

Abstract
Inactivation of voltage-gated ion channels is an intrinsic auto-regulatory process necessary to govern the occurrence and shape of action potentials and establish firing patterns in excitable tissues. Inactivation may occur from the open state (open-state inactivation, OSI) at strongly depolarized membrane potentials, or from pre-open closed states (closed-state inactivation, CSI) at hyperpolarized and modestly depolarized membrane potentials. Voltage-gated Na+, K+, Ca2+ and non-selective cationic channels utilize both OSI and CSI. Whereas there are detailed mechanistic descriptions of OSI, much less is known about the molecular basis of CSI. Here, we review evidence for CSI in voltage-gated cationic channels (VGCCs) and recent findings that shed light on the molecular mechanisms of CSI in voltage-gated K+ (Kv) channels. Particularly, complementary observations suggest that the S4 voltage sensor, the S4S5 linker and the main S6 activation gate are instrumental in the installment of CSI in Kv4 channels. According to this hypothesis, the voltage sensor may adopt a distinct conformation to drive CSI and, depending on the stability of the interactions between the voltage sensor and the pore domain, a closed-inactivated state results from rearrangements in the selectivity filter or failure of the activation gate to open. Kv4 channel CSI may efficiently exploit the dynamics of the subthreshold membrane potential to regulate spiking properties in excitable tissues.

Robert Bähring (left) trained as a biologist and did his PhD on retinal ion channels in Munich in 1994. After doing a Post-Doc with Mark L. Mayer at NIH, Bethesda, working on glutamate receptor biophysics, he returned to Germany where he entered the potassium channel field by joining the group of Olaf Pongs in Hamburg in 1998. Since 2006 he has been at the Center for Experimental Medicine of the University Clinical Center Hamburg-Eppendorf (UKE). Manuel Covarrubias (right) received his MD and PhD degrees from the Autonomous National University of Mexico (UNAM, Mexico City). He was then trained in receptor and ion channel biophysics and molecular biology at the Max Planck Institute (Dortmund, Germany) and Washington University in St Louis (MO, USA). Currently, he is Professor in the Department of Neuroscience at Jefferson Medical College of Thomas Jefferson University (Philadelphia, Pennsylvania, USA). For many years, Drs Bähring and Covarrubias have shared an interest in the intriguing mechanisms of inactivation of Kv4.x channels and their modulation by accessory β-subunits.
Introduction
Ion channels of excitable membranes employ self-regulatory mechanisms to tone down their activity whenever the activating stimuli (membrane potential change or increase in neurotransmitter concentration) are prolonged. Inactivation and desensitization are the terms typically associated with these mechanisms in voltage-gated and ligand-gated ion channels, respectively. This article reviews the inactivation mechanisms of voltage-gated cationic channels (VGCCs) typically expressed in neurons and muscle cells. Inactivation was first characterized by Hodgkin and Huxley in their classic studies of the Na+ conductance in the squid giant axon (Hodgkin & Huxley, 1952; Aldrich, 2001). Since then, biophysicists and physiologists have intently investigated inactivation of VGCCs at the functional and structural levels, and have realized that there are multiple types of inactivation involving distinct and complex molecular mechanisms. Upon changes in membrane potential (e.g. a depolarization), VGCCs may essentially inactivate from pre-open closed-states (closed-state inactivation, CSI) or from the open state(s) (open-state inactivation, OSI). Most VGCCs use both CSI and OSI. However, some VGCCs undergo more inactivation from the open state and others undergo more inactivation from pre-open closed states. We refer to these distinct behaviours as preferential OSI and preferential CSI, respectively. In either case, recovery from inactivation usually occurs when the membrane potential is returned to its initial value (e.g. repolarized). Kv4 channels, a subclass of voltage-gated K+ (Kv) channels, undergo unstable, seemingly vestigial, OSI but prominent and physiologically relevant CSI (Jerng et al. 2004). OSI mechanisms are relatively well known at the biophysical and structural levels (reviewed by Rasmusson et al. 1998; Yellen, 1998; Aldrich, 2001; Kurata & Fedida, 2006). By contrast, the molecular basis of CSI is not fully understood. CSI may involve mechanisms related to those invoked for OSI or novel mechanisms that need further investigation. Here, we review CSI in VGCCs, and discuss advances made in recent years towards understanding the mechanisms of CSI. Particularly, we entertain novel working hypotheses that may explain the molecular basis of CSI in Kv4 channels. We also discuss the general applicability of the proposed CSI mechanism and present a perspective of its structural and biological implications.
Pathways of inactivation gating in VGCCs
VGCCs may adopt four distinct conformational states: resting, partially activated, open and inactivated. The open state is conducting, whereas the resting, partially activated and inactivated states are non-conducting. In the partially activated states, the channels are still closed but eventually become opening-permissive. By contrast, in the inactivated states, the channels are either opening-reluctant despite being partially activated or the open conformation has become non-conducting. Thus, in the resting, partially activated and inactivated states ion flow is prevented by certain channel conformations, including physical occlusion of the ion-selective pore. However, the mechanisms that control channel closing and channel inactivation are thought to be structurally distinct and are, therefore, referred to as different ‘gates’. A significant implication of this concept is that at least two separate gates may co-exist in VGCCs: the activation gate and at least one inactivation gate. Later, we will discuss important exceptions to this general concept.
Figure 1 depicts general kinetic gating schemes illustrating how the conformational states might be interconnected in VGCCs. C0 is the resting conformation and C1–Cn are the partially activated conformations, which develop sequentially before the channel reaches the open conformation O. Cn is the opening-permissive conformation, and n may conventionally vary in more specific schemes between 3 and 7 (Vandenberg & Bezanilla, 1991; Ayer & Sigworth, 1997; Olcese et al. 1997; Burgess et al. 2002; Talavera & Nilius, 2006). From this basic framework, two possible pathways of inactivation emerge: (1) VGCCs may inactivate in a strictly coupled manner only after they have fully activated and opened (i.e. OSI; Scheme I); and (2) VGCCs may inactivate from pre-open closed states (partially activated but still closed), which can be reached before the channels have opened or after they have closed (i.e. CSI; Schemes II and III). Thus, inactivation is strongly coupled to activation in Schemes I and II. Scheme III is a more general case in which resting, partially activated and open conformations are inactivation-permissive. Here, activation and inactivation may proceed independently, or inactivation transitions may become more likely as voltage-dependent activation progresses, implying an allosteric mechanism. An important general feature of the schemes in Fig. 1 is that the rate constants that control activation (transitions between C0 and Cn, and between I0 and In) are strongly voltage dependent, whereas those controlling pore opening and inactivation carry little or no voltage dependence. The development of and recovery from inactivation may, however, exhibit an apparent voltage dependence if the inactivation pathways are coupled to voltage-dependent transitions in the activation pathway. Recovery from inactivation and the eventual return to the resting state (C0) may occur upon repolarization via re-opening (if channels reside in an open-inactivated state) or electrically ‘silent’, via a direct return to Cn (if channels reside in a closed-inactivated state).
Figure 1. Kinetic schemes of activation and inactivation gating in VGCCs.
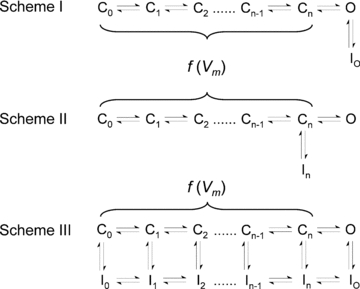
C, O and I represent closed, open and inactivated states, respectively. C0 is the resting state, C1–Cn−1 represents a set of partially activated states, and Cn is the opening-permissive state. In three hypothetical situations, VGCCs may inactivate either from the O state only (Scheme I, open-state inactivation, OSI), from the closed state Cn only (Scheme II, closed-state inactivation, CSI) or from both open and closed states (Scheme III, OSI and CSI). IO and In are open-inactivated and closed-inactivated states, respectively; and I0–In−1 represents a set of closed-inactivated states directly associated with inactivation-permissive closed states. Transitions between C states are assumed to be strongly voltage dependent (f(Vm) indicates voltage-dependent transitions). Therefore, they involve conformational changes of the channel's voltage sensors. In specific kinetic models, n may range between 3 and 7 (text). VGCCs have four voltage sensors, which upon voltage-dependent activation move sequentially and independently in their tetrameric or pseudo-tetrameric assemblies. By contrast, the opening step and inactivation transitions may carry little or no voltage dependence. To open the pore, a single activation gate undergoes a concerted conformational change following activation of four voltage sensors and additional pre-open rearrangements. Whereas inactivation is tightly coupled to voltage-dependent activation in Schemes I and II, Scheme III assumes that activation and inactivation are independent or weakly coupled.
CSI in voltage-gated ion channels
OSI and CSI may overlap in a time- and/or voltage-dependent manner, which confounds the separation of the two gating pathways and complicates the study of the underlying mechanisms. The decaying phase of the macroscopic current evoked by a step depolarization reflects inactivation of an ensemble of VGCCs. Thus, we observe macroscopic inactivation and can analyse its kinetic properties. However, the underlying state transitions cannot be defined by such a simple analysis. Let's assume a channel in which activation is fast and not rate limiting. If this channel only undergoes OSI, macroscopic inactivation reflects OSI kinetics. Alternatively, if the channel only undergoes CSI, macroscopic inactivation may reflect channel closure and subsequent CSI. Finally, if the channel undergoes both OSI and CSI, but shows preferential CSI, then macroscopic inactivation most likely reflects a complicated overlap of OSI, channel closure and CSI. In the light of these considerations and the lack of an acid test to unambiguously demonstrate CSI, researchers have typically inferred CSI by combining complementary electrophysiological approaches. Before discussing these investigations in greater detail for relevant examples of CSI more closely, we will outline typical electrophysiological approaches.
Voltage dependence of steady-state inactivation and kinetics of macroscopic inactivation
A variety of voltage pulse protocols in the voltage and time domains can test the impact of conditioning pulses on the availability of functional channels, and the kinetics of cumulative inactivation induced by repetitive brief pulses. In particular, conditioning prepulse protocols can ask whether substantial inactivation occurs at voltages that induce no perceptible channel opening, and whether the prepulse inactivation curve exhibits U-type features. The latter suggests preferential CSI because the prepulse inactivation curve displays deep inactivation at intermediate voltages and progressively less complete inactivation at more positive voltages. Notwithstanding, caution is necessary when interpreting the results from prepulse protocols because slow OSI can occur at a very low open probability. Separately, the voltage dependence of the observed time constant of macroscopic inactivation may also help infer preferential CSI.
The voltage dependence of gating charge movement
Since CSI is electrically ‘silent’, gating current measurements can yield more direct special information about the underlying mechanisms because they reflect the conformational changes of the voltage sensors before the channels open. A key criterion is the observation of apparent gating charge immobilization, which occurs when the relaxation of the gating current is very slow and, therefore, gating charge movement becomes imperceptible. This is an apparent loss of gating charge because voltage-dependent gating must obey the principle of charge conservation. At a more fundamental level, apparent gating charge immobilization is a necessary consequence of inactivation coupled to activation. Since activation promotes inactivation and vice versa, gating charge movement is favoured among closed-inactivated states. Therefore, if a modest depolarization induces CSI and the available gating charge quickly moves among closed-inactivated states, a subsequent stronger depolarization detects an apparent loss of gating charge (i.e. the gating charge appears immobilized).
