

Abstract
Cells lacking ataxia telangiectasia mutated (ATM) have impaired mitochondrial function. Furthermore, mammalian cells lacking ATM have increased levels of reactive oxygen species (ROS) as well as mitochondrial DNA (mtDNA) deletions in the region encoding for cytochrome c oxidase (COX). We hypothesized that ATM specifically influences COX activity in skeletal muscle. COX activity was ~40% lower in tibialis anterior from ATM-deficient mice than for wild-type mice (P<0.01, n=9/group). However, there were no ATM-related differences in activity of succinate dehydrogenase, isocitrate dehydrogenase, alpha-ketoglutarate dehydrogenase, mitochondrial glycerol 3-phosphate dehydrogenase, or complex III. Incubation of wild-type extensor digitorum longus muscles for 1 h with the ATM inhibitor KU55933 caused a ~50% reduction (P<0.05, n=5/group) in COX activity compared to muscles incubated with vehicle alone. Among the control muscles and muscles treated with the ATM inhibitor, COX activity was correlated (r=0.61, P<0.05) with activity of glucose 6-phosphate dehydrogenase, a key determinant of antioxidant defense through production of NADPH. Overall, the findings suggest that ATM has a protective role for COX activity.
Keywords: ataxia telangiectasia mutated, cytochrome c oxidase, mitochondria, glucose 6-phosphate dehydrogenase, KU-55933
Introduction
Deficiency of the serine/threonine kinase ATM leads to radiosensitivity, neurodegeneration, insulin resistance and metabolic syndrome, and cancer predisposition [1]. While ATM deficiency is often studied in relation to the rare human disease ataxia telangiectasia (A-T), the recent finding that a high fat diet dramatically decreases ATM levels [2] extends the relevance of ATM deficiency to the increasing world-wide prevalence of obesity and metabolic syndrome.
It has been demonstrated that lymphoblastoid cells lacking functional ATM have reduced respiratory capacity [3]. This is true both in cells from patients with A-T ( in which both alleles of ATM produce non-functional products) and in wild-type cells transfected with siRNA to reduce ATM expression [3]. Additionally, studies have indicated that ATM deficient cells not only have decreased mitochondrial function but also have increased levels of ROS and mitochondrial DNA (mtDNA) damage specifically in the region encoding for cytochrome c oxidase (COX/complex IV) [4]. Furthermore, it has been demonstrated that ionizing radiation activates ATM as well as increases mtDNA and mitochondrial mass in wild-type fibroblasts, and these effects are prevented or blunted in fibroblasts from A-T patients (21).
Given the previously-reported susceptibility of COX to reactive oxygen species [4,5] and the reports of high ROS levels in ATM-deficient cells [4,6,7], we hypothesized that ATM deficiency or inhibition of ATM would influence COX activity in skeletal muscle.
Methods
Reagents
3,3′-Diaminobenzidine tetrahydrochloride (DAB), cytochrome c, bovine liver catalase, saponin, ascorbic acid, p-iodonitrotetrazolium violet (INT), sn-glycerol 3-phosphate bis(cyclohexylammonium salt), decylubiquinone, rotenone, sodium azide, adenosine 5′ diphosphate (ADP), nicotinamide adenine dinucleotide (NAD+), nicotinamide adenine dinucleotide phosphate (NADP+), phenazine methosulfate (PMS), 2,6-dichloroindophenol (DCIP), 2-amino-2-methyl-1-propanol, disodium EDTA, bovine serum albumin (BSA), and sodium cyanide were obtained from Sigma-Aldrich (St. Louis, MO).
The ATM inhibitor KU55933 was a generous gift from KuDos Pharmaceuticals (Cambridge, UK). KU55933 was used at a concentration of 1 μM, which we have previously shown to suppress levels of p53 protein (consistent with inhibition of ATM) in cultured skeletal muscle cells [8]. Even at 10 μM, the ATM inhibitor reportedly does not interfere with any of a panel of 60 kinases tested [9]. The 1 μM concentration of KU55933 is substantially lower than the IC50s for other members of the PI3K family kinases, including PI3K, mTOR, and ATR [9]. At 1 μM, KU55933 does not affect insulin-stimulated phosphorylation of Akt in mouse skeletal muscle [10], suggesting that it does not interfere with PI3K, which is upstream of Akt.
