is generated when oxidized CcO reacts with one to five equivalents hydrogen peroxide at pH 9. This intermediate is characterized by a maximum around 610 nm in the difference absorption spectrum and a red-shifted Soret peak. It has been shown by Raman spectroscopy that PH like the F state is an oxoferryl (Fe4+ = O2-) intermediate (10, 11). Usage of EPR spectroscopy has shown that the formation of the PH state is accompanied by the appearance of a tyrosine radical (12), which is located on tyrosine 167 (Paracoccus denitrificans numbering) (13). In contrast to formation of the PH state at pH 9, the O state CcO is converted into the so-called F• state when reacting with equimolar amounts of hydrogen peroxide at pH 6 (14, 15). This F• state shows a maximum at 576 nm in the difference spectrum similar to the F state; however, a radical signal assigned to Y167 is present in the EPR spectrum as for the PH state.
A second possibility to form an artificial P state is the incubation of O state CcO with carbon monoxide and oxygen at high pH (16, 17). This PCO state is formed with high yield and does not possess an EPR-detectable amino acid radical (18). But like the other P states, the PCO state shows a difference absorption maximum at 609 nm.
Addition of an excess of H2O2 generates the F state, which is characterized by a maximum around 580 nm in the difference absorption spectrum. It is generally believed that a second molecule of hydrogen peroxide provides an electron to the PH state converting it into the F state (19, 20). However, attempts to detect the expected superoxide radical formed from hydrogen peroxide during the formation of the F state in an appropriate amount failed (18, 21).
Considering the fact that the P state as well as the F state are oxoferryl intermediates, differences in spectral properties may be caused by different CuB ligands and accompanying differences in coordination geometry and charge distribution affecting heme a3 and heme a. The amino acid radical of the PH and the F• states seems not to have a distinct influence on the spectral properties because the radical free PCO state shows a UV/visible absorption spectrum very similar to the PH state spectrum.
In this paper, we report the observation that the F state generated by addition of an excess of hydrogen peroxide can be converted, in contradiction to the usual direction of the catalytic cycle, to another P state, which we term the PN state, by the addition of ammonia at high pH. This up to now undescribed artificial intermediate is characterized by UV/visible absorption spectroscopy as well as with EPR spectroscopy. Implications for the chemical structures of F and P intermediates are discussed.
Results
To gain further information regarding the properties of the oxidized state and the intermediates formed in the catalytic cycle, different artificial intermediates were generated and studied by UV/visible absorption spectroscopy and EPR spectroscopy.
O state CcO had a Soret absorption maximum at 426 nm; the α-band had its maximum at 600 nm.
Characterization of CcO Intermediates by Absorption spectroscopy.
To form the PH state, five equivalents of hydrogen peroxide (providing formally molecular oxygen, two electrons, and two protons) were added to CcO at pH 9. A maximum in the difference absorption spectrum typically for P state formation is located at 610 nm (Fig. 2, blue curve). CO is oxidized by CcO at high pH (22) providing two reduction equivalents (CO + 2OH- → 2e- + CO2 + H2O) for P state generation. The absorption maximum of the PCO state is slightly blue-shifted to 609 nm (Fig. 2, violet curve), compared to that of the PH state. The Soret bands of both P state species are red-shifted compared to O state CcO; however, these shifts are slightly different for each species.
Fig. 2.

Difference absorption spectra (induced states minus O state) of artificial CcO intermediates at pH 9. The PH (blue curve) and F state (red curve) were prepared by addition of 5 and 500 equivalents hydrogen peroxide, respectively, and the PCO state (violet curve) was induced by aeration of the O state with CO gas. Treatment of the F state with ammonia resulted in formation of the PN state (green curve). The characteristic difference maxima of the α-band (600 nm in the O state) indicate the formation of an oxoferryl state. Soret bands (O state: 426 nm) are red-shifted compared to the O state noticeably by the minima and maxima in the Soret region. Absorption differences in the Soret region of the F and PN states are very similar.
After addition of a 500-fold molar excess of hydrogen peroxide to CcO in the PH or PCO states, the 610/609 nm peak disappeared and a new broad maximum showed up at 582 nm, indicating formation of the F state at pH 9 (Fig. 2, red curve, and Fig. 3A, red curve). In 1963 it was found that CcO also possesses catalase activity (23). The hydrogen peroxide derived F state is the most stable intermediate in which CcO catalyzes the continuous dismutation of H2O2 to water and oxygen. Oxygen formation due to the dismutation of H2O2 was measured with a Clark-type oxygen electrode: 2 μM CcO produced 26.5( ± 1.0) μM O2 min-1 at pH 9.