The stochastic properties of unitary currents
Single channel recordings (unitary currents) may provide the most conclusive electrophysiological evidence for CSI. One can investigate how the conditional probability of observing unitary activity depends on (1) the presence of unitary activity during a prepulse, and (2) the probability of observing the first opening a certain time after the onset of a voltage pulse.
Computer modelling of macroscopic and unitary currents
To test specific pathways of CSI, analytical methods and numerical modelling compare the experimental outcomes of the approaches outlined above with the calculated predictions of specific kinetic schemes. Toward this goal, quantitative global kinetic modelling is particularly powerful. This highly constrained analysis evaluates model predictions quantitatively against data sets that simultaneously examine a broad range of voltages, time scales and activities of the channels (e.g. ionic and gating currents). Furthermore, maximum likelihood methods applied to single channel recordings are also exploited to rigorously compare plausible kinetic schemes of inactivation gating.
Specific published examples of the approaches summarized above will be discussed for individual types of VGCCs throughout the remainder of the review. Also, for certain VGCCs displaying CSI or preferential CSI, we overview the inferred molecular determinants and discuss conceptual and structural models of inactivation gating.
Nav channels
Although the original studies by Hodgkin and Huxley suggested CSI in Nav channels (Hodgkin & Huxley, 1952), convincing quantitative evidence for inactivation occurring from pre-open closed states came from investigating the Nav channel in the crayfish giant axon (Bean, 1981). This study compared the current kinetics in response to a single activating pulse to the inactivation onset kinetics investigated with a prepulse protocol. The prepulse protocol essentially tested the impact of a low voltage conditioning step of variable duration on the availability of functional Nav channels activated by a constant depolarizing test pulse. Quantitative inspection of the data showed that it takes significantly longer for the currents to reach their peak than for prepulse inactivation to develop. Thus, inactivation may occur before the Nav channels open. Such a discrepancy between the probability of opening and the probability of inactivation, an indication of CSI, was directly examined in single Nav channel recordings from rat myotubes (Horn et al. 1981). Essentially, these experiments showed a direct relationship between the magnitude of inactivation and the time it takes for the first opening to occur, suggesting that inactivation could happen before opening. Further quantitative testing of multiple kinetic schemes by maximum likelihood methods confirmed the plausibility of CSI (Horn & Vandenberg, 1984). A study by Aldrich & Stevens (1983) then established the occurrence of CSI in single neuroblastoma Nav channels by showing substantially reduced open probability when the activating voltage pulse occurs after a conditioning prepulse that elicits no unitary activity. Later studies also demonstrated that the recovery of Nav channels from inactivation at hyperpolarized potentials starts with a delay and is nearly electrically silent (Kuo & Bean, 1994), suggesting that channels do not instantaneously begin to recover from inactivation by re-opening. Moreover, the recovery kinetics were strongly voltage dependent and exhibited saturation at extremely hyperpolarized membrane potentials, consistent with an allosteric mechanism in which recovery from inactivation involves pre-open closed states. Overall, these results showed that Nav channels actually deactivate before they recover from inactivation (Kuo & Bean, 1994). Altogether, these experimental results have been explained qualitatively and quantitatively by assuming significant CSI in combination with prominent OSI in Nav channels (Bean, 1981; Aldrich & Stevens, 1983; Horn & Vandenberg, 1984; Patlak, 1991; Kuo & Bean, 1994; Armstrong, 2006).
A combination of electrophysiological and modelling approaches has provided a structural framework for Nav channel CSI (Patlak, 1991; Armstrong, 2006). Nav channels are formed by a single polypeptide chain with four similar repeats of a six transmembrane segment motif (domain I–IV). Nav channel inactivation is mediated by a short segment of the channel polypeptide located between domains III and IV, which takes the form of a ‘hinged lid’ (Vassilev et al. 1988; West et al. 1992; Kellenberger et al. 1997a,b; Catterall, 2000). This inactivation particle requires voltage sensor movement to gain access to its binding site (Fig. 2A). Upon strong depolarization all Nav channel voltage sensors move outwardly, the activation gate opens and the inactivation particle plugs the pore (OSI). However, the four voltage sensors in Nav channels are not equally important in this respect (Patlak, 1991; Horn et al. 2000; Armstrong, 2006): if only the voltage sensors in domains III and IV move, the channel does not open, but this movement is thought to be sufficient to allow access of the inactivation particle to its binding site, resulting in CSI (Fig. 2A).
Figure 2. Cartoon representations of plausible mechanisms of inactivation gating in Nav channels (A) and spHCN channels (B).
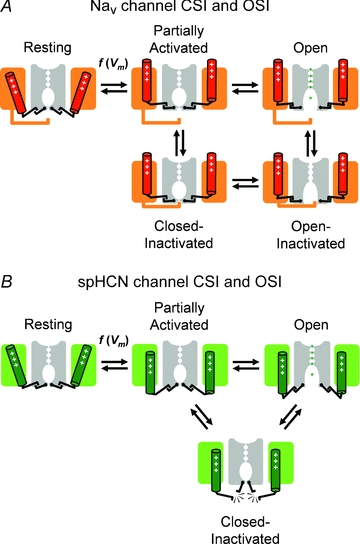
Two opposing subunits (or pseudosubunits in Nav channels) of a fourfold symmetric channel are shown. Each subunit includes a voltage sensing domain (coloured) and a pore domain (grey). In the open conformation, ions are explicitly represented in the permeation pathway. As explained in Fig. 1, the activation steps are voltage dependent. Assuming a mechanical analogy, the voltage sensors and activation gate are shown ‘engaged’ in all conformations in the Nav channel. Inactivation of this channel occurs when an intracellular inactivation gate (‘hinged lid’) occludes the pore before or after opening of the activation gate. Note that the outward movement of the voltage sensor is necessary to allow interaction of the inactivation gate with its receptor site in the pore domain. By contrast, inactivation of spHCN channels may occur when the voltage sensors and activation gates ‘disengage’. Thus, the partially activated inactivation-permissive state may undergo inactivation, and ‘reclosure’ may cause inactivation from the open state.
Cav channels
Cav channels also undergo CSI. However, despite the close topological relationship between Nav and Cav channels, CSI in the latter case exhibits specialized features. N-type (neuronal, Cav2.2) and R-type (dihydropyridine and toxin-resistant, Cav2.3) Cav channels display fairly rapid cumulative inactivation in response to repeated action potential-like pulsing, whereas a single extended voltage pulse induces a much slower onset of inactivation (Patil et al. 1998). Upon repeated brief stimulation, these Cav channels visit the open state less frequently, but run more often through pre-open (partially activated) closed states, from which they prefer to inactivate (preferential CSI). As a hallmark of preferential CSI, voltage-dependent inactivation of N- and R-type Cav channels exhibits the ‘U-type’ profile (Jones & Marks, 1989; Patil et al. 1998; also termed ‘J-type’). Accordingly, inactivation curves show a minimum at certain voltages, indicating that the rate of onset of inactivation is fastest and steady-state inactivation most pronounced at intermediate voltages. Stronger depolarizations, which favour channel opening, do not promote fast voltage-dependent inactivation of N- and R-type Cav channels. These features are not typically observed in Nav channels, which undergo a combination of CSI and prominent OSI. Preferential CSI of Cav channels is not dependent on a conducting open state because it is not influenced by the use of Ba2+ instead of Ca2+ as charge carrier, and is insensitive to internal perfusion with a Ca2+ chelator (Jones & Marks, 1989; Patil et al. 1998). Further support for the idea that Cav channel preferential CSI is solely coupled to voltage-dependent conformational changes and not to channel opening came from gating current measurements. Namely, Cav channel preferential CSI is fastest at a membrane potential where only one-third of the gating charge has moved (Jones et al. 1999).
Evidence for CSI in L-type and T-type Ca2+ channels (Cav1.x and Cav3.x, respectively) also emerged from the analysis of ionic and gating currents, and computer modelling (Shirokov et al. 1998; Burgess et al. 2002; Tadross & Yue, 2010). Resembling the behaviour of Nav and Kv channels (Bezanilla et al. 1982; Ruben et al. 1992; Fedida et al. 1996; Olcese et al. 1997; Dougherty et al. 2008), Cav1.2 channels exhibit slow gating charge immobilization indicating that the gating charge may move distinctly among partially activated closed states and among closed-inactivated states (e.g. Scheme III, Fig. 1). We will later discuss this general phenomenon further in the context of ‘voltage sensor inactivation’ (see Perspective on CSI mechanisms and the biological implications). More recently, an elegant straightforward combination of analytical expressions and electrophysiological measurements demonstrated Ca2+-dependent CSI in Cav1.3 channels (Tadross & Yue, 2010). Separately, an allosteric scheme of CSI has also provided an economical way to numerically model the complex kinetic and voltage-dependent properties of Cav3.2 channels (Burgess et al. 2002; Talavera & Nilius, 2006).
The structural determinants of the fast and voltage-dependent Cav channel inactivation including CSI differ from the ones identified for Nav channels. In Cav channels, the linker region between domains I and II and the S6 region in domain III play crucial roles in inactivation (Herlitze et al. 1997; Stotz et al. 2000; Talavera & Nilius, 2002). Despite structural differences, Cav channel CSI may in part be governed by mechanisms similar to those of Nav channels, but more work is necessary to establish this relationship (Talavera & Nilius, 2002; Tadross et al. 2010).
HCN channels
CSI has also been demonstrated for the hyperpolarization-activated and cyclic nucleotide-regulated channel of the sea urchin (spHCN; Shin et al. 2004). HCN channels are formed by the assembly of four α-subunits, each with six transmembrane segments very similar to those of Kv channels. However, HCN channels are non-selective cationic channels and their voltage dependence is reversed. Since cAMP binding prevents inactivation of spHCN channels a different experimental approach was used to demonstrate CSI. In an elegant series of experiments, Shin and coworkers showed that the access of the specific open-channel blocker ZD7288 was prevented in the absence of cAMP. In particular, with repeated hyperpolarizing voltage pulses (channel activation), the irreversible blockade of wild-type spHCN channels by ZD7288 developed much slower in the absence of cAMP (i.e. when inactivation occurs), and reversible blockade of a specific channel mutant was relieved only in the presence of cAMP (i.e. when inactivation does not occur). Thus, spHCN channel inactivation not only prevented access of an open-channel blocker, but also supported the trapping of this agent. Conversely, Shin et al. also showed that keeping spHCN channels in a ‘locked-open’ state, by a Cd2+ bridge between two engineered cysteines, prevented inactivation completely, and cAMP no longer influenced gating in these channels (Shin et al. 2004). These results strongly support the occurrence of CSI in spHCN channels. Furthermore, this study indicates that spHCN channel CSI does not arise from a separate specialized inactivation gate. Rather, it is the main activation gate, which, by having a dual role, is also responsible for CSI.
Shin and coworkers have proposed a conceptual model of CSI in cAMP-regulated spHCN channels (Shin et al. 2004). According to this model, ‘slippage’ between the voltage sensors and the channel activation gate causes inactivation by ‘re-closure’ of the same gate (Fig. 2B). If the ‘slippage’ occurs after voltage sensor movement but before pore opening, the activation gate remains shut, and the open-state can never be reached. This mechanism of CSI is likely to occur in Kv4 channels (see below, Inactivation of Kv4 channels), and may represent the basis of a more general mechanism of inactivation in VGCCs (see below, Perspective on CSI mechanisms and the biological implications).