Collection and processing of skeletal muscle
All work with live animals was approved by the Saint Louis University Institutional Animal Care and Use Committee. Transgenic mice heterozygous for a truncation mutation that results in non-functional ATM [11] were obtained from The Jackson Laboratory (Bar Harbor, ME) and used to breed wild-type (ATM +/+) and ATM-deficient (ATM −/−) animals. Mice were genotyped from tail DNA using primer sets described by the Jackson Laboratory. Mice were anesthetized with sodium pentobarbital (50 mg/kg), and tibialis anterior muscles were removed and clamp-frozen using aluminum tongs cooled in liquid nitrogen. Muscles were stored at −80 °C until analysis.
For in vitro incubation of isolated skeletal muscle, mice were anesthetized as described above, and extensor digitorum longus (EDL) were removed. These muscles are small enough that diffusion of oxygen and nutrients is not limited, and thus they are suitable for in vitro experiments generally as previously described [10,12,13]. EDL muscles were incubated for 1 h in oxygenated (95% O2: 5% CO2) Krebs Henseleit bicarbonate buffer (KHB) containing 32 mM mannitol, 8 mM glucose, and 0.1% radioimmunoassay-grade bovine serum albumin (BSA). Muscles were then incubated in the absence (0.1% DMSO vehicle) or presence of KU55933 for 1 h. At the completion of incubations, muscles were blotted, trimmed, and clamp-frozen with aluminum tongs cooled in liquid nitrogen.
Muscles were homogenized in Kontes ground glass tubes in ice-cold buffer containing phosphatase and protease inhibitors (50 mM HEPES, pH 7.4, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1 mM EDTA, 1.5 mM MgCl2, 10 mM sodium pyrophosphate, 100 mM NaF, 2 mM Na3VO4, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 0.5 μg/ml pepstatin, and 0.2 mM PMSF). Protein content of homogenates was assessed with bicinchoninic acid assays (Pierce Protein Research Products, Rockford, IL). Depending on the specific enzyme assay, approximately 10–50 μg of muscle protein was used in enzyme activity assays.
Enzyme activity assays
Muscle homogenates were assayed for activities of representative mitochondrial enzymes. Representative enzymes involved in respiratory electron transfer included cytochrome c oxidase (COX; electron transport chain complex IV), succinate dehydrogenase (SDH; complex II, also a component of the Krebs cycle), mitochondrial glycerol 3-phosphate dehydrogenase (G3PDH), and complex III. Other representative enzymes from the Krebs cycle included α-ketoglutarate dehydrogenase (KDH) and isocitrate dehydrogenase (IDH).
Cytochrome c (10 mg/ml) in 10 mM potassium phosphate buffer, pH 7.0, was reduced by addition of 0.5 mg/ml (final concentration) L-ascorbic acid and incubation for 30 min at room temperature, essentially as described by Wharton and Tzagoloff [14]. The cytochrome c solution was cleared of ascorbate by multiple washings with 10 mM phosphate buffer using Millipore Biomax Ultrafree 10 kDa cutoff spin filters. The concentration of reduced cytochrome c was assessed spectrophotometrically (EmM550 nm=29.5).
COX activity (reduced cytochrome c + DAB ⇒ oxidized cytochrome c + reduced DAB) was determined spectrophotometrically by the increase in absorbance as reduced DAB accumulated (EmM450 nm=59). The assay was generally performed as described by Chrzanowska-Lightowlers [15], and COX activity was determined as the azide inhibitable (1 mM NaN3) portion of the increase in absorbance at 450 nm for assay reagent containing 100 mM potassium phosphate, pH 7.0, 4 mM DAB, 5 μM reduced cytochrome c, 2 μg/ml catalase, and 0.0033% saponin.
Mitochondrial G3PDH (glycerol 3-phosphate + INT ⇒ dihydroxyacetone phosphate + reduced INT) activity was assessed as described by MacDonald et al [16] by monitoring the change in absorbance at 490 nm as INT was reduced. Assay solutions contained 5 mM PIPES buffer, pH 7.5, 4 mM INT, 1 mM sodium cyanide, 50 mM tricine, and 0 (background reaction) or 100 mM glycerol 3-phosphate.
SDH activity (succinate + PMS ⇒ fumarate + reduced PMS; reduced PMS + DCIP ⇒ PMS + reduced DCIP) was assessed spectrophotometrically by the decrease in absorbance related to reduction of DCIP (EmM600 nm=21) generally as described by MacDonald et al [16]. The assay reagent contained 80 mM potassium phosphate buffer, pH 7.5, 0.1 mM PMS, 1 mM NaCN, and 0.05 mM DCIP. The background reaction was monitored without succinate in the reagent, and the specific reaction was begun by addition of 20 mM sodium succinate.
