

Abstract
Fibroblast growth factors (FGFs) negatively regulate long bone development by inhibiting the proliferation of chondrocytes that accumulate in the G1 phase of the cycle following FGF treatment. Here we report that FGF also causes a striking but transient delay in mitotic entry in RCS chondrocytes by inactivating the cyclin B1-associated CDK1(CDC2) kinase. As a consequence of this inactivation, cells accumulate in the G2 phase of the cycle for the first 4–6 hours of the treatment. Cyclin B1/CDK1 activity is then restored and cells reach a G1 arrest.
The reduced cyclin B1/CDK1 activity was accompanied by increased CDK1 inhibitory phosphorylation, likely caused by increased activity and expression of the Myt1 kinase. FGF1 also caused dephosphorylation of the CDC25C phosphatase. That, however, appears due the inactivation of cyclin B1/CDK1 complex in the CDK1 feedback loop and not the activation of specific phosphatases. The inactivation of the cyclin B1/CDK1 complex is a direct effect of FGF signaling and not a consequence of the G2 arrest as can be observed also in cells blocked at mitosis by Nocodazole. The Chk1 and ATM/ATR kinase are known to play essential roles in the G2 checkpoint induced by DNA damage/genotoxic stress, but inhibition of Chk1 or ATM/ATR not only did not prevent, but rather potentiated the FGF-induced G2 arrest.
Additionally, our results indicate that the transient G2 arrest is induced by FGF in RCS cell through mechanisms that are independent of the G1 arrest, and that the G2 block is not strictly required for the sustained G1 arrest but may provide a pausing mechanism that allows the FGF response to be fully established.
Key words: fibroblast growth factor, chondrocyte, G2/M arrest, Myt1, cyclin B1, CDK1
Introduction
Fibroblast growth factors (FGFs) are master regulators of skeletal development.1 In vertebrates, long bones are developed through a process called endochondral ossification. FGF signaling regulates endochondral ossification by inhibiting chondrocyte proliferation via activating FGF receptor 3 (FGFR3), which is highly expressed in chondrocytes.2 Gain-of-function mutations of FGFR3 caused several forms of human dwarfism, including achondroplasia (ACH) and thanatophoric dysplasia (TD).3 The major effect of FGF signaling on chondrocyte proliferation is a viable G1 arrest and the induction of some aspects of chondrocyte differentiation. The inhibition of chondrocyte proliferation induced by FGF requires the function of p107 and p130 Rb proteins.4 p107 dephosphorylation, which is mediated by the PP2A phosphatase, is a key early event in FGF-induced growth arrest.5
We and others had previously noted that FGF treatment of RCS chondrocytes, in addition to causing a prolonged G1 arrest, also resulted in an early and significant arrest of the cells in the G2/M phase of the cycle that was relieved, however, within 4–5 hours of FGF treatment as cells progressively arrested in G0/G1.6–8 To better understand the mechanisms by which FGF signaling inhibits chondrocyte proliferation and the role of the transient G2/M arrest in this phenomenon, we have studied the activity and perturbations of the pathways that regulate cell entry and transition to the G2/M phase in FGF-treated RCS chondrocytes.
In the eukaryotic cell cycle, mitotic entry requires the activation of the maturation promoting factor (MPF), whose major component is the cyclin B1/CDK1(CDC2) kinase complex.9,10 Binding to cyclin B and phosphorylation at T161 by CDKactivating kinase (CAK) are required to activate CDK1.11 During G2, the cyclin B1/CDK1 complex is kept inactive by phosphorylation on Y15 and T14 of CDK1 by the Wee1 and Myt1 kinases.12,13 At the onset of mitosis, both of these residues are dephosphorylated by the dual-specificity CDC25C phosphatase.14 CDC25C is required to be activated in an initiation step prior to CDK1 activation and then is further activated in an amplification step by CDK1 itself.15,16 CDC25C is activated by hyperphosphorylation of its N-terminal regulatory domain by p42 MAPK, CDK1 and polo-like (PLK) kinases.17–19 During the normal cell cycle and after incomplete DNA replication/DNA damage, the phosphorylation of CDC25C by Chk1/2 and CTAK1 kinases creates a binding site for the 14-3-3 family of proteins, causing CDC25C sequestration in the cytoplasm during interphase and the inhibition of mitotic entry.20 Protein phosphatases PP1 and PP2A have been implicated in controlling mitotic entry by keeping CDC25 inactive during interphase.21–24
We report here that FGF1 causes a striking but transient delay in mitotic entry in RCS chondrocytes by inactivating the cyclin B1/CDK1 kinase complex. This inactivation does not require cell cycle progression and is likely dependent on FGF-induced activation of the Wee1 and Myt1 kinases that inactivate CDK1. Furthermore, the G2 arrest is induced independently from the following sustained G0/G1 arrest and was surprisingly potentiated by inhibition of Chk1 or ATM/ATR.
Results
FGF1 causes an immediate, transient delay in mitotic entry in RCS chondrocytes.
We and others previously reported that FGF treatment induces a transient G2/M arrest in RCS chondrocytes,4,6,8 but it was not determined whether this reflected a delay of mitotic entry or exit. To address this issue, RCS cells were treated with FGF1 and mitotic cells were labeled with phospho-histone H3(S10) antibody. As shown in Figure 1A, a significant reduction of mitotic cell number was observed after 1 hour of FGF1 treatment in parallel with the accumulation of cells in G2 phase. After 4–6 hours, the G2 block was released and cells progressively accumulated in G1. To confirm this conclusion, RCS cells were treated with both FGF1 and Nocodazole, which stops cell cycle progression at early mitotic phase by preventing microtubule polymerization. FGF1 caused a delay of entry into mitosis during the first 4 hours of treatment (Fig. 1B), i.e., a G2 arrest.