Fig. 3.

α-Band difference spectra (induced states minus O state) of artificial CcO intermediates series. (A and B) 6–8 μM of fully oxidized solubilized CcO in 50 mM glycine pH 9.0 or 50 mM MES pH 6.0 and 0.05% lauryl maltoside (LM) were successively mixed with 5 equivalents of H2O2 (blue curve) to induce the PH state (pH 9.0) or the F• state (pH 6.0), respectively, followed by addition of 500 equivalents of H2O2 (red curve) to form the F state, which was transformed with 20 mM ammonia to the new PN state (green curve) at pH 9.0. At pH 6.0, no reaction with ammonia was observed until the pH was raised to pH 9 (B, cyan curve). Lowering the pH from 9 to 6 resulted in reverse formation of the F/F• state (A, cyan curve). (C and D) Time-dependent development of 6–8 μM CcO in the PN (1∶500 H2O2 and 20 mM ammonia) and the F (1∶500 H2O2) state after catalase addition.
When ammonium sulfate/ammonia was added to CcO in the F state at pH 9, the 582-nm maximum disappeared and a difference absorption maximum appeared at 612 nm typical for a P state with a broad shoulder at 590 nm (Fig. 2, green curve, and Fig. 3A, green curve). Just as for the other artificial P states, the Soret band is red-shifted. Measurement of oxygen production shows that the addition of ammonia at pH 9 does not inhibit but actually enhances the decomposition of H2O2 as compared to the F state. The catalase activity of CcO increases at 2 μM CcO more than twofold to 57.9( ± 3.6) μM O2 min-1 after addition of ammonia, suggesting that ammonia binds to CuB to form a labile complex, whereas the catalase reaction takes place at heme a3.
When the pH was lowered from pH 9 to pH 6, the difference absorption maximum of the new PN state shifted back to 578 nm (Fig. 3A, cyan curve) indicating the formation of the initial F state (now at pH 6) as hydrogen peroxide is still present. If ammonia is coordinated to CuB at high pH, it will be protonated and no longer bind to CuB at low pH.
The treatment of CcO with five equivalents of hydrogen peroxide at pH 6 did not result in formation of the PH state but in formation of the F• state with a maximum in difference absorption at 577 nm (Fig. 3B, blue curve). Addition of excess H2O2 shifted this broad difference absorption peak by ∼1 nm to 578 nm (Fig. 3B, red curve). The difference absorption maximum of this F state at pH 6 is shifted by approximately 4 nm to the blue as compared to pH 9. In addition the catalase activity of CcO at pH 6 is much lower, namely 15.1( ± 0.5) μm O2 min-1 for 2 μM CcO, and even slows down with time. Addition of ammonium sulfate/ammonia at pH 6 did not result in the formation of the PN state and left the spectrum unaffected (Fig. 3B, green curve). Ammonia also had no effect on the catalase reaction at pH 6. But raising the pH to 9 induced PN state formation (Fig. 3B, cyan curve). This spectrum has a more prominent difference absorption maximum at 612 nm as compared to the PN state directly prepared at pH 9 and a smaller admixture of the F state is apparent by the absence of the broad underlying feature around 590 nm.
Provided that ammonia is coordinated to CuB, one would expect a decreased oxidase activity of CcO complementary to the enhanced catalase activity of CcO in the presence of ammonia at pH 9. Indeed the oxidase activity decreased by 22% from 401( ± 4) e-/s to 313( ± 7) e-/s in the presence of ammonia.
The measurement of oxygen production by CcO in the PN state and also the prominent shoulder in the UV/visible absorption spectrum indicate that this state still contains some CcO in the F state as long as there is hydrogen peroxide present. Adding catalase destroys the excess hydrogen peroxide thereby arresting the catalase reaction of CcO (Fig. 3C). The notable shoulder of the 612-nm maximum in the difference absorption spectrum decreases. The 612-nm PN state difference maximum itself also decreases but shifts to the blue by 4 nm within approximately 30 min and increases in intensity again. The resulting spectrum is very similar to the difference spectrum of the PH state except that the maximum is located at 608 nm instead of 610 nm. Decreasing the pH from 9 to 6 before the 612-nm maximum has moved to 608 nm results in a spectrum similar to the F state as shown in Fig. 3A (cyan curve), but lowering the pH to 6 after the shift of the maximum from 612 nm to 608 nm has no effect on the position of the difference absorption maximum; this shifted PN state remains stable. The addition of catalase to the F state without ammonia causes the decay of the 580-nm peak and results in an absorption spectrum that is similar to the spectrum of the O state but shows an increased absorption at 620–640 nm (Fig. 3D). This “degraded F state” reacts with ammonia directly forming the shifted PN state with a maximum at 608 nm, which also forms during the catalase-induced decay of the PN state (Fig. 3C).