IDH activity (isocitrate + NAD+ ⇒ α-ketoglutarate + CO2 + NADH + H+) was monitored spectrophotometrically by the rate of appearance of reduced NADH (EmM340 nm=6.22) as described by Passonneau and Lowry [17]. The assay reagent contained 50 mM potassium phosphate, pH 7.0, 18 mM citrate, 18 mM MgCl2, 4.5 mM ADP, 0.05% bovine serum albumin, 0.18% Triton X-100, and 2 mM NAD+. After monitoring the background reaction, the specific reaction was begun by addition of 2.5 mM isocitrate.
KDH activity (KDH, α-ketoglutarate + coenzyme A + NAD+ ⇒ succinyl coenzyme A + CO2 + NADH + H+) was assayed by fluorometric monitoring (excitation 360/40 nm, emission 460/40 nm) of the increase in fluorescence as NADH accumulated [17]. The assay reagent contained 30 mM potassium phosphate, pH 7.0, 50 μM coenzyme A, 10 μg/ml diaphorase, 1 mM EDTA, 0.025% Triton X-100, 2 mM β-mercaptoethanol, 5 mM MgCl2, and 1 mM NAD+. Assays were run with 1 mM α-ketoglutarate or without α-ketoglutarate (to determine the background reaction rate).
Mitochondrial complex III activity (reduced decylubiquinone + cytochrome c ⇒ decylubiquinone + reduced cytochrome c) was assessed in a spectrophotometric assay adapted from the method described by Minchenko et al [18]. Assay solutions contained 80 mM potassium phosphate buffer, pH 7.5, 2 mM sodium cyanide, 4.8 mM rotenone, 0.05% BSA, and 0.1 mM cytochrome c (non-reduced). Reduced decylubiquinone (110 μM) served as substrate, and the rate of the background reaction was monitored in reagent containing muscle samples but no decylubiquinone. Reactions were monitored at 550 nm for reduction of cytochrome c.
Glucose 6-phosphate dehydrogenase (G6PDH) activity (glucose 6-phosphate + NADP+ ⇒ 6-phosphogluconolactone + NADPH + H+) was assessed fluormetrically generally as described by Passonneau and Lowry [17]. The assay reagent included 100 mM 2-amino-2-methyl-1-propanol, pH 9.4, 50 μM NADP+, 0.5 mM EDTA, and 0.02% BSA. Muscle samples were pipetted into duplicate wells. The reaction was started by addition of 1 mM glucose 6-phosphate (G6P) to one of the two wells for each sample, and the difference in fluorescence intensity (excitation 360/40 nm, emission 460/40 nm) between wells containing G6P and those without G6P was taken as a measure of G6PDH activity, which was calculated using NADPH standards.
Results
As shown in figure 1, COX activity (per μg of muscle protein) was about ~40% lower in muscle from ATM-deficient animals than muscle from wild-type mice (P<0.01, n=9/group). When expressed per unit of muscle wet weight, COX activity was about 25% lower in transgenic animals than in wild-type mice (wild type 34.1±3.1 μmol/g/min, ATM −/− 25.0±2.8 μmol/g/min, n=9/group, P<0.05). The values for wild-type COX activity are consistent with the range of values for COX activity previously reported for mouse skeletal muscle [19–21].
Figure 1. Cytochrome c oxidase activity of skeletal muscle is lower in ATM-deficient animals than in wild-type mice.