Figure 1.

FGF causes an immediate, transient delay in mitotic entry in RCS cells. (A) FACScan™ analysis of RCS cells treated with FGF1 and harvested at the indicated periods of time. Mitotic cells were stained with anti-phospho-histone H3(S10) antibodies. Numbers on the Y-axis indicate percentage of total cells in G1, S, G2 and M phases. (B) RCS cells were treated with either Nocodazole alone or together with FGF1. Only changes in the levels of G2 and M phases are indicated. The data are representatives of several independent experiments with the same results.
FGF1 signaling downregulates cyclin B1/CD K1 activity.
In the eukaryotic cell cycle, activation of CDK1 kinase is required for cells to enter mitosis.25 We asked whether the FGF1-induced G2 arrest in RCS chondrocytes was due to the inactivation of the cyclin B1/CDK1 kinase complex. As shown in Figure 2A, cyclin B1/CDK1 kinase activity was reduced by more than 80% after 1 hour of treatment with FGF1 but then recovered by 6 hours. Binding of cyclin B1 to CDK1, as assayed by immunoprecipitation as well as the levels of cyclin B1 and CDK1 were unchanged for 10 hrs of FGF1 treatment, after which time all cell cycle progression factors are strongly downregulated and the cells become quiescent. FGF1 caused an increase in the level of tyrosine 15 phosphorylation of CDK1 in RCS chondrocytes, continuing until 6 h of treatment (Fig. 2B). This increase is much more evident in Figure 2A, when an antibody recognizing total CDK1 is used in the cyclin B1 immunoprecipitates. This antibody recognizes two forms of CDK1, and the slowly migrating form has been shown to correspond to phosphorylated CDK1. At 1–4 hours after FGF treatment, the only CDK1 form that immunoprecipitates with cyclin B1 is the slow moving form, while when cyclin B1/CDK1 activity is restored both forms can be detected in the immunoprecipitates. It is likely that the level of CDK1 phosphorylation detected in cyclin B1 immunoprecipitates reflects more faithfully the overall activity of the cyclin B1/CDK1 complex, than what can be determined by western blot of total cellular CDK1. A slight increase of phospho-CDK1(T14) was also observed starting at 1 h and continuing until 6 h of FGF treatment (Fig. 2B). These data indicate that the FGF-induced G2 arrest in chondrocytes is due to the downregulation of cyclin B1/CDK1 activity, possibly mediated by increased phosphorylation on Y15 and T14 of CDK1 and that kinase inactivation is not due to the dissociation of CDK1 from cyclin B1 or to protein degradation.
Figure 2.

FGF signaling strongly downregulates activity of cyclin B1/CDK1 complexes in chondrocytes. RCS cells were treated with FGF1 for indicated times. (A and C) Kinase activity of immunoprecipitated cyclin B1/CDK1 complexes was assayed in vitro. The cyclin B1/CDK1 complexes were isolated from 1 mg of total cellular protein using anti-cyclin B1 antibodies, and histone H1 was used as a substrate. Antimouse IgG was used as a negative control. An equal amount of protein loading was confirmed by immunodetection of cyclin B1 and CDK1 in immunoprecipitated complexes. (B) 20 µg of total cellular protein were analyzed by SDS-PAGE followed by immunoblotting for anti-phospho-CDK1(Y15), anti-CDK1 and anti-phospho-CDK1(T14) antibodies. Equal amount of protein loading was confirmed by α-tubulin and α-actin immunodetection. (C) RCS cells were pretreated with Nocodazole for 12 hrs and then FGF1 was added for indicated periods of time.
FGF-mediated inactivation of cyclin B1/CD K1 is a direct effect of FGF signaling.
In order to determine whether FGF-induced inactivation of CDK1 was dependent on cell cycle progression, cells were synchronized at M phase by treatment with Nocodazole for 12 h, then FGF was added and cyclin B1/CDK1 activity was monitored. As shown in Figure 2C, FGF caused a strong reduction of cyclin B1/CDK1 activity in M-phase synchronized cells as well. These data indicate that the inhibition cyclin B1/CDK1 activity is a direct result of FGF signaling and not a consequence of the G2 arrest.
FGF1 mediates CDC 25C inactivation in RCS chondrocytes.
The CDC25C phosphatase dephosphorylates Y15 and T14 on CDK1 to activate CDK1 at the onset of mitosis.14 Phosphorylation at specific sites on CDC25C is required for the activation of CDC25C at G2/M transition. Active CDK1 then phosphorylates CDC25C, creating an amplifying feedback loop.25 Treatment of RCS chondrocytes with Nocodazole for 4 h accumulated cells in M-phase and induced hyperphosphorylation of CDC25C, which migrated at higher molecular mass. Treatment with λ-phosphatase greatly decreased the abundance of the slowly migrating form of CDC25C, in line with the notion that it represents hyperphosphorylated CDC25C (Fig. 3A). In exponentially growing cells, active pCDC25C accounts for only about 5% of total CDC25C (Fig. 3B), reflecting the small fraction of cells in mitotic phase. FGF1 signaling caused a reduction of CDC25C phosphorylation during the first 4 h of treatment (Fig. 3B) concomitantly with CDK1 inactivation and G2 arrest, suggesting that FGF1 mediated CDC25C inactivation. When RCS cells were simultaneously treated with FGF1 and Nocodazole, FGF1 caused a reduction in the levels of active CDC25C (Fig. 3C), strengthening the hypothesis that FGF1 negatively regulates the activity of CDC25C. Furthermore, FGF1 accelerated the reduction in the levels of active pCDC25C that follows cell release from the Nocodazole block (Fig. 3D).
Figure 3.