Formation of a PN state from the F state is also observed after addition of other small amines such as hydrazine (which is oxidized to diazene by hydrogen peroxide) and methylamine. Therefore we named this previously undescribed artificial intermediate the PN state, with N standing for nitrogen.
EPR Studies of CcO.
For all EPR experiments, cells were grown in a manganese-free medium to prevent signal overlap from the Mn2+ center of CcO. Manganese is replaced by magnesium without any effect on the activity of CcO or optical properties of the artificial intermediates (12). Fig. 4A shows the EPR spectrum of the pulsed O state (black curve) at pH 9. The binuclear heme a3-CuB center is magnetically coupled and therefore EPR silent; hence only EPR signals from the dinuclear CuA and the low-spin heme a center are observed.
Fig. 4.

EPR spectra of CcO artificial intermediates. Just as for the absorption difference spectra CcO samples were successively mixed with H2O2 and NH3 at pH 9 and pH 6 to prepare different intermediates. (A) EPR spectra of CcO intermediates recorded at 20 K, 2 mW, and a modulation amplitude of 1 mT at 9.6 GHz microwave frequency. The O state CcO (200 μM in 50 mM glycine pH 9 and 0.05% LM) is shown as the black curve. The PH (blue solid curve), prepared at pH 9, and F• (blue dotted curve), prepared at pH 6, show an additional radical signal. The F state (red curve) shows no radical at either pH (pH 9 is shown). Both catalase and ammonia in any order were added to prepare the PN state (green curve) from the F state, resulting in the reappearance of the Y167 radical at pH 9, which vanishes after lowering the pH to pH 6. Addition of catalase or ammonia individually resulted in no change from the F state. (B) EPR spectra of CcO intermediates were recorded at 20 K, 2 mW, and a modulation amplitude of 0.4 mT at 9.6 GHz microwave frequency. These spectra are difference spectra (spectrum of respective intermediate minus O state spectrum) to study the radical signal in more detail. The PH state (blue solid curve), prepared at pH 9, and F• state (blue dotted curve), prepared at pH 6, contain a radical signal that is formed again in the PN state prepared from the F state at pH 9 with ammonia and catalase (green solid curve) but not at pH 6 (green dotted curve). When the PN state is prepared at pH 6 and the pH is then raised to pH 9, the same radical signal does reappear (cyan curve).
In addition to these EPR signals, a narrow g ∼ 2 radical signal is observed after addition of one to five equivalents of H2O2 at pH 9 (PH state, Fig. 4A, blue curve) and pH 6 (F• state, Fig. 4A, dotted blue curve), respectively. This signal has been identified previously through isotope labeling and site-directed mutagenesis and is attributed to a tyrosyl radical located on residue Y167 (12, 13, 24). The yield of this radical signal appears to be pH dependent and is always more prominent in the F• state at pH 6 than in the PH state at pH 9. The yield in the F• state has previously been determined to be ∼20% (as compared to CuA) (12).
In the F state, prepared with an excess (500-fold) of hydrogen peroxide, the Y167 radical species is no longer present (Fig. 4A, red curve); however, a very small yield (< 0.5%) of a different much narrower, unresolved EPR signal was observed as has been seen previously (25).
Upon treatment of the F state with ammonia/ammonium sulfate to form the PN state (equivalent to that detected optically) at pH 9, no reappearance of a signal attributable to the Y167 radical was observed in contrast to the PH or F• states. However, upon further addition of catalase to terminate CcO’s underlying catalase reaction, this amino acid radical signal does reappear (Fig. 4A, green curve). Reversing the order in which ammonia and catalase are added to the F state does not alter the fact that only once both are added does the signal attributed to Y167 reappear. Close inspection of this amino acid radical signal formed after ammonia/catalase addition reveals that the overall linewidth and line shape as well as the partially resolved hyperfine structure are very similar to the signal observed in both the PH and F• states. The very slight variations in the intensities of the resolved hyperfine structure may be caused by slight variations in pH or the relative orientation of the tyrosine head group (possibly due to a slightly different conformation or environment due to the ammonia bound in the active center). However, this unique EPR signature clearly suggests that it originates from the residue Y167.