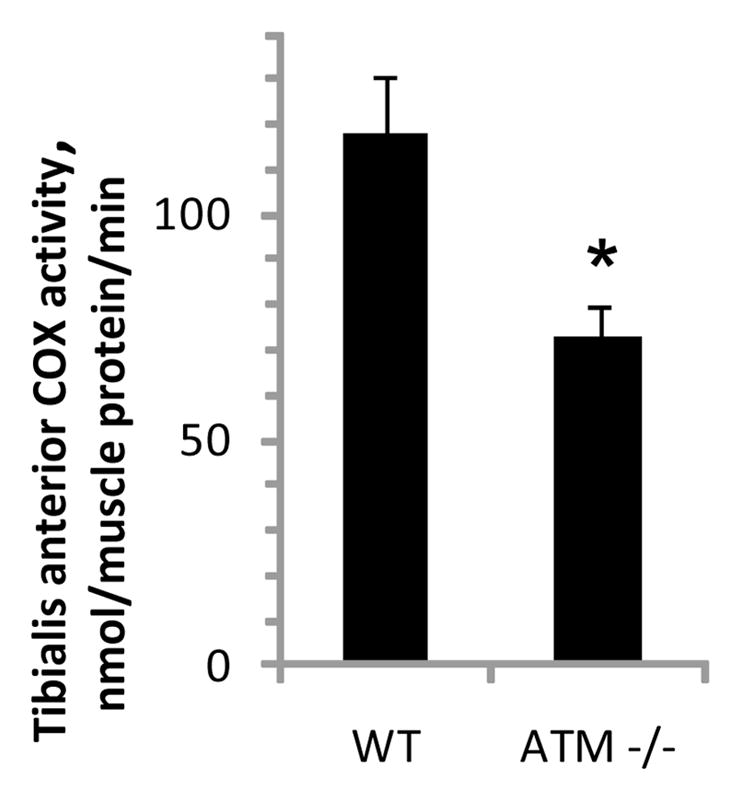
Tibialis anterior muscles from wild-type and ATM-deficient (ATM −/−) were homogenized in a buffer containing phosphatase and protease inhibitors and spectrophotometrically assayed for cytochrome c oxidase (COX) activity. *indicates a significant difference (P<0.01, n=9/group). Bars represent means with SEs.
To determine if the decrease in COX activity was a result of a general depression of mitochondrial function, activities of several other representative enzymes of respiratory electron transfer and the Krebs cycle were assayed as well. As shown in figure 2, SDH, IDH, KDH, mitochondrial G3PDH, and Complex III activities were similar in muscles from wild-type and ATM-deficient animals. Thus, the depressed COX activity in ATM-deficient mice appears to be a specific effect on COX (at least among the pool of enzymes assessed), as opposed to a general, coordinated downregulation of mitochondrial enzyme activity.
Figure 2. There is no general reduction in activities of mitochondrial enzymes in skeletal muscle of ATM-deficient mice.

Tibialis anterior muscle homogenates from wild-type and ATM-deficient (ATM −/−) mice (n=6/group) were assayed for activities of succinate dehydrogenase (SDH), isocitrate dehydrogenase (IDH), α-ketoglutarate dehydrogenase (KDH), and mitochondrial glycerol 3-phosphate dehydrogenase (G3PDH). Soleus muscles (n=3/group) were assayed for activity of respiratory chain Complex III. Bars represent means with SEs.
Although it has been reported that cells that are deficient in ATM have decreased mitochondrial function, it is unknown whether this effect of ATM deficiency is acute (i.e. involving post-translational regulation of mitochondrial proteins) or chronic (i.e. involving changes in gene expression). To test the possibility that there could be an acute role of ATM in control of COX activity, EDL muscles were incubated in vitro for 1 h in the presence of 1 μM KU55933 (an ATM inhibitor) or 0.1% DMSO (vehicle) before assessment of COX activity. Given the a priori hypothesis that the ATM inhibitor would decrease COX activity, mean comparisons were made using a one-tailed t test. As shown in figure 3A, the ATM inhibitor decreased COX activity by about 50% (P<0.05, n=5/group).
Figure 3. Acute inhibition of ATM decreases cytochrome c oxidase activity.

Extensor digitorum longus muscles were removed from anesthetized wild-type mice and incubated for 1 h in the absence (0.1% DMSO vehicle) or presence of 1 μM KU55933 (KU, a specific inhibitor of ATM) before assays for cytochrome c oxidase (COX) and glucose 6-phosphate dehydrogenase (G6PDH) activity. A) COX activity (n=5/group). B) correlation between COX activity (relative to DMSO control) and G6PDH activity. *indicates a significant difference or correlation (P<0.05). Bars represent means with SEs.
It has recently been reported that ATM plays a role in maintenance of antioxidant defenses through stimulation of G6PDH [22], which provides for much of cellular production of NADPH (a key co-factor in antioxidant processes). Indeed, silencing of G6PDH causes an increase of ROS levels in human fibroblasts [22], and ROS have previously been shown to selectively inhibit COX among constituents of the electron transport chain [5]. Thus, we hypothesized that COX activity and G6PDH activity would be positively correlated in the muscle samples exposed to KU (ATM inhibitor) or vehicle (DMSO). As shown in figure 3B, COX activity and G6PDH activity were significantly correlated (r=0.61, r2=0.37, P<0.05).