Kv and BK channels
The prepulse inactivation curves of Kv2.1 (Klemic et al. 1998) and Kv3.1 channels (Klemic et al. 2001) exhibit a U-type profile, and the channels undergo strong cumulative inactivation with repeated brief voltage pulses. These features are consistent with preferential CSI occurring during voltage-dependent activation and deactivation, and closely resemble Cav channel behaviour. These typical inactivation properties become even more prominent when Kv2.1 channels form heteromers with otherwise ‘silent’α-subunits of the Kv5, Kv6, Kv8 or Kv9 subfamily (Salinas et al. 1997; Kramer et al. 1998; Kerschensteiner & Stocker, 1999; Kerschensteiner et al. 2003). Moreover, U-type features of prepulse inactivation have also been observed for N-terminally truncated Shaker channels (ShΔ; Klemic et al. 2001) and a naturally occurring Kv1.5 N-terminal deletion mutant (Kv1.5ΔN209; Kurata et al. 2001). Originally, Aldrich (1981) demonstrated cumulative inactivation possibly involving pre-open closed states in delayed-rectifier Kv channels from molluscan neurons. He showed that, after a voltage pulse, the maximum current evoked by a second pulse was less than the current at the end of the first pulse, indicating that inactivation must have occurred during the interpulse interval at a negative voltage. Separately, computer modelling of non-Shaker A-type K+ currents in neurons from Drosophila (Solc & Aldrich, 1990) and of the transient outward current (Ito) in ferret ventricular myocytes (Campbell et al. 1993) suggested that inactivation coupled to partially activated states in the channel's activation pathway is necessary to explain the observed gating kinetics. Importantly, prepulse inactivation of the ferret Ito occurs at voltages where no opening was detectable (Campbell et al. 1993). Kv4 channels, which also undergo preferential CSI, contribute to the Ito (Dixon et al. 1996). However, Kv4 channels are different in this respect because they show preferential CSI at all relevant voltages, meaning that, even in response to strong depolarizations that induce considerable channel opening, Kv4 channels end up in pre-open closed-inactivated states (Bähring et al. 2001a). This may also explain why U-type features are not so obvious in Kv4 channels (but see Barghaan & Bähring, 2009). The absence of tail currents following a strong depolarization long enough to inactivate all channels suggests preferential CSI at all relevant voltages in Kv4 channels (Bähring et al. 2001a). Thus, contrary to a prediction of Scheme I (Fig. 1; inactivation strictly coupled to channel opening) and in marked contrast to Shaker Kv channels (Demo & Yellen, 1991; Ruppersberg et al. 1991), Kv4 channels do not re-open during recovery from inactivation. Furthermore, Kv4 channel recovery kinetics are identical, no matter if this recovery follows strong (with channel opening) or moderate depolarizations (without channel opening). This observation also strongly suggests preferential CSI at positive voltages. In addition, recovery from inactivation is likely to involve voltage-dependent pre-open closed-state transitions because the recovery kinetics exhibit steep voltage dependence (Bähring et al. 2001a; Dougherty et al. 2008). Intriguingly, isochronal Kv4 channel inactivation curves show different midpoint voltages and different slope factors depending on the prepulse length: shorter prepulses yield more positive midpoint voltages and shallower curves; longer prepulses yield more negative midpoint voltages and steeper curves. These results support the notion that Kv4 channels may transiently visit open-inactivated states, but with longer depolarizing prepulses they end-up in closed-inactivated states (Wang et al. 2005).
Big conductance voltage- and Ca2+-dependent potassium channels (BK or KvCa channels) are closely related to Kv channel proteins, but have seven instead of six transmembrane segments with an extracellular N-terminus. In addition, Ca2+ binding promotes activation of BK channels by shifting the voltage-dependent activation curve toward more negative voltages (Cui et al. 2009). BK channels in chromaffin cells and recombinant BK channels co-expressed with their β2 subunits also exhibit inactivation prior to opening (Ding & Lingle, 2002). At the single-channel level, Ding and Lingle showed that conditioning prepulses without opening events may cause inactivation (i.e. a reduced open probability in response to a test pulse). Thus, channel opening is not required for inactivation of BK channels. Also, BK channel CSI is indirectly dependent on the concentration of cytosolic Ca2+: at low concentrations, where voltage-dependent activation proceeds slower, inactivation from pre-open closed states is more likely (Ding & Lingle, 2002).
Most structure–function studies of Kv channel inactivation have focused on the archetypical Shaker channel and its relatives, and various key structural determinants have been identified. Shaker Kv channel inactivation involves occlusion of the open pore by the cytoplasmic N-terminal region of the channel (Zagotta et al. 1990). This mechanism was termed ‘N-type inactivation’ to describe the involvement of the N-terminal inactivation domain (NT-ID; Hoshi et al. 1990; Zagotta et al. 1990). Shaker Kv channels also exhibit a distinct and typically slower form of inactivation, which is influenced by alternative splicing within the C-terminal region of the channel protein (Hoshi et al. 1991), and was therefore termed ‘C-type inactivation’ (Choi et al. 1991; Hoshi et al. 1991). The pore region and the selectivity filter itself are most significantly involved in this form of inactivation, which has led to the frequent adoption of the term ‘P/C-type inactivation’ (Pardo et al. 1992; De Biasi et al. 1993; Lopez-Barneo et al. 1993; Kurata & Fedida, 2006; see also Fig. 6A). P/C-type inactivation results from a concerted conformational change at the selectivity filter (Ogielska et al. 1995; Panyi et al. 1995; Liu et al. 1996; Kiss & Korn, 1998; Yellen, 1998; Zhou et al. 2001b; Kurata & Fedida, 2006; Ahern et al. 2009), and electrostatic interactions involving H-bonds between the pore helix and the selectivity filter underlie this conformational change (Cordero-Morales et al. 2006, 2007).
Figure 6. Conceptual cartoon representations of plausible general mechanisms of non-N-type inactivation gating in Kv channels.
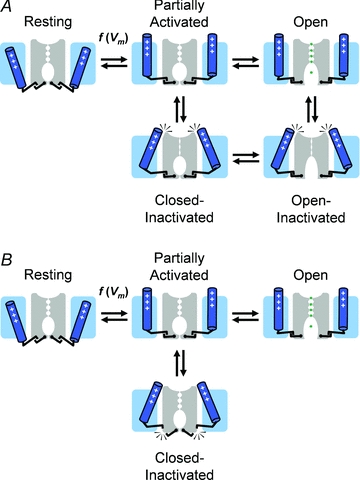
A, P/C-type mechanism of CSI and OSI. B, putative mechanism of CSI. Labelling and representations are as explained in Fig. 2. The voltage sensors play a central role in these mechanisms. Note that the P/C-type mechanism occurs when the channel's selectivity filter ‘collapses’ and this conformation is directly stabilized by the voltage sensors as they adopt a ‘relaxed’ conformation. Alternatively, the voltage sensors adopt a similar ‘relaxed’ conformation but the pore does not ‘collapse’ because there is no stable interaction between the voltage sensors and the selectivity filter. Instead, inactivation results when the voltage sensors and the activation gate ‘disengage’ as a result of a weak interaction (Fig. 2B). This mechanism can thus be seen as a ‘failure to open’ (Fig. 5).
The N- and P/C-type inactivation mechanisms are canonically considered OSI mechanisms. To explain CSI in Kv channels, Zhou et al. (2001a) suggested that the NT-ID may reside in a ‘pre-inactivated’ conformation when bound to a site outside the pore of closed channels. Then, the actual inactivation step may, in some Kv channels and/or under certain conditions (Murrell-Lagnado & Aldrich, 1993a,b;), be so rapid that the channel is unable to conduct when the activation gate opens (Zhou et al. 2001a). We may consider this phenomenon ‘apparent’ CSI, which we must clearly distinguish from ‘real’ CSI. The latter occurs from pre-open partially activated closed states in the voltage-dependent activation pathway (Fig. 1). Apparent CSI is unlikely to underlie the U-type features observed in many Kv channels, because this inactivation phenotype is actually favoured by N-terminal deletions (Klemic et al. 2001; Kurata et al. 2002). Fast P/C-type inactivation as the mechanism underlying the U-type phenomenon in certain Kv channels also seems unlikely because, contrary to a key prediction of the P/C-type mechanism, elevated external K+ concentrations actually accelerate CSI in these Kv channels (Klemic et al. 1998, 2001; Kurata et al. 2002, 2005). These findings support the notion that OSI and CSI in some Kv channels are mediated by distinct mechanisms and that the development of CSI models and identification of the structures involved requires novel concepts.
Enhanced CSI has been observed in N-terminally truncated and L382I point-mutated ShakerB channels (Ayer & Sigworth, 1997). These channels exhibit a positive shift in the voltage dependence of activation of ∼45 mV with only a 9 mV shift in the voltage dependence of steady-state inactivation relative to wild-type. Interestingly, the L382I mutation lies at the S4 end of the S4S5 linker, and mutations in this region had been previously reported to critically influence Shaker channel inactivation (Isacoff et al. 1991). Moreover, the S4S5 linker is critically involved in the coupling of the voltage sensor to the S6 gate in Kv channels (Lu et al. 2002; Yifrach & MacKinnon, 2002; Long et al. 2005). Mutations in the S6 PVP motif of silent α-subunits are also critical for their favourable effects on CSI and U-type features in heteromeric Kv2.1 channels (see above; Kerschensteiner et al. 2003). We will further discuss the roles of the voltage sensor, the S4S5 linker and the S6 gate in the context of Kv4 channel CSI in the next two sections.
Inactivation of Kv4 channels
The Kv4 subfamily of Kv channels is related to the Shal gene of Drosophila and has three members: Kv4.1, Kv4.2 and Kv4.3, with a short and a long Kv4.3 splice variant (Baldwin et al. 1991; Pak et al. 1991; Serodio et al. 1994; Serodio et al. 1996). Kv4 channels have attracted significant attention in cardiovascular physiology and neuroscience because they represent the molecular substrate of both the Ito in cardiac myocytes and the subthreshold-operating somatodendritic A-type K+ current (ISA) in the brain (see Jerng et al. 2004; Patel & Campbell, 2005 for review and see below, Perspective on CSI mechanisms and the biological implications). In contrast to the phenotypic diversity of Kv1 and Kv3 channels, Kv4 channels generate rapidly inactivating A-type K+ currents across the animal kingdom. As we will see, Kv4 channel CSI is prominent among other mechanisms of inactivation that may exist in vestigial forms.
Initial studies of recombinant Kv4 channels showed that a sum of three exponential terms is necessary to adequately describe macroscopic current decay (Baldwin et al. 1991; Jerng & Covarrubias, 1997; Beck & Covarrubias, 2001; Bähring et al. 2001a). Such complex inactivation kinetics may reflect two or more independent inactivation mechanisms overlapping in time, eventually leading to a steady-state distribution of channels among different inactivated states. Alternatively, multiple gating transitions may lead to a single inactivated state. Supporting the latter, the kinetics of recovery from inactivation in Kv4 channels are, despite the multi-exponential onset kinetics, adequately described in general by a single-exponential function (Baldwin et al. 1991; Jerng & Covarrubias, 1997; Bähring et al. 2001a; Dougherty et al. 2008). Moreover, there is strong collective evidence indicating that this final state is closed-inactivated (see above, CSI in voltage-gated ion channels). Irrespective of the depolarization magnitude, Kv4 channels end up in a closed-inactivated state at steady-state; thus, they are ideally suited to study CSI mechanisms. To review our current understanding of Kv4 channel inactivation, the remainder of this section will entertain two central questions: (1) what are the relative contributions of OSI and CSI to the multiphasic current decay? and (2) what are the structural determinants of CSI?