The EPR spectrum of the PCO state of P. denitrificans CcO at pH 9 shows no induced amino acid radical species as was also shown for bovine CcO (18, 26, 27).
Discussion
Our experiments clearly demonstrate the formation of a previously undescribed artificial P state from the hydrogen peroxide derived F state of CcO in the presence of ammonia, with a difference absorption maximum at 612 nm and a red-shifted Soret absorption peak, both characteristic for a P state. This reaction is not restricted to ammonia but is also possible, albeit with lower yield, by using other small amines such as methylamine and hydrazine (oxidized to diazene by hydrogen peroxide). Hence the crucial point for this F to P transition appears to be the amino group.
The formation of this previously undescribed P-like state from the F state appears to be not compatible with the usual forward direction of the catalytic cycle. Wikström (4) has shown previously that the reaction of CcO could be partially reversed in mitochondria by creating an electrochemical proton gradient by reversing the ATP-synthase reaction at high pH. However, the reaction conditions used here are not sufficient to explain the reversal of the natural CcO reaction. Also, the intermediates investigated here are artificial intermediates showing similar UV/visible spectroscopic properties as the natural ones.
In order to understand the formation of the PN state one has to precisely know the original F state. This artificial F state is observed under conditions when CcO catalyzes the dismutation of H2O2. Assuming that the mechanism of hydrogen peroxide decomposition by CcO is similar to the mechanism in heme-containing catalases, one can expect that the dismutation of hydrogen peroxide in CcO is catalyzed by the heme moiety of the active center. Thus this artificial F state is not a static but a dynamic state that due to the catalase activity leads to permanent oxygen formation. The broad maximum in the difference absorption spectra (Fig. 3) also indicates a mixture of different intermediates in the catalase cycle of CcO whereby the most stable intermediate of the catalase reaction dominates. By comparison with the mechanism of human catalase (28), the intermediate with the longest lifetime is likely to be the oxoferryl intermediate (compound I), which is the state formed when the initial hydrogen peroxide molecule is decomposed and the enzyme is ready to take a subsequent molecule of H2O2. CcO is lacking the asparagine and histidine residues of catalase that ensure H2O2 supply to the active site, thus possibly prolonging the lifetime of this intermediate. The fourth ligand of CuB (besides the three histidine residues) may be unaffected by this catalase reaction. The properties of the H2O2-derived F state at pH 6 differ from those at pH 9: The difference absorption maximum is shifted by 4 nm to the blue and the catalase activity is reduced and even slows down during the reaction. This observation indicates that the protonation state of the protein has an influence on this catalase-like reaction.
The continuing and even enhanced decomposition of hydrogen peroxide in the presence of ammonia (PN state) demonstrates that the enzyme still has the chemical properties of the artificial F state, although the visible absorption spectrum is shifted due to the interaction of ammonia with CuB. There are several possibilities how ammonia could enhance the catalase reaction; ligation to CuB might alter the water network in and/or near the active center leading to an improved hydrogen peroxide supply for the catalase reaction, or it might accelerate the catalase reaction by interacting with hydrogen peroxide at the active center and polarize it in a favorable way for the reaction.
The idea of ammonia interacting directly with CuB is also supported by the observation that the cytochrome c oxidase activity of the enzyme is lowered by the presence of ammonia because CuB is required to catalyze the reduction of molecular oxygen in the natural enzymatic cycle of CcO.
The formation of PN is strongly pH dependent and there is no PN state formation at pH < 8 (pKa of ammonium in aqueous solution is 9.2). This result indicates that it is ammonia and not the ammonium ion that interacts with CcO. Furthermore, by shifting the pH to 6 it is also revealed that the reaction of the F to the PN state is fully reversible suggesting binding and unbinding of ammonia without any further reaction of ammonia with CcO.
After addition of catalase, the PN state initially exhibits a decrease in difference absorption with time, until the 612-nm absorption shifts by 4 nm and increases again after ca. 30 min, resulting in formation of another P-like state with a difference absorption maximum at 608 nm and a shoulder at around 570 nm. This response of the PN state to the addition of catalase (Fig. 3C), i.e., the decay of the PN state analogous to the F state, indicates that the optical manifestation of ammonia coordination to CuB is dependent on the oxoferryl ligand at heme a3.
The F state alone decomposes after catalase addition to a state that exhibits a spectrum similar to the O state as has been described by Vygodina and Konstantinov (20, 29) previously. Nevertheless, this decomposed F state differs from the O state by the increased absorption around 635 nm pointing eventually to a reduction of CuB (30). This “reduced F state” is still able to react with ammonia albeit in a different way and the shifted PN state (608 nm) forms, although a clear explanation for this finding is still lacking, and further experiments are necessary to explain this observation.