Discussion
The present study’s data indicate that among the several mitochondrial enzymes assessed in skeletal muscle, COX activity is specifically suppressed in ATM deficient animals. This is in-line with previous findings indicating COX mtDNA dysfunction in ATM deficient animals [4]. Furthermore, the finding that a brief incubation with an ATM inhibitor decreases COX activity indicates that ATM’s influence on COX activity is likely to be acute. Previously, Eaton et al [23] showed a depletion of mtDNA associated with 24 hours treatment with an ATM inhibitor, and Ambrose et al [3] found complete restoration of mitochondrial respiratory capacity in ATM −/− cells after 72 h of incubation with the antioxidant α-lipoic acid. The important information provided by the current study is that that the time span (1 h) over which inhibition of ATM suppresses COX activity is too short for transcriptional and translational regulation of COX in skeletal muscle, suggesting a post-translational mechanism for ATM’s involvement in COX modulation.
In mammalian cells lacking ATM, ROS levels are substantially increased. For example, ROS concentrations are elevated in cerebellum, striatum, and astrocytes of ATM −/− mice [6,7] and in vascular smooth muscle cells of ATM+/− mice on ApoE−/− backgrounds [4]. Interestingly, ATM-deficient cells are also hypersensitive to agents inducing oxidative stress [24], suggesting a decrease in antioxidant defense in ATM-deficient cells. Consistent with this, Consentino et al [22] have shown that the activity of G6PDH is stimulated by ionizing radiation in an ATM-dependent fashion. Likewise, addition of DNA with double stranded breaks to Xenopus egg extracts stimulates G6PDH activity in an ATM-dependent manner [22]. Thus, ATM appears to stimulate G6PDH, the gatekeeper enzyme of the pentose phosphate pathway, which is a major source of reducing equivalents for antioxidant pathways. Consistent with a role of ATM in activation of antioxidant defenses, ionizing radiation stimulates G6PDH activity in control human fibroblasts to a much greater extent than in fibroblasts from patients with A-T [22]. Finally, siRNA-mediated knockdown of G6PDH in human fibroblasts was sufficient to increase ROS levels [22]. Thus, it seems that ATM plays an important role in antioxidant defenses by stimulating G6PDH and the pentose phosphate pathway.
As the site of most ROS production in the cell is the mitochondrion, findings of increased ROS levels in ATM-deficient cells [4,6,7] firms up the connection between ATM and mitochondria. While the work of Consentino et al [22] suggests that increased ROS in ATM-deficient cells could be a result of decreased antioxidant defenses, it is also possible that ROS production per se (as opposed to the net result of ROS production and ROS destruction) could be increased in ATM-deficient cells. ROS themselves can lead to further decline in mitochondrial function by modifying mitochondrial enzymes. For example, 4–6 h treatment of astrocytes with the NO donor deta NONOate causes an inhibition of COX but not Complex I, Complexes II/III, or citrate synthase [5]. In turn, inhibition of the electron transport chain at complex I, complex III, or complex IV (COX) causes an 80% increase in superoxide levels in astrocytes [5].
Together, previous reports and the current findings suggest the hypothesis that there could be a feed forward effect in which an initial surge in ROS levels after inhibition of ATM could cause a decrease in electron transport chain efficiency (e.g. by inhibition of COX), causing a subsequent further increase in ROS and resulting in mitochondrial dysfunction (figure 4). This model is consistent with recent findings implicating ATM as a sensor of oxidative stress that activates compensatory signaling (including mitochondrial biogenesis) in response to ROS [25].
Figure 4. Proposed model of mitochondrial dysfunction in the setting of ATM deficiency.

ATM deficiency results in decreased G6PDH activity [22] and a concomitant increase in ROS levels [4,6,7]. ROS are known to acutely decrease COX activity, and it has been shown that inhibition of COX increases ROS [5]. In the current study, a brief exposure of skeletal muscle to an ATM inhibitor decreased COX activity, and COX activity and G6PDH activity were correlated. Together, the previous and current findings suggest an acute feed-forward model, initiated by inhibition of or deficiency of ATM, in which increased levels of ROS inhibit COX, stimulating further ROS production and decrease in COX activity, resulting in general mitochondrial dysfunction.