Classical OSI mechanisms in Kv4 channels
To investigate the structure-function relationship of Kv4 channel inactivation, researchers examined the contribution of N- and P/C-type inactivation mechanisms. It became clear early on that classical N-type inactivation, as described for Shaker Kv channels, plays a minor role in Kv4 channel inactivation. Although N-terminal deletions cause modest slowing of Kv4 current decay, the transient A-type character of the current is not abolished (Baldwin et al. 1991; Pak et al. 1991; Jerng & Covarrubias, 1997; Bähring et al. 2001a; Pourrier et al. 2004; Barghaan et al. 2008; Kaulin et al. 2008), as in the case of Shaker Kv channels (Hoshi et al. 1990). Nevertheless, the Kv4 N-terminus confers functional features consistent with N-type inactivation (Gebauer et al. 2004; Pourrier et al. 2004), and Kv4 channels appear to possess a typical NT-ID receptor site because current decay of N-terminally truncated Kv4 cannels is influenced not only by Kv4 N-terminal peptide but also by the ShakerB NT-ID peptide (Gebauer et al. 2004). Despite these features, N-type inactivation may not underlie the final inactivated state in Kv4 channels because their voltage-dependent recovery from inactivation is not affected by N-terminal deletions (Bähring et al. 2001a; Barghaan et al. 2008). Moreover, N-type inactivation is virtually eliminated in native Kv4 channels by integral association with Kv channel-interacting proteins (KChIPs). These accessory β-subunits sequester the proximal NT-ID of Kv4 channels (An et al. 2000; Bähring et al. 2001b; Kim et al. 2004a,b; Scannevin et al. 2004; Zhou et al. 2004; Callsen et al. 2005; Pioletti et al. 2006; Wang et al. 2007). Surprisingly, however, another type of Kv4-specific β-subunit (DPP10a, a dipeptidyl aminopeptidase-like protein) confers robust N-type OSI to certain Kv4 channel complexes under physiological conditions (Jerng et al. 2009).
Although proceeding slowly in most other channels, a fast form of P/C-type inactivation involving the selectivity filter of Kv4 channels may contribute to macroscopic Kv4 channel inactivation. Kv4.3 currents decay more quickly upon complete removal of external K+ (Eghbali et al. 2002). Similar to the K+ conductance of the squid giant axon (Almers & Armstrong, 1980), Kv4 channels seemingly lose their functionality in response to K+ deprivation. A distortion of the selectivity filter, possibly related to classical P/C-type inactivation, is thought to be responsible for this loss of function. However, external TEA, which interacts with the external mouth of the selectivity filter, has no effect on Kv4 channel inactivation (Jerng & Covarrubias, 1997); and elevated external K+ concentrations actually accelerate inactivation without significantly affecting recovery from inactivation of both recombinant Kv4 channels and native ISA (Jerng & Covarrubias, 1997; Kirichok et al. 1998; Bähring et al. 2001a; Shahidullah & Covarrubias, 2003; Kaulin et al. 2008). Conversely, both external TEA and elevated external K+ concentrations prevent P/C-type inactivation; and elevated external K+ accelerates recovery from inactivation in Shaker Kv channels (Choi et al. 1991; Lopez-Barneo et al. 1993; Levy & Deutsch, 1996). Moreover, Kv4 channel recovery from inactivation is complete within tens to hundreds of milliseconds, especially in the presence of accessory β-subunits, whereas recovery of ShakerB and Kv1 channels from P/C-type inactivation usually takes tens of seconds. It is also noteworthy that the T449V mutation eliminates P/C-type inactivation in the ShakerB channel (Lopez-Barneo et al. 1993), but valine already occupies the equivalent position in all Kv4 channels. Thus, except for the drastic effects of a complete removal of external K+, there is no evidence supporting P/C-type inactivation as a major player in Kv4 channel inactivation under physiological conditions.
The contributions of N- and P/C-type mechanisms to Kv4 channel inactivation can be summarized as follows: both mechanisms may exist in vestigial forms. The interaction between the NT-ID and its receptor site in the pore is thought to be much weaker in Kv4 channels than in ShakerB and Kv1 channels. This view is supported by mutational analyses of the respective domains in the Kv4.2 channel (Gebauer et al. 2004) and the inactivation-removed Shaker-related channel Kv1.4IR in combination with its inactivation-conferring Kvβ1-subunit (Zhou et al. 2001a). The results of these two studies demonstrate the involvement of N-terminal residues and pore-lining S6 residues in both channel types. However, much larger thermodynamic coupling coefficients in double-mutant cycle analysis were obtained for Kv1.4IR/Kvβ1 than for Kv4.2 channels. The weaker thermodynamic coupling reflects the vestigial role of OSI mediated by an NT-ID in the Kv4.2 channel. Vestigial P/C-type inactivation may also occur in Kv4 channels. For instance, under physiological ionic conditions and in the presence of auxiliary β-subunits, the selectivity filter of the Kv4.3 channel may enter the P/C-type inactivated state quickly but may never adopt a stable non-conducting conformation. However, this rapid and unstable rearrangement is sufficient to reduce the open probability of Kv4.3 channels. Consistent with this idea, when vestigial P/C-type inactivation is eliminated by exposing the channels to elevated external K+, the maximum open probability increases substantially and, furthermore, channels can now only leave the open state by closing (Kaulin et al. 2008). Increasing the effective closing rate also increases the probability of populating the pre-open inactivation-permissive closed state (Cn, Fig. 1). Thus, inactivation appears accelerated.
Preferential CSI at all relevant voltages in Kv4 channels
The relative contributions of OSI and CSI to Kv4 channel gating have been incorporated in kinetic models similar to Scheme III in Fig. 1 (see also Beck & Covarrubias, 2001; Bähring et al. 2001a; Barghaan et al. 2008; Kaulin et al. 2008). These specific models have two important features: (1) voltage-independent inactivation coupled to voltage-dependent activation; and (2) the opening transition is weakly voltage dependent. Coupling implies that, upon voltage-dependent activation, inactivation occurs preferentially from partially activated closed states that precede the open state. The underlying mechanism may, thus, involve long-range allosteric interactions between the voltage-sensing and pore domains, which gradually influence the probability of entering inactivated states as the homologous subunits of the channel tetramer activate sequentially (Dougherty et al. 2008; Fig. 1). To explain preferential CSI, the weak voltage dependence of the opening step is a key feature of the model. A weak depolarization favours closing, deactivation and CSI from intermediate and late closed states. In contrast, a strong depolarization increases the opening probability and decreases the CSI probability. Consequently, the time constant of macroscopic inactivation appears faster at low voltages and slower at high voltages. In the latter case, OSI is expected to be more likely. However, since N-type and/or P/C-type OSI mechanisms are not significant (see above), Kv4 channels end-up in the most stable and possibly absorbing closed-inactivated states after sufficient time regardless of the depolarization's strength (due to preferential CSI). These properties are enhanced by auxiliary β-subunits (KChIPs and DPPs) and have been observed and successfully modelled quantitatively for Kv4.2 and Kv4.3 channels expressed natively and heterologously (Amarillo et al. 2008; Barghaan et al. 2008; Dougherty et al. 2008; Kaulin et al. 2008). Early kinetic models of Kv4 channel CSI assumed an opening step with a slow forward transition and a fast backward transition over a certain voltage range (‘reverse-biased’ opening step; Jerng et al. 1999; Beck & Covarrubias, 2001; Bähring et al. 2001a; Beck et al. 2002; Barghaan et al. 2008). This assumption accounted for the apparently low maximum open probability of Kv4 channels. However, if fast and unstable OSI involving vestigial P/C-type inactivation is mostly responsible for lowering the maximum open probability of Kv4 channels, the ‘reverse-biased’ opening step assumption is unnecessary (Kaulin et al. 2008).
The closing step plays a critical role in Kv4 channel inactivation at positive membrane potentials. If channel closing is prevented or significantly slowed, any OSI mechanism would be favoured as a direct result of an increased open probability and, consequently, current decay would be accelerated. However, this is not the case in Kv4 channels, because, instead of a prominent OSI, they display preferential CSI. This has been shown by testing different permeant cations. Replacing K+ with Rb+, which has a longer residency time in the pore, not only slows Kv4 deactivation tail currents but also macroscopic inactivation (Bähring et al. 2001a; Shahidullah & Covarrubias, 2003; Barghaan et al. 2008). Also, point mutations, which cause a slowing of tail current decay, at the same time slow macroscopic inactivation in Kv4 channels (Jerng et al. 1999). Theoretically, at high voltages Kv4 channels could also reach a closed-inactivated state through a direct transition from IO to In, with no need for reopening (IO to O) and a subsequent transition to Cn (Fig. 1, Scheme III). However, gating models emphasizing such a direct transition are not capable of reproducing the experimentally observed Rb+ effect on macroscopic inactivation (Wang et al. 2005; Barghaan et al. 2008). Taken together, to explain the effect of Rb+ on Kv4 channel inactivation, it appears that open Kv4 channels have to close in order to reach their most stable inactivated state. We will see below that a strict distinction between ‘closing’ and ‘inactivation’, as generally understood for VGCCs, may not be entirely appropriate for Kv4 channels.
Insights into the molecular mechanism of Kv4 channel CSI
Although the data collectively support transitions from closed to closed-inactivated states in Kv4 channels, the main question still is: how do closed Kv4 channels become opening-reluctant? A conceptual model, originally proposed for spHCN channels (see CSI in voltage-gated ion channels, and Fig. 2B), assumes that CSI may result from a temporary separation of the voltage sensor and the activation gate (Shin et al. 2004). Barghaan & Bähring (2009) tested this hypothesis by applying scanning mutagenesis and double-mutant cycle analysis to the Kv4.2 S4S5 linker and the distal S6 region (Fig. 3). The strategy was based on previous Shaker/KcsA mutagenesis (Lu et al. 2002) and Kv1.2 crystal structure data (Long et al. 2005) indicating that these two regions are critically involved in voltage sensor–activation gate coupling. Furthermore, double-mutant cycle analysis has previously identified pairs of amino acids within these two domains, which, by direct coupling, mediate the voltage-dependent opening of the activation gate in the ShakerB channel (Yifrach & MacKinnon, 2002). In corresponding Kv4.2 single and double mutants, these sites were tested for their involvement in CSI by applying prepulse inactivation protocols. In some double mutants, the effects on CSI resulted from the independent additive energetic effects of the individual mutations. In others, however, amino acid exchange at one site influenced the mutational effect at the second site. The latter results suggest that the respective native amino acid side chains may come in close spatial proximity and interact with each other (Fig. 3A and B). Thus, pairs of amino acids, one in the S4S5 linker and the other in the distal S6, not only interact with each other to open the activation gate, but are dynamic interaction partners in CSI (Barghaan & Bähring, 2009). Structural homology modelling based on the Kv1.2 crystal structure (Long et al. 2005; Fig. 3C) and selective redox modulation of Kv4.2 mutants with substituted cysteine residues at the identified sites (e.g. E323C:V404C) supported this hypothesis further (Barghaan & Bähring, 2009).