FGF mediates CDC25C inactivation in RCS cells. (A) Western blot analysis of extracts (50 µg of total cellular protein) from RCS cells; untreated, Nocodazole-treated for 4 h or treated with lambda phosphatase after lysis. An upper band that is sensitive to lambda phosphatase treatment corresponds to a hyperphosphorylated form of CDC25C. (B–D) RCS cells were treated with FGF1 for the indicated times either in absence (B) or presence of Nocodazole (C and D). 50 µg of total cellular protein was analyzed by SDS-PAGE followed by immunoblotting with anti-CDC25C antibodies. Equal amount of protein loading was confirmed by α-actin immunodetection.
We investigated the hypothesis that FGF signaling could have directly affected the phosphorylation of CDC25C, possibly by the activation of the PP2A phosphatase, that has been shown to play an important role in CDC25C dephosphorylation as well as in mediating dephosphorylation of p107 in FGF-treated chondrocytes.5,22,24 Activation of the PP2A phosphatase would have provided a unitary mechanism responsible for both the G1 and G2 arrest induced by FGF in chondrocytes.
We thus studied whether the FGF-induced dephosphorylation of CDC25C was caused by activation of a phosphatase. However, as shown in Figure 4A, inhibition of phosphatases by okadaic acid did not prevent CDC25C dephosphorylation caused by FGF1. Specific inhibition of PP2A by overexpression of SV40 small T antigen, previously shown to allow cells to escape FGF mediated growth inhibition, did not prevent cyclin B1/CDK1 inactivation (Fig. 4B) or the decrease in CDC25C phosphorylation mediated by FGF1 (not shown), and the cells still underwent a transient G2 arrest.
Figure 4.

FGF-induced inhibition of CDC25C phosphorylation is not mediated by activation of a phosphatase. (A) RCS cells were pre-incubated with okadaic acid (OA) at the indicated concentrations for 1 hour and either treated or not with FGF1 for one more hour. 50 µg of total cellular protein were analyzed by SDS-PAGE, followed by immunoblotting for anti-CDC25C antibodies. Equal amount of protein loading was confirmed by α-actin immunodetection. (B) RCS cells were infected with adenoviruses expressing either GFP or SV40 ST antigen following FGF1 treatment as indicated. Kinase activity of immunoprecipitated cyclin B1/CDK1 complexes was assayed in vitro. The cyclin B1/CDK1 complexes were isolated from 0.5 mg of total cellular protein using anti-cyclin B1 antibodies. Histone H1 was used as a substrate. Antimouse IgG was used as a negative control. RCS cells infected with SV40 ST routinely exhibited lower basal levels of cyclin B1-associated kinase activity in untreated cells as assayed in several independent experiments. (C) RCS cells were treated with Nocodazole and either with CDK1 inhibitor (Alsterpaullone), MEK1/2 inhibitor (U0126) or PLK1/3 inhibitor (GW843682X), as marked, and harvested at the indicated times. 50 µg of total cellular protein were analyzed by SDS-PAGE followed by immunoblotting for anti-CDC25C antibodies.
In eukaryotic cells, activating hyperphosphorylation of CDC25C is mediated by ERK1/2, PLK1/3 and CDK1 kinases. In RCS cells, inhibition of either ERK1/2 (by U0126) or PLK1/3 (by GW843682X26) did not prevent the induction of CDC25C phosphorylation induced by Nocodazole, which instead was greatly reduced when the CDK1 kinase was inhibited by Alsterpaullone, a CDK1 inhibitor27 (Fig. 4C). This suggests that under our experimental conditions, the major increase in phosphorylation of CDC25C we observe is mediated by CDK1.
In conclusion, the dephosphorylation of CDC25C induced by FGF signaling is unlikely to be caused by activation of PP2A since both okadaic acid and SV40 small T-Antigen did not prevent such a process and an association between CDC25C and PP2A was not induced by FGF treatment (not shown). We believe, therefore, that it is likely that the reduction in CDC25C phosphorylation is initially a result of the inhibition cyclin B1/CDK1 activity, and then the resulting low activity of the CDC25C phosphatase contributes to the inhibition cyclin B1/CDK1 activity by failing to reactivate CDK1.
The FGF-induced G2 arrest is independent of the sustained G0/G1 arrest.
The major effect of FGF in chondrocytes is a G0/G1 cell cycle arrest, detectable in RCS cells after 10–12 h of treatment.4 To investigate whether the transient G2 block was required for cells to reach a G1 arrest, RCS cells were synchronized at G0/G1 stage by serum starvation and then treated with FGF1 before being released into medium with 10% serum. Nocodazole was added at the releasing time to stop cell cycle progression at M phase. As shown in Figure 5A, without FGF1 treatment, around 70% of cells moved to mitosis after 22 hours of release from G1 arrest. However, pre-incubation of cells with FGF for 8 hours before releasing cells into full medium containing FGF caused approximately 80% cells to arrest in G0/G1 (Fig. 5D); this effect was lessened if G0/G1 synchronized cells were treated with FGF simultaneously or for 4 hours before cells were released into medium containing 10% serum (Fig. 5B and C). These data indicate that the G0/G1 cycle block does not require the G2 arrest. In line with this conclusion, overexpression of cyclin D1/CDK4 complexes previously shown to rescue RCS cells from the FGF-induced G0/G1 arrest5 did not affect cyclin B1/CDK1 inactivation (Fig. 5E), and the cells still experienced a transient G2 arrest. Similarly, RCS cells constitutively expressing Polyoma virus large T antigen, which inhibits the repressing function of the Rb proteins,28 are resistant to the FGF-induced inhibition of proliferation but still show a transient G2 arrest and downregulation of cyclin B1/CDK1 activity (not shown). Together, these data indicate that the mechanisms leading to chondrocytes G1 or G2 arrest are distinct and independent of each other and that the G2 arrest is not required for cessation of proliferation, although it may provide a “pausing” mechanism to allow FGF to cause its maximum effect on chondrocyte proliferation.