While we have found an acute effect of ATM inhibition on COX activity in skeletal muscle, it is likely that chronic ATM deficiency results in additional mitochondrial deficits in other tissue or cell types. For example, Mercer et al [4] noticed an approximate decrease of 15–20% in Complex I activity in ATM+/−/ApoE−/− livers (though not in heart muscle). Furthermore, Wei et al [26] have reported that a 4977-bp mtDNA deletion is strongly correlated with increased ROS levels, thus supporting a model wherein ATM deficiency leads to increased ROS and acute and chronic decreases in mitochondrial function, mediated by COX inhibition or mtDNA deletion, respectively.
In conclusion, ATM-deficient skeletal muscle has decreased COX activity compared to wild-type muscle. Furthermore, inhibition of ATM decreases COX activity within an hour, and COX activity is correlated with G6PDH activity among the group of control muscles and muscles incubated with the ATM inhibitor. Further study is necessary to fully understand the interplay among ATM, ROS, COX, and their possible relation to deficits of ATM-deficiency, such as reduced protein synthesis [27], metabolic syndrome [28], impaired insulin secretion [29], insulin resistance in skeletal muscle [28] or muscle cells [2,8,10], or predisposition to cancer [1].
Acknowledgments
This work was supported by the U.S. Public Health service through National Institutes of Health grants R15DK080437 and R15DK080437-01S2 (American Recovery and Reinvestment Act). The grant sponsor played no role in study design; in the collection, analysis and interpretation of data; in the writing of the report; or in the decision to submit the paper for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9:759–769. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 2.Halaby MJ, Hibma JC, He J, Yang DQ. ATM protein kinase mediates full activation of Akt and regulates glucose transporter 4 translocation by insulin in muscle cells. Cell Signal. 2008;20:1555–1563. doi: 10.1016/j.cellsig.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 3.Ambrose M, Goldstine JV, Gatti RA. Intrinsic mitochondrial dysfunction in ATM-deficient lymphoblastoid cells. Hum Mol Genet. 2007;16:2154–2164. doi: 10.1093/hmg/ddm166. [DOI] [PubMed] [Google Scholar]
- 4.Mercer JR, Cheng KK, Figg N, Gorenne I, Mahmoudi M, Griffin J, Vidal-Puig A, Logan A, Murphy MP, Bennett M. DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ Res. 2010;107:1021–1031. doi: 10.1161/CIRCRESAHA.110.218966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacobson J, Duchen MR, Hothersall J, Clark JB, Heales SJ. Induction of mitochondrial oxidative stress in astrocytes by nitric oxide precedes disruption of energy metabolism. J Neurochem. 2005;95:388–395. doi: 10.1111/j.1471-4159.2005.03374.x. [DOI] [PubMed] [Google Scholar]
- 6.Quick KL, Dugan LL. Superoxide stress identifies neurons at risk in a model of ataxia-telangiectasia. Ann Neurol. 2001;49:627–635. [PubMed] [Google Scholar]
- 7.Kim J, Wong PK. Oxidative stress is linked to ERK1/2-p16 signaling-mediated growth defect in ATM-deficient astrocytes. J Biol Chem. 2009;284:14396–14404. doi: 10.1074/jbc.M808116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ching JK, Rajguru P, Marupudi N, Banerjee S, Fisher JS. A role for AMPK in increased insulin action after serum starvation. Am J Physiol Cell Physiol. 2010;299:C1171–C1179. doi: 10.1152/ajpcell.00514.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 10.Jeong I, Patel AY, Zhang Z, Patil PB, Nadella ST, Nair S, Ralston L, Hoormann JK, Fisher JS. Role of ataxia telangiectasia mutated in insulin signaling of muscle-derived cell lines and mouse soleus. Acta Physiologica. 2010;198:465–475. doi: 10.1111/j.1748-1716.2009.02069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 12.Ju JS, Smith JL, Oppelt PJ, Fisher JS. Creatine feeding increases GLUT4 expression in rat skeletal muscle. Am J Physiol Endocrinol Metab. 2004;288:E347–E352. doi: 10.1152/ajpendo.00238.2004. [DOI] [PubMed] [Google Scholar]