Figure 3. A dynamic interaction between the S4S5 linker and the distal portion of the S6 segment underlies CSI in Kv4.2 channels (adapted fromBarghaan & Bähring, 2009).
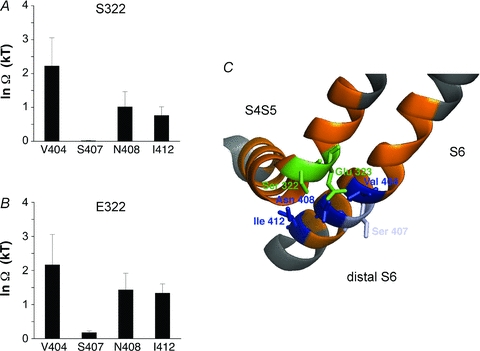
Barghaan and Bähring used prepulse voltage protocols to asses CSI and thermodynamic double mutant cycle analysis to probe the combined role of the S4S5 linker and the S6 segment in Kv4.2 channel CSI. A, there is a significant energetic coupling (Ω= coupling coefficient) between residues S322 in the S4S5 linker and V404 in the S6 segment. B, there is also a similarly significant coupling between E323 and V404. Note that in such experiments the S6 residue S407 showed little or no coupling to the S4S5 linker residues examined. C, molecular model of the spatial relationships between the S4S5 linker and the distal portion of the S6 segment in Kv4.2 channels. Note a possible direct crosstalk between Glu 323 (E323) and Val 404 (V404). ©Barghaan & Bähring, 2009. Originally published in The Journal of General Physiology– doi:10.1085/jgp.200810073.
The model discussed above predicts an important role of the voltage sensor in Kv4 channel CSI. To test this prediction, another study directly examined the relationship between CSI and gating charge movement in Kv4.2 channels (Dougherty et al. 2008). Under conditions that induce CSI at negative membrane potentials (between −100 and −70 mV), the biophysical properties of ionic and gating currents were measured separately. In addition, ionic and gating currents were measured simultaneously by manipulating the Cs+ reversal potential (Fig. 4A). The study by Dougherty et al. demonstrated slow and profound gating charge (Q)-immobilization. This phenomenon was originally characterized for Nav channel inactivation (Bezanilla & Armstrong, 1975; Armstrong & Bezanilla, 1977) and refers to a condition where there is an apparent loss of Q. Essentially, Q movements are dramatically slowed as a result of actively hindering the return of the voltage sensor to its resting conformation, or allowing the voltage sensor to slowly adopt a distinct and more stable conformation. In Kv channels, Q-immobilization has been observed for both N-type (Bezanilla et al. 1991; Perozo et al. 1992; Roux et al. 1998) and P/C-type inactivation (Fedida et al. 1996; Olcese et al. 1997). However, the slow Q-immobilization in Kv4.2 channels is independent of the NT-ID associated with OSI and occurs over a narrow range of negative membrane potentials (−110 and −75 mV; Dougherty et al. 2008). Consistent with CSI, membrane potential changes in this range move ∼10% of the gating charge associated with gate opening. Also, the gating charge experiences two distinct energy landscapes depending on whether it is associated with the activation or inactivation pathways of the channel (Fig. 1, Scheme III). Most significantly, simultaneous observations of slow Q-immobilization and ionic current decay show that these processes are equivalent manifestations of CSI (Fig. 4B). The authors explained the observed tight correlation between ionic current inactivation and Q-immobilization quantitatively by employing a global kinetic modelling approach, based on a general allosteric scheme of CSI coupled to voltage-dependent activation (Dougherty et al. 2008). These findings suggest that the voltage sensor is involved in the mechanism of CSI, and help explain why CSI is influenced by neutralizing positive charges (Skerritt &; Campbell, 2007, 2009) or introducing non-native positive charges (Skerritt & Campbell, 2008) in the Kv4.3 S4 segment. A second implication is that the Kv4.2 voltage sensor undergoes two types of movement with distinct consequences (Fig. 5): fast rearrangements tightly coupled to the opening of the activation gate, and additional rearrangements responsible for an apparent loss of gating charge. The latter may occur independently of pore opening as the channel slowly transitions from a set of partially activated (closed but opening-permissive) states to a set of closed-inactivated states (Fig. 1, Scheme III; Fig. 4C). Thus, a closed-inactivated state may correspond to the failure of the voltage sensors to actively open the activation gate. One must bear in mind, however, that, independently of the molecular mechanism of inactivation, the apparent loss of gating charge is bound to occur when inactivation and activation are coupled: as dictated by the thermodynamic constraints of an allosteric system, activation favours inactivation, and inactivation favours fast gating charge movement. Consequently, once the channel undergoes CSI, a test depolarization reports a decrease in the available gating charge.
Figure 4. Slow gating charge immobilization precisely mirrors inactivation in Kv4.2 channels.
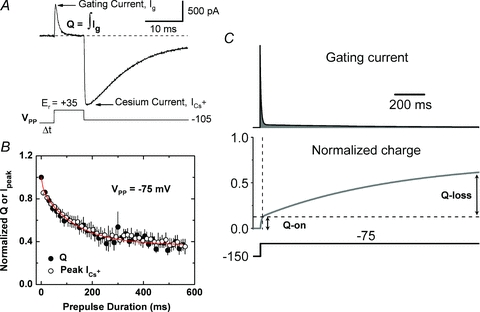
A, simultaneous measurement of gating and ionic currents recorded under whole-cell patch-clamping conditions (adapted from Dougherty et al. 2008). The pulse protocol displayed below the current trace evoked the gating current (Ig) by stepping the voltage to the reversal potential (Er) of the ionic current, and the ionic Cs+ current (ICs) is subsequently observed by applying a large inward driving force (repolarization to −105 mV). In this experiment, VPP=−145 mV, a voltage insufficient to induce inactivation. Under these conditions, inactivation of gating and ionic currents is probed simultaneously by varying the duration and voltage of a conditioning prepulse (VPP) preceding the test pulse to the Er. B, the gating charge (Q=∫Ig) and ICs against the duration of VPP at −75 mV. Note, that the slow processes follow virtually identical trajectories; however, Q kinetics are biphasic (double exponential fit, continuous red line), indicating fast Q movement associated with voltage-dependent activation and a slow apparent Q-loss directly associated with inactivation. C, simulated gating current (top) evoked by a step depolarization from −150 to −75 mV and the corresponding Q kinetics (bottom). The shaded area under the gating current trace (i.e. integral, see above) is the corresponding Q. This simulation assumed a modified Zagotta–Hoshi–Aldrich model (Zagotta et al. 1994) as reported by Dougherty et al. (2008). The traces clearly show the fast observable component reflecting voltage-dependent activation (Q-on), and the slow undetectable component (Q-loss) reflecting apparent Q-immobilization precisely correlated with slow inactivation. Panels B and C are also adapted from Dougherty et al. (2008). ©Dougherty et al. 2008. Originally published in The Journal of General Physiology– doi:10.1085/jgp.200709938.
Figure 5. Working structural model of slow inactivation in Kv4 channels.
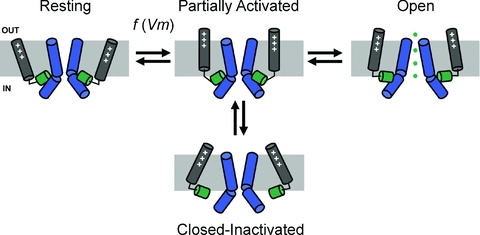
This hypothesis emerged from the studies by Dougherty et al. (2008) and Barghaan & Bähring (2009) (Figs 3 and 4). The simplified cartoons illustrating the significant states depict the relevant segments of two opposing channel subunits in the tetramer. Positively charged S4 voltage sensors are coloured grey; the S4S5 linkers are coloured green; and the S6 segments (bent at the PVPV motif) are coloured blue. Note that channel opening depends on maintaining physical contact between the S4S5 linkers and the distal portion of the S6 segments (post PVP). This contact is broken when the S4 segments slowly adopt an alternate ‘relaxed’ conformation. Consequently, the channel fails to open and becomes inactivated. Additional details can be found in the text.
In a plausible physical model of CSI (Barghaan & Bähring, 2009), the S4S5 linker residues may fail to engage tightly with their S6 counterparts (Figs 3C and 5). Consequently, the voltage sensor freely moves, physically uncoupled from the closed activation gate. CSI in Kv4 channels may resemble desensitization in ligand-gated ion channels, where the transmitter remains bound but the ion channel is closed. Functional and crystallographic studies of glutamate receptor channels (Sun et al. 2002) showed that the dimeric ligand binding cores in their bound state initially adopt a tense conformation, which allows channel opening. Eventually, however, the binding cores adopt a relaxed conformation, which leads to channel closure. Glutamate receptor desensitization then results from rearrangements at the dimer interface. Such a mechanism is conceptually comparable to the proposed uncoupling between the S4S5 linker and distal S6 in Kv4 channels (Fig. 5). Thus, CSI may be described by ‘voltage sensor inactivation’ with the activation gate being ‘desensitized’ to voltage in the closed-inactivated state (Shin et al. 2004). In the next section, we shall discuss a working hypothesis that attempts to explain how the voltage sensor might be generally implicated in slow inactivation processes in Kv channels (i.e. CSI and P/C-type inactivation) at a more fundamental level.
Perspective on CSI mechanisms and the biological implications
CSI is a common and physiologically relevant phenomenon in VGCCs. In particular, Kv4 channels may have evolved to undergo preferential CSI by exploiting variations in the cross-talk between the voltage-sensing domain (VSD) and the pore domain (PD). Two recent studies underscore the roles of the pore and voltage sensor in CSI and offer a preview of how we may shed more light on the molecular mechanisms of CSI in Kv channels. In the first study, Claydon and coworkers (Claydon et al. 2007, 2008) employed an elegant combination of traditional electrophysiology and voltage-clamp fluorimetry to directly suggest CSI in Shaker Kv channels. Essentially, acidic pH promotes rearrangements in the outer mouth of the selectivity filter, which are typically associated with P/C-type inactivation. These conformational changes were inferred by observing the fluorescence changes reported by flourophores carefully placed on the extracellular S3-S4 loop and an external pore site. Thus, the authors confirmed the effect of acidic pH on P/C-type inactivation and examined channel availability under conditions that uncouple pore opening from voltage-dependent activation to unveil CSI more directly. A parsimonious interpretation of the results and additional diagnostic testing strongly suggest that the P/C-type mechanism is responsible for both OSI and CSI, which are known to co-exist in Kv1.5 channels (Kurata et al. 2005). In the second study, Schmidt et al. (2009) opened opportunities to study the structural basis of CSI in Kv channels. They discovered that the bacterial KvAP channel, which has a known crystal structure (Lee et al. 2005), may also inactivate from a partially activated pre-open closed state, and the authors considered a mechanism of dynamic coupling analogous to desensitization in glutamate receptors (Sun et al. 2002), as proposed earlier in this review and previous studies (Shin et al. 2004; Dougherty et al. 2008; Kaulin et al. 2008; Barghaan & Bähring, 2009). It is especially interesting that the recovery from inactivation in KvAP channels is sensitive to lipid composition and that the voltage sensor toxin VSTx1, which directly binds to the VSD, favours the inactivated state of the channel. Thus, the proposed role of the voltage sensor in Kv4 channel CSI (Dougherty et al. 2008) is plausible.