Figure 5.

FGF-induced G2 arrest is independent from the sequential sustained G0/G1 block. (A–D) Cell cycle arrest of RCS cells was induced by serum starvation for 32 h followed by release into medium containing 10% FBS and Nocodazole. (B–D) RCS cells were treated with FGF1 as indicated and subjected to cell cycle analysis. (E) Cyclin D1 and CDK4 were stably introduced into RCS cells, and this cell line and parental RCS cells were treated with FGF1 for the indicated periods of time. Kinase activity of immunoprecipitated cyclin B1/CDK1 complexes was assayed in vitro. The cyclin B1/CDK1 complexes were isolated from 1 mg of total cellular protein using anti-cyclin B1 antibodies. Histone H1 was used as a substrate.
FGF signaling modulates the activity of the Myt1 kinase.
As mentioned earlier, the activity of CDK1 is regulated by inhibitory phosphorylation on Y15 and T14. Phosphorylation of both residues was increased in the first hours of FGF treatment (Fig. 2B), suggesting that these inhibitory post-translation modifications could be responsible for FGF-induced downregulation of cyclin B1/CDK1 activity. T14 and Y15 phosphorylation of CDK1 is controlled by the balance between Wee1/Myt1 kinases and the CDC25C phosphatase,29 but the reduction in the amount of active CDC25C caused by FGF is likely the result, rather than the cause, of reduced cyclin B1/CDK1 activity (Fig. 3B). We studied the expression and activity of the Wee1 and Myt1 kinases following FGF treatment of RCS cells. As shown on Figure 6A, FGF treatment modestly upregulated the expression of both kinases during the first 4 hours. We therefore investigated the interaction of the Wee1/Myt1 kinases with CDK1. RCS cells were treated with FGF, and Myt1 and Wee1 were immunoprecipitated using specific antibodies. Figure 6B shows that in FGF-treated and untreated cells we could detect an association between Myt1 and CDK1. While FGF treatment induced only a modest increase in the amount of CDK1 associated with Myt1, the amount of phosphorylated CDK1 in Myt1 immunoprecipitates was increased strongly by FGF treatment. This FGF-induced interaction between Myt1 and pCDK1(Y15) therefore correlated with the kinetics of cyclin B1/CDK1 inactivation. We could not detect any association between Wee1 and CDK1. Thus, Myt1 might be the major kinase that inhibits CDK1 in RCS cells, as Myt1 can phosphorylate both Y15 and T14 residues.30 To confirm this hypothesis, we immunoprecipitated Myt1 from FGF-treated RCS cells and used recombinant cyclin B1/CDK1 complexes as a substrate in this kinase reaction. As shown in Figure 6C, the ability of Myt1 immunoprecipitates to phosphorylate CDK1 is strongly stimulated by FGF during the first 2–4 hours. Increased expression of Myt1 upon FGF treatment might be partially responsible for this effect, but other mechanisms likely contribute to FGF-induced upregulation of Myt1 activity.
Figure 6.
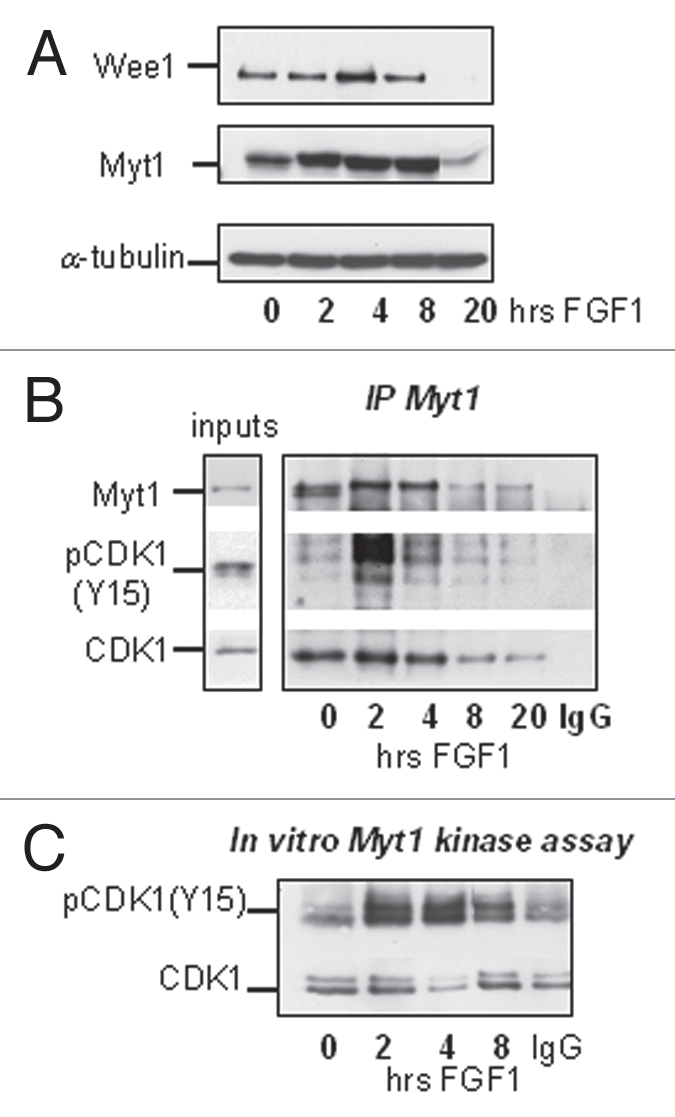
FGF signaling modulates the activity of Myt1 kinase. RCS cells were treated with FGF1 for indicated periods of times. (A) 20 µg of total cellular protein were analyzed by SDS-PAGE followed by immunoblotting for anti-Wee1 and anti-Myt1 antibodies. Equal amount of protein loading was confirmed by α-tubulin immunodetection. (B and C) Lysates were prepared and normalized by the amount of total cellular protein and subjected to immunoprecipitation with anti-Myt1 antibody as described in Materials and Methods. 10% of input and 50% of immunoprecipitated samples were analyzed by immunoblotting with anti-Myt1, anti-CDK1 or anti-phospho-CDK1(Y15) antibody. Control immunoprecipitation (*) was done using agarose A beads. (C) Kinase activity of immunoprecipitated Myt1 complexes was assayed in vitro. Recombinant cyclin B1/CDK1 complex was used as a substrate for Myt1 immunoprecipitates and phosphorylation of CDK1 was monitored by immunoblotting using anti-phospho-CDK1(Y15) antibody. Note that under these conditions endogenous CDK1 and phospho-CDK1(Y15) were not detectable in Myt1 immunoprecipitates.