The decline of the F-state admixture to the PN state appears to be slower than the decay of the F state alone. Ammonia might stabilize the oxoferryl group at heme a3 due to a possible interaction of ammonia and the oxoferryl ligand. This feature of ammonia potentially bridging the metal ions in the active center is discussed in more detail below as a possible reason for the differences of P and F state spectra.
The optical characterization of this previously undescribed PN state is complemented by EPR spectroscopy. In the F state, a narrow, unresolved EPR signal is observed. However, its yield (< 0.5%) is much lower than that of the optically detectable intermediate. This observation suggests that this radical is not related to the F state but may be the result of a side reaction of CcO in the presence of such high hydrogen peroxide concentrations, as has been shown previously. This signal has been termed the “6-ms radical” and was attributed to an organic radical or a main-chain radical (25).
Formation of the PN state by addition of ammonia to the F state does not result in stable tyrosyl radical formation in contrast to the PH state but, because the PN state is a mixture with the dynamic F state, it might not be possible to form a stable radical species as long as there is excess hydrogen peroxide present. The P as well as the F state of CcO are oxoferryl intermediates (31, 32). In contrast to the PH state neither the PCO nor the PN state contains an EPR-detectable radical. Therefore the difference in absorption properties of the P and F states does not result from the presence of a radical species in or near the active center.
P and F states were originally defined on the basis of their optical signatures in the UV/visible spectral range (5), and the P state was believed to represent the two-electron reduced state after the reaction with molecular oxygen, whereas the F state was supposed to be the oxoferryl state after the third reduction step. This scheme had to be modified because a three-electron reduced P state was observed during the reaction of fully reduced four-electron CcO with molecular oxygen (33), and by the observation that the PH state is converted to the F• state upon lowering the pH (15). Here we provide additional evidence that the UV/visible spectra do not reflect the overall redox state of the enzyme as a simple and facile correlation because the presence of a previously undescribed ammonia ligand, presumably at CuB, can cause the difference in absorption properties of the F and PN states without comprising any redox reaction or radical formation. It is obvious that it is not understood which structural conditions lead to the typical P state difference absorption maximum at approximately 610 nm. This spectral property is apparently not inevitably connected to the two-electron reduced form of CcO that reacted with molecular oxygen resulting in an oxoferryl group at heme a3. There is also another example of an artificial intermediate that resembles the spectrum of a physiological intermediate; the mixed valence state with CO bound to reduced heme a3 has a difference absorption maximum at 592 nm, whereas the oxygen adduct of reduced CcO (intermediate A) also shows a difference maximum of the α-band at 591 nm (34). Thus the CO ligand appears to cause the same spectral shifts as the physiological oxygen ligand.
What can be learned about the fourth CuB ligand in the P and F states from these experiments? We propose that ammonia has to replace this CuB ligand present in the F state (and is able to replace it in contrast to other ligands present in other intermediates) as well as mimicking the properties of the ligand that is present in the P state. It is generally assumed that a water molecule is the fourth CuB ligand in the F state, and, when compared to water, ammonia is a more nucleophilic ligand with a free electron pair resembling the properties of a hydroxyl ion, which has been expected to be the CuB ligand in the P state. Thus it might be possible that the neutral ammonia is able to replace a water molecule bound to CuB in the F state and resembles with its nucleophilic character the properties of a hydroxyl ion bound to CuB in the P state.
However, this interpretation does not explain the reappearance of the tyrosine radical observed upon treating CcO in the F state with ammonia and catalase. But which ligand would require another electron upon replacement by ammonia? And which ligand could explain the decomposition of the F state after adding catalase? Electron densities observed by X-ray crystallography of the CcO from bovine heart (35), from Rhodobacter sphaeroides (36), and from P. denitrificans (37) show the existence of a peroxide bridging the metal ions in the oxidized form of CcOs. Because CcO in the F state contains one electron less than in the O state, the presence of a superoxide as the fourth CuB ligand in the F state makes good sense: This superoxide would be able to form a peroxide bridge between the heme a3 iron and CuB once the oxoferryl oxygen has been converted to a water molecule and released upon the final fourth reduction step in the catalytic cycle of CcO. The removal of a possibly exchangeable superoxide as the fourth ligand in the F state as hydrogen peroxide would require another electron, which could be provided by a tyrosine residue. Exactly such a sequence of events would explain the reappearance of the tyrosine radical observed upon treating CcO in the F state with ammonia and catalase, provided that there is a rapid exchange of ammonia and peroxide as CuB ligands. The reappearance of the radical signal only after addition of both ammonia and catalase would be explained, too, because there would be no stable radical as long as there is peroxide present in the sample. The question, which ligand in the P state is mimicked by ammonia and could be transformed to a superoxide ligand in the F state, remains. However, one has to keep in mind that the events upon addition of catalase are not understood and that further experiments are necessary.