Voltage sensor inactivation in Kv channels
The cross-talk between the VSD and PD, which is essential for channel activation, is also central to our working hypothesis of CSI (Fig. 5). The VSD communicates with two distinct gates in the PD: the P-gate (pore-gate) in the selectivity filter and the A-gate (activation gate) involving the S6 bundle crossing. There is ample evidence for interactions between the voltage sensor and these gates (Loots & Isacoff, 2000; Elinder et al. 2001; Lu et al. 2002; Lainéet al. 2003; Webster et al. 2004; Soler-Llavina et al. 2006; Barghaan & Bähring, 2009). In addition, it is well established that the voltage sensor of Kv channels undergoes significant movement in response to changes in the transmembrane voltage (Horn, 2002; Ruta et al. 2005; Tombola et al. 2005; Bezanilla, 2008; Swartz, 2008; Caterall, 2010); and other studies have shown that the voltage sensor of Kv channels may experience distinct energy landscapes as it first undergoes rapid activation and subsequently finds its most stable conformation upon a prolonged depolarization (Olcese et al. 1997; Dougherty et al. 2008). At depolarized membrane potentials, the voltage sensor may adopt at least two distinct sets of conformations. First, it moves quickly and strives to ‘shake hands’ with the A-gate to open it. Then, if a depolarization is sustained, the voltage sensor slowly drifts toward its most stable conformation. As a result, it may encounter favourable strong interactions with the P-gate and may stabilize it in its non-conducting conformation (Loots & Isacoff, 2000; Elinder et al. 2001). This mechanism would correspond to P/C-type inactivation, which may eventually lead either to a closed-inactivated or an open-inactivated state (Fig. 6A). C-type inactivation may, however, involve interruption of the permeation pathway at other locations (e.g. the A-gate). Let's consider another scenario in which the voltage sensor interacts poorly with the P-gate. In this case, the P-gate may still undergo rearrangements; however, the lack of a strong interaction with the voltage sensor prevents stabilization of a non-conducting selectivity filter (i.e. P-type inactivation is effectively absent). Moreover, the dynamic interaction between VSD and PD at the A-gate may not be equal in strength and stability among Kv channels. Thus, as the voltage sensor starts to move and reaches for the A-gate, the ‘hand shake’ may fail or be short-lived as a result of a ‘slippery’ A-gate. This corresponds to a different CSI mechanism where the voltage sensor experiences a distinct energy landscape and fails to establish strong stable interactions with both the A-gate and the P-gate (Fig. 6B). Kv4 channels and possibly other VGCCs exhibiting prominent U-type features of inactivation may utilize this mechanism.
From the scenarios discussed above, it is conceivable that the voltage sensor is actively behind the installation of CSI in Kv channels and other VGCCs. In contrast, the A-gate is only passively involved in CSI and simply remains closed. Another recent study clearly demonstrates that S4 voltage sensors intrinsically adopt a ‘relaxed’ conformation in response to prolonged depolarization, which may be related to slow inactivation in VGCCs (Villalba-Galea et al. 2008). Voltage sensor inactivation thus refers to the transition of the voltage sensors from a conformation that supports pore opening and ion conduction to a ‘relaxed’ conformation that, under wild-type conditions, cannot support pore opening. Certain Kv channel mutations can dissociate this ‘relaxed’ voltage sensor conformation from inactivation (Olcese et al. 2001; Piper et al. 2003); however, voltage sensor inactivation under native conditions may drive ‘slow’ inactivation exhibiting two distinct manifestations (Fig. 6). This is consistent with the tight correlation between slow gating charge immobilization and slow inactivation of the ionic currents in both ShakerB and Kv4 channels (Olcese et al. 1997; Dougherty et al. 2008) (Fig. 4A), even though the former undergo P/C-type inactivation but the latter do not. We propose a more general mechanistic framework: upon experiencing a prolonged depolarization, voltage sensors in VGCCs are prone to enter a stable state that may not be optimally poised to support pore opening. Whether a particular form of inactivation is prevalent in a VGCC (Fig. 5), or whether multiple mechanisms overlap or even compete, depends on the stability of the interactions between the voltage sensor and the two gates in the PD. From an evolutionary perspective, this is an economical solution where relatively small structural variations can provide diverse pathways of autoregulation in VGCCs. Further investigations are, however, necessary to determine whether the proposed interactions truly involve direct or indirect physical coupling between voltage sensors and gates. Moreover, it is also necessary to show that the A-gate is actually closed when VGCCs reside in the closed-inactivated state.
Physiological and evolutionary implications
The impact of CSI in excitable tissues is widespread and more than one specific mechanism may underlie this important functional trait. Kv4 channels are particularly relevant in this regard because they undergo preferential CSI at all relevant voltages and play vital roles in the heart and the nervous system (Johnston et al. 2003; Jerng et al. 2004; Nerbonne & Kass, 2005; Patel & Campbell, 2005; Maffie & Rudy, 2008). In the heart, Kv4.2 and Kv4.3 channels contribute significantly to the transient outward K+ current Ito (Dixon et al. 1996; Guo et al. 1999), which causes an early notch in the repolarization trajectory of the cardiac action potential. In the soma and dendrites of central neurons, Kv4.2 and Kv4.3 channels underlie the subthreshold-operating A-type K+ current ISA (Serodio et al. 1994; Malin & Nerbonne, 2000), which regulates excitability in diverse ways. Acting as shock absorbers and gate keepers, neuronal Kv4 channels in dendrites regulate backpropagating action potentials (Hoffman et al. 1997) and the propagation of plateau potentials at branching points (Cai et al. 2004). These activities impact synaptic plasticity by influencing synaptic scaling and coincidence detection, and are critically dependent on the keen sensitivity of Kv4 channels to subthreshold membrane potential changes induced by synaptic transmission (Schoppa & Westbrook, 1999; Ramakers & Storm, 2002; Chen et al. 2006; Hu et al. 2006; Andrásfalvy et al. 2008). Furthermore, by influencing the subthreshold dynamics of the membrane potential, neuronal Kv4 channels act as brakes that regulate the latency to the first spike and the interspike interval during slow repetitive spiking (Song et al. 1998; Shibata et al. 2000; Liss et al. 2001). Separately, and similar to cardiac Ito, neuronal Kv4 channels may also regulate the repolarization of the action potential (Kim et al. 2005).
What common aspects of these physiological activities may explain the evolution of preferential CSI in Kv4 channels? Possibly, the need for an energetically efficient self-regulatory mechanism in increasingly complex excitable tissues was the selective force that favoured preferential CSI in Kv4 channels. If inactivation and recovery from inactivation are not associated with passive ion flux across the membrane, regulation of excitability can occur without dissipating the ion gradients actively maintained by the Na+/K+ pump. Moreover, small changes in membrane potential (low energetic cost) are sufficient to trigger preferential CSI; leaving most of the membrane potential energy to generate the action potential. The intrinsic ability of all S4 voltage sensors to slowly adopt a low energy ‘relaxed’ conformation upon modest depolarization (see above) may have primed the evolution of energy efficient preferential CSI. It is then conceivable that relatively minor variations in the interactions between VSD and PD (resulting from mutations) led to the implementation of preferential CSI as proposed above. In coming years, we shall see further investigations of this hypothesis at the biophysical and structural levels and the mechanisms underlying the regulation of CSI in Kv channels by auxiliary β-subunits and post-translational modifications (Covarrubias et al. 2008; Maffie & Rudy, 2008; Xie et al. 2009).
Acknowledgments
We thank Drs Richard Horn (Thomas Jefferson University, Philadelphia, PA) and Henry Jerng (Baylor College of Medicine, Houston, TX) for their critical reading of the manuscript. This work was supported by NIH grant R01 NS032337-14, and grant BA 2055/1 from the Deutsche Forschungsgemeinschaft (DFG).
Glossary
Abbreviations
- CSI
closed-state inactivation
- OSI
open-state inactivation
- VGCC
voltage-gated cationic channel
References
- Ahern CA, Eastwood AL, Dougherty DA, Horn R. An electrostatic interaction between TEA and an introduced pore aromatic drives spring-in-the-door inactivation in Shaker potassium channels. J Gen Physiol. 2009;134:461–469. doi: 10.1085/jgp.200910260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich RW. Inactivation of voltage-gated delayed potassium current in molluscan neurons. A kinetic model. Biophys J. 1981;36:519–532. doi: 10.1016/S0006-3495(81)84750-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich RW. Fifty years of inactivation. Nature. 2001;411:643–644. doi: 10.1038/35079705. [DOI] [PubMed] [Google Scholar]
- Aldrich RW, Stevens CF. Inactivation of open and closed sodium channels determined separately. Cold Spring Harb Symp Quant Biol. 1983;48:147–153. doi: 10.1101/sqb.1983.048.01.017. [DOI] [PubMed] [Google Scholar]
- Almers W, Armstrong CM. Survival of K+ permeability and gating currents in squid axons perfused with K+-free media. J Gen Physiol. 1980;75:61–78. doi: 10.1085/jgp.75.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amarillo Y, De Santiago-Castillo JA, Dougherty K, Maffie J, Kwon E, Covarrubias M, Rudy B. Ternary Kv4.2 channels recapitulate voltage-dependent inactivation kinetics of A-type K+ channels in cerebellar granule neurons. J Physiol. 2008;586:2093–2106. doi: 10.1113/jphysiol.2007.150540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, Hinson JW, Mattsson KI, Strassle BW, Trimmer JS, Rhodes KJ. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 2000;403:553–556. doi: 10.1038/35000592. [DOI] [PubMed] [Google Scholar]
- Andrásfalvy BK, Makara JK, Johnston D, Magee JC. Altered synaptic and non-synaptic properties of CA1 pyramidal neurons in Kv4.2 knockout mice. J Physiol. 2008;586:3881–3892. doi: 10.1113/jphysiol.2008.154336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM. Na channel inactivation from open and closed states. Proc Natl Acad Sci U S A. 2006;103:17991–17996. doi: 10.1073/pnas.0607603103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM, Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. J Gen Physiol. 1977;70:567–590. doi: 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayer RK, Jr, Sigworth FJ. Enhanced closed-state inactivation in a mutant Shaker K+ channel. J Membr Biol. 1997;157:215–230. doi: 10.1007/s002329900230. [DOI] [PubMed] [Google Scholar]