Discussion
The response of chondrocyte to FGF signaling is a unique growth inhibitory response that distinguishes these cells from most other cell types, which respond to FGF signaling with increased proliferation and protection from apoptosis. This response is not receptor-specific but appears to depend on specific signal transduction mechanisms that direct the cell response towards growth inhibitory pathways.
We and others have identified several molecules that play a key role in determining the growth-inhibitory response of chondrocytes to FGF signaling. Notably, the inhibition of chondrocyte proliferation induced by FGF requires the activity of the p107 and p130 Rb proteins but not that of pRb itself, and p107 dephosphorylation, which is induced by PP2A, is a key event in the FGF-induced growth arrest.5 Constitutive phosphorylation of p107 as well as PP2A inactivation prevent growth arrest. All of these studies, however, had focused on the sustained G1 arrest that is induced by FGF treatment of primary chondrocytes or chondrocytic cell lines. The observation that a transient but very significant pausing of cells in the G2/M phase of the cycle preceded the G1 arrest has never been investigated. We thought that a detailed study of this phenomenon could have provided further clues on the mechanisms by which FGF signaling regulates chondrocyte proliferation an differentiation.
The results presented here show that FGF treatment of RCS chondrocytes induces a strong downregulation of cyclin B1/CDK1 activity, the kinase complex required for entry of cells into mitosis, leading to a G2 arrest in a sizable portion of the cell population. Surprisingly, the inhibition of cyclin B1/CDK1 activity only lasts for 3–4 hours, and then it is restored to high levels as the cells move through G2/M to reach a sustained G1 arrest.
It is interesting to note that our original interpretation of the transient nature of the G2 arrest was that the G2/M transit block was leaky, and thus cells slowly traversed the G2/M phase to eventually accumulate in G1. However, the data shown here clearly show that cyclin B1/CDK1 kinase activity is depressed for the first 3–4 hours of FGF treatment and then shows a strong rebound to levels higher than those shown by untreated cells. Thus the G2 block is quite stringent, but it is clearly reversed at later times by mechanisms that are presently not understood.
The downregulation of cyclin B1/CDK1 activity is a direct result of FGF signaling and not a consequence of G2 arrest, since cells arrested in mitosis by Nocodazole still show inhibition of cyclin B/CDK1 activity in response to FGF. Furthermore, it does not appear to require activation of gene transcription, since it is detectable also in cells treated with Actinomycin D (data not shown). We could also show that the G2 and G1 blocks induced by FGF occur through independent mechanisms, as relieving the G1 arrest response by constitutive expression of the cyclin D1/CDK4 complex, which prevents p107 dephosphorylation, did not affect the transient downregulation of cyclin B1/CDK1 activity and consequent G2 block and similar results were obtained by inhibiting the activity of the PP2A phosphatase by expressing the SV40 small T antigen. G2 arrest and downregulation of cyclin B1/CDK1 activity are well known features of DNA damage response in mammalian cells. Activation of the G2 check point by DNA damage/genotoxic stress is mediated by ATM/ATR and Chk1/2 kinases and leads to CDK1 inhibition.31 This scenario is very similar to what we observe in the RCS response to FGF. We therefore tested whether inhibition of the ATM/ATR kinases by caffeine, which can abrogate DNA damage-induced G2 arrest,32 would have prevented the FGF-induced G2 arrest. Caffeine did not prevent the G2 arrest or the downregulation of cyclin B1/CDK1 activity in FGF treated RCS cells, and similar results were obtained with UCN-01,33 an inhibitor of Chk1. Indeed, these two inhibitors slightly potentiated the effect of FGF (Sup. Fig. 1). Furthermore, although FGF decreases phosphorylation of CDC25C, this process was not affected by Chk1 or ATM inhibitors. Chk1 independence of FGF-induced G2 arrest in chondrocytes may also reflect the fact that rodent homologs of CDC25C do not have S216, the Chk1/2 phosphorylation site that has been shown in human and Xenopus to be important for binding with the 14-3-3 protein and retaining CDC25C in inactive form in the cytoplasm.34
The balance between CDC25C and Wee1/Myt1 activities determines the inhibitory phosphorylation of CDK1. However, the activity of CDC25C that is reflected by its phosphorylation status does not seem to be an important player in FGF treatment of chondrocytes. According to our data, the mediators of FGF-growth-inhibitory response, such as the ERK1/2 pathway and the PP2A phosphatase, are not responsible for dephosphorylation of CDC25C upon FGF treatment and neither is PLK1/3 that has been shown to phosphorylate CDC25C in other systems. Only inhibition of CDK1 by Alsterpaullone resulted in accumulation of hyperphosphorylated form of CDC25C. Thus, CDC25C dephosphorylation upon FGF treatment likely results from reduced CDK1 activity.