- 13.Smith JL, Ju JS, Saha BM, Racette BA, Fisher JS. Levodopa with carbidopa diminishes glycogen concentration, glycogen synthase activity, and insulin-stimulated glucose transport in rat skeletal muscle. J Appl Physiol. 2004;97:2339–2346. doi: 10.1152/japplphysiol.01219.2003. [DOI] [PubMed] [Google Scholar]
- 14.Wharton DC, Tzagoloff A. Cytochrome oxidase from beef heart mitochondria. Methods Enzymol. 1967;10:245–250. [Google Scholar]
- 15.Chrzanowska-Lightowlers ZM, Turnbull DM, Lightowlers RN. A microtiter plate assay for cytochrome c oxidase in permeabilized whole cells. Anal Biochem. 1993;214:45–49. doi: 10.1006/abio.1993.1454. [DOI] [PubMed] [Google Scholar]
- 16.MacDonald MJ, Warner TF, Mertz RJ. High activity of mitochondrial glycerol phosphate dehydrogenase in insulinomas and carcinoid and other tumors of the amine precursor uptake decarboxylation system. Cancer Res. 1990;50:7203–7205. [PubMed] [Google Scholar]
- 17.Passonneau JV, Lowry OH. Enzymatic Analysis: A Practical Guide. Humana Press; Totowa, NJ: 1993. [Google Scholar]
- 18.Minchenko J, Williams AJ, Christodoulou J. Adaptation of a mitochondrial complex III assay for automation: examination of reproducibility and precision. Clin Chem. 2003;49:330–332. doi: 10.1373/49.2.330. [DOI] [PubMed] [Google Scholar]
- 19.Adhihetty PJ, Uguccioni G, Leick L, Hidalgo J, Pilegaard H, Hood DA. The role of PGC-1alpha on mitochondrial function and apoptotic susceptibility in muscle. Am J Physiol Cell Physiol. 2009;297:C217–C225. doi: 10.1152/ajpcell.00070.2009. [DOI] [PubMed] [Google Scholar]
- 20.Houle-Leroy P, Garland T, Jr, Swallow JG, Guderley H. Effects of voluntary activity and genetic selection on muscle metabolic capacities in house mice Mus domesticus. J Appl Physiol. 2000;89:1608–1616. doi: 10.1152/jappl.2000.89.4.1608. [DOI] [PubMed] [Google Scholar]
- 21.Saleem A, Adhihetty PJ, Hood DA. Role of p53 in mitochondrial biogenesis and apoptosis in skeletal muscle. Physiol Genomics. 2009;37:58–66. doi: 10.1152/physiolgenomics.90346.2008. [DOI] [PubMed] [Google Scholar]
- 22.Cosentino C, Grieco D, Costanzo V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. doi: 10.1038/emboj.2010.330. (epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eaton JS, Lin ZP, Sartorelli AC, Bonawitz ND, Shadel GS. Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J Clin Invest. 2007;117:2723–2734. doi: 10.1172/JCI31604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stern N, Hochman A, Zemach N, Weizman N, Hammel I, Shiloh Y, Rotman G, Barzilai A. Accumulation of DNA damage and reduced levels of nicotine adenine dinucleotide in the brains of Atm-deficient mice. J Biol Chem. 2002;277:602–608. doi: 10.1074/jbc.M106798200. [DOI] [PubMed] [Google Scholar]
- 25.Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010;330:517–521. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 26.Wei YH, Lu CY, Lee HC, Pang CY, Ma YS. Oxidative damage and mutation to mitochondrial DNA and age-dependent decline of mitochondrial respiratory function. Ann N Y Acad Sci. 1998;854:155–170. doi: 10.1111/j.1749-6632.1998.tb09899.x. [DOI] [PubMed] [Google Scholar]
- 27.Yang DQ, Kastan MB. Participation of ATM in insulin signalling through phosphorylation of eIF-4E-binding protein 1. Nat Cell Biol. 2000;2:893–898. doi: 10.1038/35046542. [DOI] [PubMed] [Google Scholar]
- 28.Schneider JG, Finck BN, Ren J, Standley KN, Takagi M, Maclean KH, Bernal-Mizrachi C, Muslin AJ, Kastan MB, Semenkovich CF. ATM-dependent suppression of stress signaling reduces vascular disease in metabolic syndrome. Cell Metab. 2006;4:377–389. doi: 10.1016/j.cmet.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 29.Miles P, Treuner K, Latronica M, Olefsky JM, Barlow C. Impaired insulin secretion in a mouse model of Ataxia Telangiectasia. Am J Physiol Endocrinol Metab. 2007;293:E70–74. doi: 10.1152/ajpendo.00259.2006. [DOI] [PubMed] [Google Scholar]