- Bähring R, Boland LM, Varghese A, Gebauer M, Pongs O. Kinetic analysis of open- and closed-state inactivation transitions in human Kv4.2 A-type potassium channels. J Physiol. 2001a;535:65–81. doi: 10.1111/j.1469-7793.2001.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähring R, Dannenberg J, Peters HC, Leicher T, Pongs O, Isbrandt D. Conserved Kv4 N-terminal domain critical for effects of Kv channel-interacting protein 2.2 on channel expression and gating. J Biol Chem. 2001b;276:23888–23894. doi: 10.1074/jbc.M101320200. [DOI] [PubMed] [Google Scholar]
- Baldwin TJ, Tsaur ML, Lopez GA, Jan YN, Jan LY. Characterization of a mammalian cDNA for an inactivating voltage-sensitive K+ channel. Neuron. 1991;7:471–483. doi: 10.1016/0896-6273(91)90299-f. [DOI] [PubMed] [Google Scholar]
- Barghaan J, Bähring R. Dynamic coupling of voltage sensor and gate involved in closed-state inactivation of Kv4.2 channels. J Gen Physiol. 2009;133:205–224. doi: 10.1085/jgp.200810073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barghaan J, Tozakidou M, Ehmke H, Bähring R. Role of N-terminal domain and accessory subunits in controlling deactivation-inactivation coupling of Kv4.2 channels. Biophys J. 2008;94:1276–1294. doi: 10.1529/biophysj.107.111344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP. Sodium channel inactivation in the crayfish giant axon. Must channels open before inactivating? Biophys J. 1981;35:595–614. doi: 10.1016/S0006-3495(81)84815-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck EJ, Bowlby M, An WF, Rhodes KJ, Covarrubias M. Remodelling inactivation gating of Kv4 channels by KChIP1, a small-molecular-weight calcium-binding protein. J Physiol. 2002;538:691–706. doi: 10.1113/jphysiol.2001.013127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck EJ, Covarrubias M. Kv4 channels exhibit modulation of closed-state inactivation in inside-out patches. Biophys J. 2001;81:867–883. doi: 10.1016/S0006-3495(01)75747-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F. How membrane proteins sense voltage. Nat Rev Mol Cell Biol. 2008;9:323–332. doi: 10.1038/nrm2376. [DOI] [PubMed] [Google Scholar]
- Bezanilla F, Armstrong CM. Kinetic properties and inactivation of the gating currents of sodium channels in squid axon. Philos Trans R Soc Lond B Biol Sci. 1975;270:449–458. doi: 10.1098/rstb.1975.0022. [DOI] [PubMed] [Google Scholar]
- Bezanilla F, Perozo E, Papazian DM, Stefani E. Molecular basis of gating charge immobilization in Shaker potassium channels. Science. 1991;254:679–683. doi: 10.1126/science.1948047. [DOI] [PubMed] [Google Scholar]
- Bezanilla F, Taylor RE, Fernándes JM. Distribution and kinetics of membrane dielectric polarization. 1. Long-term inactivation of gating currents. J Gen Physiol. 1982;79:21–40. doi: 10.1085/jgp.79.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess DE, Crawford O, Delisle BP, Satin J. Mechanism of inactivation gating of human T-type (low-voltage activated) calcium channels. Biophys J. 2002;82:1894–1906. doi: 10.1016/S0006-3495(02)75539-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Liang CW, Muralidharan S, Kao JP, Tang CM, Thompson SM. Unique roles of SK and Kv4.2 potassium channels in dendritic integration. Neuron. 2004;44:351–364. doi: 10.1016/j.neuron.2004.09.026. [DOI] [PubMed] [Google Scholar]
- Callsen B, Isbrandt D, Sauter K, Hartmann LS, Pongs O, Bähring R. Contribution of N- and C-terminal Kv4.2 channel domains to KChIP interaction. J Physiol. 2005;568:397–412. doi: 10.1113/jphysiol.2005.094359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DL, Rasmusson RL, Qu Y, Strauss HC. The calcium-independent transient outward potassium current in isolated ferret right ventricular myocytes. I. Basic characterization and kinetic analysis. J Gen Physiol. 1993;101:571–601. doi: 10.1085/jgp.101.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- Caterall WA. Ion channel voltage sensors: structure, function and pathophysiology. Neuron. 2010;67:915–928. doi: 10.1016/j.neuron.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Yuan LL, Zhao C, Birnbaum SG, Frick A, Jung WE, Schwarz TL, Sweatt JD, Johnston D. Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurosci. 2006;26:12143–12151. doi: 10.1523/JNEUROSCI.2667-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KL, Aldrich RW, Yellen G. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proc Natl Acad Sci U S A. 1991;88:5092–5095. doi: 10.1073/pnas.88.12.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claydon TW, Kehl SJ, Fedida D. Closed-state inactivation induced in KV1 channels by extracellular acidification. Channels. 2008;2:139–142. doi: 10.4161/chan.2.2.6231. [DOI] [PubMed] [Google Scholar]
- Claydon TW, Vaid M, Rezazadeh S, Kwan DC, Kehl SJ, Fedida D. A direct demonstration of closed-state inactivation of K+ channels at low pH. J Gen Physiol. 2007;129:437–455. doi: 10.1085/jgp.200709774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero-Morales JF, Cuello LG, Zhao Y, Jogini V, Cortes DM, Roux B, Perozo E. Molecular determinants of gating at the potassium-channel selectivity filter. Nat Struct Mol Biol. 2006;13:311–318. doi: 10.1038/nsmb1069. [DOI] [PubMed] [Google Scholar]
- Cordero-Morales JF, Jogini V, Lewis A, Vasquez V, Cortes DM, Roux B, Perozo E. Molecular driving forces determining potassium channel slow inactivation. Nat Struct Mol Biol. 2007;14:1062–1069. doi: 10.1038/nsmb1309. [DOI] [PubMed] [Google Scholar]
- Covarrubias M, Bhattacharji A, De Santiago-Castillo JA, Dougherty K, Kaulin YA, Na-Phuket TR, Wang G. The neuronal Kv4 channel complex. Neurochem Res. 2008;33:1558–1567. doi: 10.1007/s11064-008-9650-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuello LG, Jogini V, Cortes DM, Perozo E. Structural mechanism of C-type inactivation in K+ channels. Nature. 2010;466:203–208. doi: 10.1038/nature09153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Yang H, Lee US. Molecular mechanisms of BK channel activation. Cell Mol Life Sci. 2009;66:852–875. doi: 10.1007/s00018-008-8609-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Biasi M, Hartmann HA, Drewe JA, Taglialatela M, Brown AM, Kirsch GE. Inactivation determined by a single site in K+ pores. Pflügers Arch. 1993;422:354–363. doi: 10.1007/BF00374291. [DOI] [PubMed] [Google Scholar]
- Demo SD, Yellen G. The inactivation gate of the Shaker K+ channel behaves like an open-channel blocker. Neuron. 1991;7:743–753. doi: 10.1016/0896-6273(91)90277-7. [DOI] [PubMed] [Google Scholar]
- Ding JP, Lingle CJ. Steady-state and closed-state inactivation properties of inactivating BK channels. Biophys J. 2002;82:2448–2465. doi: 10.1016/S0006-3495(02)75588-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JE, Shi W, Wang HS, McDonald C, Yu H, Wymore RS, Cohen IS, McKinnon D. Role of the Kv4.3 K+ channel in ventricular muscle. A molecular correlate for the transient outward current. Circ Res. 1996;79:659–668. doi: 10.1161/01.res.79.4.659. [DOI] [PubMed] [Google Scholar]
- Dougherty K, De Santiago-Castillo JA, Covarrubias M. Gating charge immobilization in Kv4.2 channels: the basis of closed-state inactivation. J Gen Physiol. 2008;131:257–273. doi: 10.1085/jgp.200709938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eghbali M, Olcese R, Zarei MM, Toro L, Stefani E. External pore collapse as an inactivation mechanism for Kv4.3 K+ channels. J Membr Biol. 2002;188:73–86. doi: 10.1007/s00232-001-0173-3. [DOI] [PubMed] [Google Scholar]
- Elinder F, Männikkö R, Larsson HP. S4 charges move close to residues in the pore domain during activation in a K channel. J Gen Physiol. 2001;118:1–10. doi: 10.1085/jgp.118.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedida D, Bouchard R, Chen FS. Slow gating charge immobilization in the human potassium channel Kv1.5 and its prevention by 4-aminopyridine. J Physiol. 1996;494:377–387. doi: 10.1113/jphysiol.1996.sp021499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebauer M, Isbrandt D, Sauter K, Callsen B, Nolting A, Pongs O, Bähring R. N-type inactivation features of Kv4.2 channel gating. Biophys J. 2004;86:210–223. doi: 10.1016/S0006-3495(04)74097-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Xu H, London B, Nerbonne JM. Molecular basis of transient outward K+ current diversity in mouse ventricular myocytes. J Physiol. 1999;521:587–599. doi: 10.1111/j.1469-7793.1999.00587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlitze S, Hockerman GH, Scheuer T, Catterall WA. Molecular determinants of inactivation and G protein modulation in the intracellular loop connecting domains I and II of the calcium channel α1A subunit. Proc Natl Acad Sci U S A. 1997;94:1512–1516. doi: 10.1073/pnas.94.4.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. The dual effect of membrane potential on sodium conductance in the giant axon of Loligo. J Physiol. 1952;116:497–506. doi: 10.1113/jphysiol.1952.sp004719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- Horn R. Coupled movements in voltage-gated ion channels. J Gen Physiol. 2002;120:449–453. doi: 10.1085/jgp.20028658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn R, Ding S, Gruber HJ. Immobilizing the moving parts of voltage-gated ion channels. J Gen Physiol. 2000;116:461–476. doi: 10.1085/jgp.116.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn R, Patlak J, Stevens CF. Sodium channels need not open before they inactivate. Nature. 1981;291:426–427. doi: 10.1038/291426a0. [DOI] [PubMed] [Google Scholar]
- Horn R, Vandenberg CA. Statistical properties of single sodium channels. J Gen Physiol. 1984;84:505–534. doi: 10.1085/jgp.84.4.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- Hu HJ, Carrasquillo Y, Karim F, Jung WE, Nerbonne JM, Schwarz TL, Gereau RWI. The Kv4.2 potassium channel subunit is required for pain plasticity. Neuron. 2006;50:89–100. doi: 10.1016/j.neuron.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Isacoff EY, Jan YN, Jan LY. Putative receptor for the cytoplasmic inactivation gate in the Shaker K+ channel. Nature. 1991;353:86–90. doi: 10.1038/353086a0. [DOI] [PubMed] [Google Scholar]