Very recently Muramoto et al. (38) used “O2 analogues” such as CO, NO, and CN- as artificial ligands to probe structural changes in the active center of bovine CcO. From their results, they deduced a structure for the P state where a water molecule bridges the oxoferryl oxygen atom at the heme a3 iron and the oxygen atom of the tyrosine residue (Y244 in bovine CcO and Y280 in P. denitrificans), which is covalently linked to one of the CuB coordinating histidines. Furthermore, the oxygen atom of Y244 is hydrogen-bonded to the OH group of the hydroxyethylfarnesyl group of heme a3. Our results clearly suggest that the ammonia ligand at CuB can cause the optical differences between the P and F states. The influence of the CuB ligand on heme a3 might be increased by a connection of the metal complexes in the active center, which is absent in the F state. It might be possible that ammonia bound to CuB either interacts with the oxoferryl ligand of heme a3 directly or promotes the binding of a water molecule in the active center mimicking the connection of the metal ions in the natural intermediate. This bridge between the metal complexes in the active center might be the reason for the UV/visible spectral shift of the α-band in the P state compared to the F state although both states possess an oxoferryl ligated heme a3.
Materials and Methods
All experiments were performed with CcO from P. denitrificans. CcO was purified by affinity chromatography in a chloride-free buffer using strep-tagged Fv fragments of a monoclonal antibody as described previously (39).
The preparation of artificial intermediates and measurement conditions for UV/visible spectroscopy, activity measurements, and EPR experiments are given in Results and in the legends of Figs. 2–4.
More details related to sample preparation, UV/visible and EPR spectroscopy, and activity measurements are provided in SI Text.
Supplementary Material
Acknowledgments.
We are grateful to Hannelore Müller and Arunas Damijonaitis for excellent technical assistance and to Myles Cheesman (University of East Anglia) for critical discussions. This work was supported by the Deutsche Forschungsgemeinschaft (SFB 472), the Center for Biomolecular Magnetic Resonance, the Max-Planck-Gesellschaft, the Cluster of Excellence “Macromolecular Complexes” Frankfurt, European Cooperation in Science and Technology Action CM0902, and the Wellcome Trust. F.M. is a Wolfson Research Merit Award holder of the Royal Society, which is gratefully acknowledged for its financial support.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1100950108/-/DCSupplemental.
References
- 1.Brzezinski P, Gennis RB. Cytochrome c oxidase: Exciting progress and remaining mysteries. J Bioenerg Biomembr. 2008;40:521–531. doi: 10.1007/s10863-008-9181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gennis RB. Coupled proton and electron transfer reactions in cytochrome oxidase. Front Biosci. 2004;9:581–591. doi: 10.2741/1237. [DOI] [PubMed] [Google Scholar]
- 3.Brzezinski P, Larsson G. Redox-driven proton pumping by heme-copper oxidases. Biochim Biophys Acta. 2003;1605:1–13. doi: 10.1016/s0005-2728(03)00079-3. [DOI] [PubMed] [Google Scholar]
- 4.Wikström M. Energy-dependent reversal of the cytochrome oxidase reaction. Proc Natl Acad Sci USA. 1981;78:4051–4054. doi: 10.1073/pnas.78.7.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wikström M, Morgan JE. The dioxygen cycle. J Biol Chem. 1992;267:10266–10273. [PubMed] [Google Scholar]
- 6.Morgan JE, Verkhovsky MI, Wikström M. Observation and assignment of peroxy and ferryl intermediates in the reduction of dioxygen to water by cytochrome c oxidase. Biochemistry. 1996;35:12235–12240. doi: 10.1021/bi961634e. [DOI] [PubMed] [Google Scholar]
- 7.Sucheta A, Georgiadis KE, Einarsdottir O. Mechanism of cytochrome c oxidase-catalyzed reduction of dioxygen to water: Evidence for peroxy and ferryl intermediates at room temperature. Biochemistry. 1997;36:554–565. doi: 10.1021/bi962422k. [DOI] [PubMed] [Google Scholar]