Increased Myt1 activity and expression are likely to play a role in the increased inhibitory phosphorylation of CDK1 upon FGF treatment. The mechanism responsible for Myt1 activation is currently unknown. It is interesting to note that the significant changes in Myt1 kinase activity are not reflected by changes in its phosphorylation status, as was observed in human cell lines and Xenopus extracts.35,36 This could be either a feature of the rodent homolog or an as of yet unidentified mechanism that regulates Myt1 activity. Strong downregulation of cyclin B1/CDK1 activity might also be mediated by FGF-induced binding of an inhibitory molecule. Although genes such as GADD45 and p21 are induced by FGF, these molecules, which have been also shown to play a role in activating the G2 check point,37,38 are unlikely to play a major role as their induction occurs at later times while inhibition of cyclin B1/CDK1 activity takes place within 1 hour of FGF treatment. We could not detect any binding of GADD45 to CDK1 as assayed by immunoprecipitation using anti-CDK1 antibody. Binding of p21 with cyclin B1/CDK1 complexes was indeed detected in FGF-treated RCS cells, but only after 12 h of FGF treatment after the G2 block was released and most of the cells had accumulated in G1/G0.
In conclusion, FGF signaling rapidly activates mechanisms that lead to inhibition of cyclin B1/CDK1 activity and a consequent G2 arrest in RCS chondrocytes that appears to be mediated by increased activity of the Myt1 kinase that performs inhibitory phosphorylation of CDK1 at Y151 and T14. The relevance of the transient G2 arrest to the sustained G1 block that follows it is unclear. We tried to counteract the downregulation of cyclin B1/CDK1 activity induced by FGF by constitutively expressing cyclin B1 and CDK1 in RCS cells, but we could never obtain cells detectably overexpressing cyclin B1, suggesting a negative effect on cell viability. On the other hand, as shown here, RCS cells can arrest in G1 following FGF treatment without traversing the G2 phase of the cell cycle. However, the maximum effect of FGF on G1 synchronized cells requires several hours of pre- exposure before release. It is therefore possible that the transient G2 block serves as a “pausing” mechanism that allows the growth-inhibitory FGF response to be fully established. It is also conceivable that, while G2 arrest may not be required per se, the inactivation of the cyclin B1/CDK1 complex affects as yet unidentified substrates or pathways which play a role the growth-inhibitory response of chondrocytes to FGF.
Materials and Methods
Reagents and antibodies.
All chemicals were from Sigma-Aldrich (St. Louis, MO), ocadaic acid from Calbiochem (La Jolla, CA), ATP from New England BioLabs (Ipswich, MA), γP32 ATP from PerkinElmer (Waltham, MA). The following antibodies were used: anti-phospho-histone H3(S10), anti-phospho-CDC2(T14) (Abcam), anti-cyclin B1, anti-phospho-CDC2 (Y15), anti-phospho-CDC2(T161), anti-Myt1 (Cell Signaling Technology), anti-CDC2, anti-CDC25C, anti-Wee1, agarose-conjugated anticyclin B1 (Santa Cruz Biotechnology), anti-α-tubulin (clone B-5-1-2) and anti-actin (Sigma-Aldrich).
Cell culture and FACS analysis.
Rat chondrosarcoma (RCS) cells were maintained in DMEM supplemented with 10% fetal calf serum at 37°C and 9% CO2. Cells were treated with FGF1 (5 ng/ml) (a kind gift from M. Mohamadi, NYU) and heparin (5 µg/ml) in the presence or absence of Nocodazole (400 ng/ml), Alsterpaullone (10 uM), U0126 (50 uM) and GW843682X (10 uM) as indicated in figure legends.
For cell cycle analysis, cells were fixed with 70% ethanol, washed with PBS, permeabilized with 0.25% Triton X-100 for 5 min, washed with PBS and stained with antibodies against phospho-histone H3(S10) for 1 h. Then, the cells were washed with PBS, incubated with FITC-conjugated secondary antibodies for 30 min, washed twice, resuspended in PBS supplemented with propidium iodide (50 µg/ml) and RNase A (100 µg/ml) and incubated at 37°C for 2 hours. Flow cytometry was performed using FACScan™ (Becton Dickinson) and analyzed using either ModFit LT™ (Verity Software House) or CellQuest software.
Adenoviral infection.
For adenoviral infection, chondrocytes were trypsinized, resuspended in TRIS buffer at a final concentration of 2 × 106 cells/ml and exposed to 10 pfu/cell of Ad-GFP or Ad-ST SV40 in suspension for 1 h at room temperature. After 20 h, cells were treated with FGF1 and heparin as indicated.
Immunoprecipitation, western blot analysis and in vitro kinase assay.
Protein lysates were prepared using modified RIPA buffer (50 mM Tris HCl pH 7.4, 150 mM NaCl, 10 mM KCl, 1% NP-40, 1 mM EDTA) in the presence of phosphatase inhibitors (1 mM Na3VO4, 10 mM NaF 10 mM Na4P2O7) and protease inhibitors (leupeptin, pepstatin and aprotinin 1 µg/ml each). For immunoprecipitation, 1 mg of total protein was pre-cleared by incubation for 30 min at 4°C with Protein G-Sepharose® 4B Conjugate (ZYMED) and then incubation with 4 µg of agarose-conjugated anti-cyclin B1 antibody overnight at 4°C. For Myt1 immunoprecipitates, 5 µg of anti-Myt1 antibodies were used and Protein G-Sepharose was added for 1 h at 4°C. Agarose-conjugated mouse or rabbit IgG as used as a negative control. The immune complexes were washed three times with 1 ml of modified RIPA buffer, resolved on SDS-PAGE and analyzed by immunoblotting. The determination of cyclin B1/CDK1 activity on histone H1 was done as described previously for cyclin E/CDK2 complexes. For Myt1, kinase assay immunoprecipitates from 0.2 mg of total cellular protein were washed twice with buffer A (50 mM Tris HCl pH 7.5, 150 mM NaCl, 1% NP-4, 1 mM DTT) and two times more with buffer B (50 mM Hepes pH 7.5, 10 mM MgCl2, 1 mM DTT, 25 mM β-glycerophosphate). Both buffers were supplemented with phosphatase and protease inhibitors. After the final wash, immunoprecipitates were resuspended in 40 µl of buffer B and supplemented with 200 µM ATP and 0.4 µg of cyclin B1/CDK1 complexes (Millipore). Reaction mixtures were incubated for 30 min at 30°C, stopped by adding 10 µl of 5 X SDS loading buffer and analyzed by immunoblotting against phospho-CDK1(Y15). λ-phosphatase assay was performed according to a manufacture protocol (New England Biolabs).