- 8.Verkhovsky MI, Morgan JE, Wikström M. Redox transitions between oxygen intermediates in cytochrome c oxidase. Proc Natl Acad Sci USA. 1996;93:12235–12239. doi: 10.1073/pnas.93.22.12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tweedle MF, Wilson LJ, Garcia-Iñiguez L, Babcock GT, Palmer G. Electronic state of heme in cytochrome oxidase III. J Biol Chem. 1978;253:8065–8071. [PubMed] [Google Scholar]
- 10.Proshlyakov DA, Ogura T, Shinzawa-Itoh K, Yoshikawa S, Kitagawa T. Microcirculating system for simultaneous determination of Raman and absorption spectra of enzymatic reaction intermediates and its application to the reaction of cytochrome c oxidase with hydrogen peroxide. Biochemistry. 1996;35:76–82. doi: 10.1021/bi9511705. [DOI] [PubMed] [Google Scholar]
- 11.Proshlyakov DA, Ogura T, Shinzawa-Itoh K, Yoshikawa S, Kitagawa T. Resonance Raman/absorption characterization of the oxo intermediates of cytochrome c oxidase generated in its reaction with hydrogen peroxide: pH and H2O2 concentration dependence. Biochemistry. 1996;35:8580–8586. doi: 10.1021/bi952096t. [DOI] [PubMed] [Google Scholar]
- 12.MacMillan F, Kannt A, Behr J, Prisner T, Michel H. Direct evidence for a tyrosine radical in the reaction of cytochrome c oxidase with hydrogen peroxide. Biochemistry. 1999;38:9179–9184. doi: 10.1021/bi9911987. [DOI] [PubMed] [Google Scholar]
- 13.Budiman K, et al. Tyrosine 167: The origin of the radical species observed in the reaction of cytochrome c oxidase with hydrogen peroxide in Paracoccus denitrificans. Biochemistry. 2004;43:11709–11716. doi: 10.1021/bi048898i. [DOI] [PubMed] [Google Scholar]
- 14.Brittain T, Little RH, Greenwood C, Watmough NJ. The reaction of Escherichia coli cytochrome bo with H2O2: Evidence for the formation of an oxyferryl species by two distinct routes. FEBS Lett. 1996;399:21–25. doi: 10.1016/s0014-5793(96)01253-7. [DOI] [PubMed] [Google Scholar]
- 15.Jünemann S, Heathcote P, Rich PR. The reactions of hydrogen peroxide with bovine cytochrome c oxidase. Biochim Biophys Acta. 2000;1456:56–66. doi: 10.1016/s0005-2728(99)00105-x. [DOI] [PubMed] [Google Scholar]
- 16.Nicholls P. A new carbon monoxide-induced complex of cytochrome c oxidase. Biochem J. 1978;175:1147–1150. doi: 10.1042/bj1751147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nicholls P, Chanady GA. Interactions of cytochrome aa3 with oxygen and carbon monoxide: The role of the 607 nm complex. Biochim Biophys Acta. 1981;634:256–265. doi: 10.1016/0005-2728(81)90144-4. [DOI] [PubMed] [Google Scholar]
- 18.Fabian M, Palmer G. The interaction of cytochrome oxidase with hydrogen peroxide: The relationship of compounds P and F. Biochemistry. 1995;34:13802–13810. doi: 10.1021/bi00042a011. [DOI] [PubMed] [Google Scholar]
- 19.Witt SN, Chan SI. Evidence for a ferryl Fea3 in oxygenated cytochrome c oxidase. J Biol Chem. 1987;262:1446–1448. [PubMed] [Google Scholar]
- 20.Vygodina TV, Konstantinov AA. H2O2-induced conversion of cytochrome-c-oxidase peroxy complex to oxoferryl state. Ann NY Acad Sci. 1988;550:124–138. doi: 10.1111/j.1749-6632.1988.tb35329.x. [DOI] [PubMed] [Google Scholar]
- 21.Ksenzenko MY, Vygodina TV, Berka V, Ruuge EK, Konstantinov AA. Cytochrome oxidase-catalyzed superoxide generation from hydrogen peroxide. FEBS Lett. 1992;297:63–66. doi: 10.1016/0014-5793(92)80328-e. [DOI] [PubMed] [Google Scholar]
- 22.Tzagoloff A, Wharton DC. Studies on the electron transfer system. J Biol Chem. 1965;240:2628–2633. [PubMed] [Google Scholar]
- 23.Orii Y, Okunuki K. Studies on cytochrome a. J Biochem. 1963;54:207–213. doi: 10.1093/oxfordjournals.jbchem.a127773. [DOI] [PubMed] [Google Scholar]
- 24.Svistunenko DA, Wilson MT, Cooper CE. Tryptophan or tyrosine? On the nature of the amino acid radical formed following hydrogen peroxide treatment of cytochrome c oxidase. Biochim Biophys Acta. 2004;1655:372–380. doi: 10.1016/j.bbabio.2003.06.006. [DOI] [PubMed] [Google Scholar]
- 25.Wiertz FGM, Richter O-MH, Ludwig B, de Vries S. Kinetic resolution of a tryptophan-radical intermediate in the reaction cycle of Paracoccus denitrificans cytrochrome c oxidase. J Biol Chem. 2007;282:31580–31591. doi: 10.1074/jbc.M705520200. [DOI] [PubMed] [Google Scholar]
- 26.Clore GM, Andréasson L-E, Karlsson B, Aasa R, Malmström BG. Characterization of the intermediates in the reaction of mixed-valence-state soluble cytochrome oxidase with oxygen at low temperatures by optical and electron-paramagnetic-resonance spectroscopy. Biochem J. 1980;185:155–167. doi: 10.1042/bj1850155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rich PR, Rigby SEJ, Heathcote P. Radicals associated with the catalytic intermediates of bovine cytochrome c oxidase. Biochim Biophys Acta. 2002;1554:137–146. doi: 10.1016/s0005-2728(02)00228-1. [DOI] [PubMed] [Google Scholar]
- 28.Putman CD, Arvai AS, Bourne Y, Tainer JA. Active and inhibited human catalase structures: Ligand and NADPH binding and catalytic mechanism. J Mol Biol. 2000;296:295–309. doi: 10.1006/jmbi.1999.3458. [DOI] [PubMed] [Google Scholar]
- 29.Vygodina T, Konstantinov AA. Evidence for two H2O2-binding sites in ferric cytochrome c oxidase. FEBS Lett. 1987;219:387–392. doi: 10.1016/0014-5793(87)80258-2. [DOI] [PubMed] [Google Scholar]
- 30.Mitchell R, Mitchell P, Rich PR. The assignment of the 655 nm spectral band of cytochrome oxidase. FEBS Lett. 1991;280:321–324. doi: 10.1016/0014-5793(91)80321-s. [DOI] [PubMed] [Google Scholar]
- 31.Kitagawa T, Ogura T. Oxygen activation mechanism at the binuclear site of heme-copper oxidase superfamily as revealed by time-resolved resonance Raman spectroscopy. In: Karlin KD, editor. Progress in Inorganic Chemistry. Vol 45. New York: Wiley; 1997. pp. 431–479. [Google Scholar]
- 32.Ogura T, Kitagawa T. Resonance Raman characterization of the P intermediate in the reaction of bovine cytochrome c oxidase. Biochim Biophys Acta. 2004;1655:290–297. doi: 10.1016/j.bbabio.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 33.Karpefors M, Ädelroth P, Namslauer A, Zhen Y, Brzezinski P. Formation of the “peroxy” intermediate in cytochrome c oxidase is associated with internal proton/hydrogen transfer. Biochemistry. 2000;39:14664–14669. doi: 10.1021/bi0013748. [DOI] [PubMed] [Google Scholar]
- 34.Chance B, Saronio C, Leigh JS. Functional intermediates in reaction of cytochrome oxidase with oxygen. Proc Natl Acad Sci USA. 1975;72:1635–1640. doi: 10.1073/pnas.72.4.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshikawa S, et al. Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science. 1998;280:1723–1729. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 36.Qin L, Hiser C, Mulichak A, Garavito RM, Ferguson-Miller S. Identification of conserved lipid/detergent-binding sites in a high-resolution structure of the membrane protein cytochrome c oxidase. Proc Natl Acad Sci USA. 2006;103:16117–16122. doi: 10.1073/pnas.0606149103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koepke J, et al. High resolution crystal structure of Paracoccus denitrificans cytochrome c oxidase: New insights into the active site and the proton transfer pathways. Biochim Biophys Acta. 2009;1787:635–645. doi: 10.1016/j.bbabio.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 38.Muramoto K, et al. Bovine cytochrome c oxidase structures enable O2 reduction with minimization of reactive oxygens and provide a proton-pumping gate. Proc Natl Acad Sci USA. 2010;107:7740–7745. doi: 10.1073/pnas.0910410107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kleymann G, Ostermeier C, Ludwig B, Skerra A, Michel H. Engineered Fv fragments as a tool for the one-step purification of integral multisubunit membrane-protein complexes. Biotechnology. 1995;13:155–160. doi: 10.1038/nbt0295-155. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.