Acknowledgements
We wish to thank Dr. Alka Mansukhani and Dr. Lisa Dailey for advice and critical reading of the manuscript, Jeff Kraynek for technical assistance and Dr. M. Pagano for advice and for providing us with valuable plasmids. We also thank K. Rundell and R. Schneider for providing us with recombinant adenovirus vectors. This investigation was supported by PHS Grant DE013745 from the NIDCR.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/13671
Supplementary Material
References
- 1.Dailey L, Ambrosetti D, Mansukhani A, Basilico C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 2005;16:17–19. doi: 10.1016/j.cytogfr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 2.Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16:1446–1465. doi: 10.1101/gad.990702. [DOI] [PubMed] [Google Scholar]
- 3.Goldfarb M. Functions of fibroblast growth factors in vertebrate development. Cytokine Growth Factor Rev. 1996;7:311–325. doi: 10.1016/s1359-6101(96)00039-1. [DOI] [PubMed] [Google Scholar]
- 4.Laplantine E, Rossi F, Sahni M, Basilico C, Cobrinik D. FGF signaling targets the pRb-related p107 and p130 proteins to induce chondrocyte growth arrest. J Cell Biol. 2002;158:741–750. doi: 10.1083/jcb.200205025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kolupaeva V, Laplantine E, Basilico C. PP2A-mediated dephosphorylation of p107 plays a critical role in chondrocyte cell cycle arrest by FGF. PLoS One. 2008;3:e3447. doi: 10.1371/journal.pone.0003447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aikawa T, Segre GV, Lee K. Fibroblast growth factor inhibits chondrocytic growth through induction of p21 and subsequent inactivation of cyclin E-Cdk2. J Biol Chem. 2001;276:29347–29352. doi: 10.1074/jbc.M101859200. [DOI] [PubMed] [Google Scholar]
- 7.Dailey L, Laplantine E, Priore R, Basilico C. A network of transcriptional and signaling events is activated by FGF to induce chondrocyte growth arrest and differentiation. J Cell Biol. 2003;161:1053–1066. doi: 10.1083/jcb.200302075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krejci P, Bryja V, Pachernik J, Hampl A, Pogue R, Mekikian P, et al. FGF2 inhibits proliferation and alters the cartilage-like phenotype of RCS cells. Exp Cell Res. 2004;297:152–164. doi: 10.1016/j.yexcr.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 9.Gautier J, Minshull J, Lohka M, Glotzer M, Hunt T, Maller JL. Cyclin is a component of maturation-promoting factor from Xenopus. Cell. 1990;60:487–494. doi: 10.1016/0092-8674(90)90599-a. [DOI] [PubMed] [Google Scholar]
- 10.Dessev G, Iovcheva-Dessev C, Bischoff JR, Beach D, Goldman R. A complex containing p34cdc2 and cyclin B phosphorylates the nuclear lamin and disassembles nuclei of clam oocytes in vitro. J Cell Biol. 1991;112:523–533. doi: 10.1083/jcb.112.4.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krek W, Nigg EA. Cell cycle regulation of vertebrate p34cdc2 activity: Identification of Thr161 as an essential in vivo phosphorylation site. New Biol. 1992;4:323–329. [PubMed] [Google Scholar]
- 12.Booher RN, Holman PS, Fattaey A. Human Myt1 is a cell cycle-regulated kinase that inhibits Cdc2 but not Cdk2 activity. J Biol Chem. 1997;272:22300–22306. doi: 10.1074/jbc.272.35.22300. [DOI] [PubMed] [Google Scholar]
- 13.Parker LL, Piwnica-Worms H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science. 1992;257:1955–1957. doi: 10.1126/science.1384126. [DOI] [PubMed] [Google Scholar]
- 14.Kumagai A, Dunphy WG. The cdc25 protein controls tyrosine dephosphorylation of the cdc2 protein in a cell-free system. Cell. 1991;64:903–914. doi: 10.1016/0092-8674(91)90315-p. [DOI] [PubMed] [Google Scholar]
- 15.Izumi T, Maller JL. Elimination of cdc2 phosphorylation sites in the cdc25 phosphatase blocks initiation of M-phase. Mol Biol Cell. 1993;4:1337–1350. doi: 10.1091/mbc.4.12.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poon RY, Chau MS, Yamashita K, Hunter T. The role of Cdc2 feedback loop control in the DNA damage checkpoint in mammalian cells. Cancer Res. 1997;57:5168–5178. [PubMed] [Google Scholar]
- 17.Hoffmann I, Clarke PR, Marcote MJ, Karsenti E, Draetta G. Phosphorylation and activation of human cdc25-C by cdc2—cyclin B and its involvement in the self-amplification of MPF at mitosis. EMBO J. 1993;12:53–63. doi: 10.1002/j.1460-2075.1993.tb05631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang R, He G, Nelman-Gonzalez M, Ashorn CL, Gallick GE, Stukenberg PT, et al. Regulation of Cdc25C by ERK-MAP kinases during the G2/M transition. Cell. 2007;128:1119–1132. doi: 10.1016/j.cell.2006.11.053. [DOI] [PubMed] [Google Scholar]
- 19.Roshak AK, Capper EA, Imburgia C, Fornwald J, Scott G, Marshall LA. The human polo-like kinase, PLK, regulates cdc2/cyclin B through phosphorylation and activation of the cdc25C phosphatase. Cell Signal. 2000;12:405–411. doi: 10.1016/s0898-6568(00)00080-2. [DOI] [PubMed] [Google Scholar]
- 20.Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501–1505. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- 21.Mochida S, Ikeo S, Gannon J, Hunt T. Regulated activity of PP2A-B55 delta is crucial for controlling entry into and exit from mitosis in Xenopus egg extracts. EMBO J. 2009;28:2777–2785. doi: 10.1038/emboj.2009.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Margolis SS, Perry JA, Forester CM, Nutt LK, Guo Y, Jardim MJ, et al. Role for the PP2A/B56delta phosphatase in regulating 14-3-3 release from Cdc25 to control mitosis. Cell. 2006;127:759–773. doi: 10.1016/j.cell.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Margolis SS, Perry JA, Weitzel DH, Freel CD, Yoshida M, Haystead TA, et al. A role for PP1 in the Cdc2/Cyclin B-mediated positive feedback activation of Cdc25. Mol Biol Cell. 2006;17:1779–1789. doi: 10.1091/mbc.E05-08-0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forester CM, Maddox J, Louis JV, Goris J, Virshup DM. Control of mitotic exit by PP2A regulation of Cdc25C and Cdk1. Proc Natl Acad Sci USA. 2007;104:19867–19872. doi: 10.1073/pnas.0709879104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindqvist A, Rodriguez-Bravo V, Medema RH. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J Cell Biol. 2009;185:193–202. doi: 10.1083/jcb.200812045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lansing TJ, McConnell RT, Duckett DR, Spehar GM, Knick VB, Hassler DF, et al. In vitro biological activity of a novel small-molecule inhibitor of polo-like kinase 1. Mol Cancer Ther. 2007;6:450–459. doi: 10.1158/1535-7163.MCT-06-0543. [DOI] [PubMed] [Google Scholar]
- 27.Lahusen T, De Siervi A, Kunick C, Senderowicz AM. Alsterpaullone, a novel cyclin-dependent kinase inhibitor, induces apoptosis by activation of caspase-9 due to perturbation in mitochondrial membrane potential. Mol Carcinog. 2003;36:183–194. doi: 10.1002/mc.10114. [DOI] [PubMed] [Google Scholar]
- 28.DeCaprio JA, Ludlow JW, Figge J, Shew JY, Huang CM, Lee WH, et al. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell. 1988;54:275–283. doi: 10.1016/0092-8674(88)90559-4. [DOI] [PubMed] [Google Scholar]
- 29.O'Farrell PH. Triggering the all-or-nothing switch into mitosis. Trends Cell Biol. 2001;11:512–519. doi: 10.1016/s0962-8924(01)02142-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mueller PR, Coleman TR, Kumagai A, Dunphy WG. Myt1: A membrane-associated inhibitory kinase that phosphorylates Cdc2 on both threonine-14 and tyrosine-15. Science. 1995;270:86–90. doi: 10.1126/science.270.5233.86. [DOI] [PubMed] [Google Scholar]
- 31.Niida H, Nakanishi M. DNA damage checkpoints in mammals. Mutagenesis. 2006;21:3–9. doi: 10.1093/mutage/gei063. [DOI] [PubMed] [Google Scholar]
- 32.Zhou BB, Chaturvedi P, Spring K, Scott SP, Johanson RA, Mishra R, et al. Caffeine abolishes the mammalian G(2)/M DNA damage checkpoint by inhibiting ataxia-telangiectasia-mutated kinase activity. J Biol Chem. 2000;275:10342–10348. doi: 10.1074/jbc.275.14.10342. [DOI] [PubMed] [Google Scholar]
- 33.Graves PR, Yu L, Schwarz JK, Gales J, Sausville EA, O'Connor PM, et al. The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. J Biol Chem. 2000;275:5600–5605. doi: 10.1074/jbc.275.8.5600. [DOI] [PubMed] [Google Scholar]
- 34.Takizawa CG, Morgan DO. Control of mitosis by changes in the subcellular location of cyclin-B1-Cdk1 and Cdc25C. Curr Opin Cell Biol. 2000;12:658–665. doi: 10.1016/s0955-0674(00)00149-6. [DOI] [PubMed] [Google Scholar]
- 35.Ruiz EJ, Vilar M, Nebreda AR. A Two-Step Inactivation Mechanism of Myt1 Ensures CDK1/Cyclin B Activation and Meiosis I Entry. Curr Biol. 2010:31. doi: 10.1016/j.cub.2010.02.050. [DOI] [PubMed] [Google Scholar]
- 36.Nakajima H, Yonemura S, Murata M, Nakamura N, Piwnica-Worms H, Nishida E. Myt1 protein kinase is essential for Golgi and ER assembly during mitotic exit. J Cell Biol. 2008;181:89–103. doi: 10.1083/jcb.200708176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin S, Antinore MJ, Lung FD, Dong X, Zhao H, Fan F, et al. The GADD45 inhibition of Cdc2 kinase correlates with GADD45-mediated growth suppression. J Biol Chem. 2000;275:16602–16608. doi: 10.1074/jbc.M000284200. [DOI] [PubMed] [Google Scholar]
- 38.Hitomi M, Shu J, Agarwal M, Agarwal A, Stacey DW. p21Waf1 inhibits the activity of cyclin dependent kinase 2 by preventing its activating phosphorylation. Oncogene. 1998;17:959–969. doi: 10.1038/sj.onc.1202005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.