- Jerng HH, Covarrubias M. K+ channel inactivation mediated by the concerted action of the cytoplasmic N- and C-terminal domains. Biophys J. 1997;72:163–174. doi: 10.1016/S0006-3495(97)78655-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerng HH, Dougherty K, Covarrubias M, Pfaffinger PJ. A novel N-terminal motif of dipeptidyl peptidase-like proteins produces rapid inactivation of Kv4.2 channels by a pore-blocking mechanism. Channels. 2009;3:1–14. doi: 10.4161/chan.3.6.10216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerng HH, Pfaffinger PJ, Covarrubias M. Molecular physiology and modulation of somatodendritic A-type potassium channels. Mol Cell Neurosci. 2004;27:343–369. doi: 10.1016/j.mcn.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Jerng HH, Shahidullah M, Covarrubias M. Inactivation gating of Kv4 potassium channels: molecular interactions involving the inner vestibule of the pore. J Gen Physiol. 1999;113:641–660. doi: 10.1085/jgp.113.5.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston D, Christie BR, Frick A, Gray R, Hoffman DA, Schexnayder LK, Watanabe S, Yuan LL. Active dendrites, potassium channels and synaptic plasticity. Philos Trans R Soc Lond B Biol Sci. 2003;358:667–674. doi: 10.1098/rstb.2002.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LP, DeMaria CD, Yue DT. N-type calcium channel inactivation probed by gating-current analysis. Biophys J. 1999;76:2530–2552. doi: 10.1016/S0006-3495(99)77407-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SW, Marks TN. Calcium currents in bullfrog sympathetic neurons. II. Inactivation. J Gen Physiol. 1989;94:169–182. doi: 10.1085/jgp.94.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaulin YA, De Santiago-Castillo JA, Rocha CA, Covarrubias M. Mechanism of the modulation of Kv4:KChIP-1 channels by external K+ Biophys J. 2008;94:1241–1251. doi: 10.1529/biophysj.107.117796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger S, West JW, Catterall WA, Scheuer T. Molecular analysis of potential hinge residues in the inactivation gate of brain type IIA Na+ channels. J Gen Physiol. 1997a;109:607–617. doi: 10.1085/jgp.109.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger S, West JW, Scheuer T, Catterall WA. Molecular analysis of the putative inactivation particle in the inactivation gate of brain type IIA Na+ channels. J Gen Physiol. 1997b;109:589–605. doi: 10.1085/jgp.109.5.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner D, Monje F, Stocker M. Structural determinants of the regulation of the voltage-gated potassium channel Kv2.1 by the modulatory α-subunit Kv9.3. J Biol Chem. 2003;278:18154–18161. doi: 10.1074/jbc.M213117200. [DOI] [PubMed] [Google Scholar]
- Kerschensteiner D, Stocker M. Heteromeric assembly of Kv2.1 with Kv9.3: Effect on the state dependence of inactivation. Biophys J. 1999;77:248–257. doi: 10.1016/S0006-3495(99)76886-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Wei DS, Hoffman DA. Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. J Physiol. 2005;569:41–57. doi: 10.1113/jphysiol.2005.095042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim LA, Furst J, Butler MH, Xu S, Grigorieff N, Goldstein SA. Ito channels are octomeric complexes with four subunits of each Kv4.2 and K+ channel-interacting protein 2. J Biol Chem. 2004a;279:5549–5554. doi: 10.1074/jbc.M311332200. [DOI] [PubMed] [Google Scholar]
- Kim LA, Furst J, Gutierrez D, Butler MH, Xu S, Goldstein SA, Grigorieff N. Three-dimensional structure of Ito; Kv4.2-KChIP2 ion channels by electron microscopy at 21 Å resolution. Neuron. 2004b;41:513–519. doi: 10.1016/s0896-6273(04)00050-9. [DOI] [PubMed] [Google Scholar]
- Kirichok YV, Nikolaev AV, Krishtal OA. [K+]out accelerates inactivation of Shal-channels responsible for A-current in rat CA1 neurons. Neuroreport. 1998;9:625–629. doi: 10.1097/00001756-199803090-00012. [DOI] [PubMed] [Google Scholar]
- Kiss L, Korn SJ. Modulation of C-type inactivation by K+ at the potassium channel selectivity filter. Biophys J. 1998;74:1840–1849. doi: 10.1016/S0006-3495(98)77894-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemic KG, Kirsch GE, Jones SW. U-type inactivation of Kv3.1 and Shaker potassium channels. Biophys J. 2001;81:814–826. doi: 10.1016/S0006-3495(01)75743-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemic KG, Shieh CC, Kirsch GE, Jones SW. Inactivation of Kv2.1 potassium channels. Biophys J. 1998;74:1779–1789. doi: 10.1016/S0006-3495(98)77888-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer JW, Post MA, Brown AM, Kirsch GE. Modulation of potassium channel gating by coexpression of Kv2.1 with regulatory Kv5.1 or Kv6.1 α-subunits. Am J Physiol Cell Physiol. 1998;274:C1501–C1510. doi: 10.1152/ajpcell.1998.274.6.C1501. [DOI] [PubMed] [Google Scholar]
- Kuo CC, Bean BP. Na+ channels must deactivate to recover from inactivation. Neuron. 1994;12:819–829. doi: 10.1016/0896-6273(94)90335-2. [DOI] [PubMed] [Google Scholar]
- Kurata HT, Doerksen KW, Eldstrom JR, Rezazadeh S, Fedida D. Separation of P/C- and U-type inactivation pathways in Kv1.5 potassium channels. J Physiol. 2005;568:31–46. doi: 10.1113/jphysiol.2005.087148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurata HT, Fedida D. A structural interpretation of voltage-gated potassium channel inactivation. Prog Biophys Mol Biol. 2006;92:185–208. doi: 10.1016/j.pbiomolbio.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Kurata HT, Soon GS, Eldstrom JR, Lu GW, Steele DF, Fedida D. Amino-terminal determinants of U-type inactivation of voltage-gated K+ channels. J Biol Chem. 2002;277:29045–29053. doi: 10.1074/jbc.M111470200. [DOI] [PubMed] [Google Scholar]
- Kurata HT, Soon GS, Fedida D. Altered state dependence of C-type inactivation in the long and short forms of human Kv1.5. J Gen Physiol. 2001;118:315–332. doi: 10.1085/jgp.118.3.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lainé M, Lin MC, Bannister JP, Silverman WR, Mock AF, Roux B, Papazian DM. Atomic proximity between S4 segment and pore domain in Shaker potassium channels. Neuron. 2003;39:467–481. doi: 10.1016/s0896-6273(03)00468-9. [DOI] [PubMed] [Google Scholar]
- Lee SY, Lee A, Chen J, MacKinnon R. Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane. Proc Natl Acad Sci U S A. 2005;102:15441–15446. doi: 10.1073/pnas.0507651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DI, Deutsch C. Recovery from C-type inactivation is modulated by extracellular potassium. Biophys J. 1996;70:798–805. doi: 10.1016/S0006-3495(96)79619-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liss B, Franz O, Sewing S, Bruns R, Neuhoff H, Roeper J. Tuning pacemaker frequency of individual dopaminergic neurons by Kv4.3L and KChip3.1 transcription. EMBO J. 2001;20:5715–5724. doi: 10.1093/emboj/20.20.5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Jurman ME, Yellen G. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 1996;16:859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- Long SB, Campbell EB, Mackinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005;309:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- Loots E, Isacoff EY. Molecular coupling of S4 to a K+ channel's slow inactivation gate. J Gen Physiol. 2000;116:623–636. doi: 10.1085/jgp.116.5.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- Lu Z, Klem AM, Ramu Y. Coupling between voltage sensors and activation gate in voltage-gated K+ channels. J Gen Physiol. 2002;120:663–676. doi: 10.1085/jgp.20028696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffie J, Rudy B. Weighing the evidence for a ternary protein complex mediating A-type K+ currents in neurons. J Physiol. 2008;586:5609–5623. doi: 10.1113/jphysiol.2008.161620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin SA, Nerbonne JM. Elimination of the fast transient in superior cervical ganglion neurons with expression of KV4.2W362F: molecular dissection of IA. J Neurosci. 2000;20:5191–5199. doi: 10.1523/JNEUROSCI.20-14-05191.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell-Lagnado RD, Aldrich RW. Energetics of Shaker K channels block by inactivation peptides. J Gen Physiol. 1993a;102:977–1003. doi: 10.1085/jgp.102.6.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell-Lagnado RD, Aldrich RW. Interactions of amino terminal domains of Shaker K channels with a pore blocking site studied with synthetic peptides. J Gen Physiol. 1993b;102:949–975. doi: 10.1085/jgp.102.6.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205–1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- Ogielska EM, Zagotta WN, Hoshi T, Heinemann SH, Haab J, Aldrich RW. Cooperative subunit interactions in C-type inactivation of K channels. Biophys J. 1995;69:2449–2457. doi: 10.1016/S0006-3495(95)80114-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olcese R, Latorre R, Toro L, Bezanilla F, Stefani E. Correlation between charge movement and ionic current during slow inactivation in Shaker K+ channels. J Gen Physiol. 1997;110:579–589. doi: 10.1085/jgp.110.5.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olcese R, Sigg D, Latorre R, Bezanilla F, Stefani E. A conducting state with properties of a slow inactivated state in a shaker K+ channel mutant. J Gen Physiol. 2001;117:149–163. doi: 10.1085/jgp.117.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak MD, Baker K, Covarrubias M, Butler A, Ratcliffe A, Salkoff L. mShal, a subfamily of A-type K+ channel cloned from mammalian brain. Proc Natl Acad Sci U S A. 1991;88:4386–4390. doi: 10.1073/pnas.88.10.4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panyi G, Sheng Z, Deutsch C. C-type inactivation of a voltage-gated K+ channel occurs by a cooperative mechanism. Biophys J. 1995;69:896–903. doi: 10.1016/S0006-3495(95)79963-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo LA, Heinemann SH, Terlau H, Ludewig U, Lorra C, Pongs O, Stühmer W. Extracellular K+ specifically modulates a rat brain K+ channel. Proc Natl Acad Sci U S A. 1992;89:2466–2470. doi: 10.1073/pnas.89.6.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Campbell DL. Transient outward potassium current, ‘Ito’, phenotypes in the mammalian left ventricle: underlying molecular, cellular and biophysical mechanisms. J Physiol. 2005;569:7–39. doi: 10.1113/jphysiol.2005.086223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil PG, Brody DL, Yue DT. Preferential closed-state inactivation of neuronal calcium channels. Neuron. 1998;20:1027–1038. doi: 10.1016/s0896-6273(00)80483-3. [DOI] [PubMed] [Google Scholar]
- Patlak J. Molecular kinetics of voltage-dependent Na+ channels. Physiol Rev. 1991;71:1047–1080. doi: 10.1152/physrev.1991.71.4.1047. [DOI] [PubMed] [Google Scholar]
- Perozo E, Papazian DM, Stefani E, Bezanilla F. Gating currents in Shaker K+ channels. Implications for activation and inactivation models. Biophys J. 1992;62:160–171. doi: 10.1016/S0006-3495(92)81802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pioletti M, Findeisen F, Hura GL, Minor DL., Jr Three-dimensional structure of the KChIP1-Kv4.3 T1 complex reveals a cross-shaped octamer. Nat Struct Mol Biol. 2006;13:987–995. doi: 10.1038/nsmb1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper DR, Varghese A, Sanguinetti MC, Tristani-Firouzi M. Gating currents associated with intramembrane charge displacement in HERG potassium channels. Proc Natl Acad Sci U S A. 2003;100:10534–10539. doi: 10.1073/pnas.1832721100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourrier M, Herrera D, Caballero R, Schram G, Wang Z, Nattel S. The Kv4.2 N-terminal restores fast inactivation and confers KChlP2 modulatory effects on N-terminal-deleted Kv1.4 channels. Pflugers Arch. 2004;449:235–247. doi: 10.1007/s00424-004-1328-8. [DOI] [PubMed] [Google Scholar]
- Ramakers GM, Storm JF. A postsynaptic transient K+ current modulated by arachidonic acid regulates synaptic integration and threshold for LTP induction in hippocampal pyramidal cells. Proc Natl Acad Sci U S A. 2002;99:10144–10149. doi: 10.1073/pnas.152620399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmusson RL, Morales MJ, Wang S, Liu S, Campbell DL, Brahmajothi MV, Strauss HC. Inactivation of voltage-gated cardiac K+ channels. Circ Res. 1998;82:739–750. doi: 10.1161/01.res.82.7.739. [DOI] [PubMed] [Google Scholar]
- Roux MJ, Olcese R, Toro L, Bezanilla F, Stefani E. Fast inactivation in Shaker K+ channels. Properties of ionic and gating currents. J Gen Physiol. 1998;111:625–638. doi: 10.1085/jgp.111.5.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruben PC, Starkus JG, Rayner MD. Steady-state availability of sodium channels. Interaction between activation and slow inactivation. Biophys J. 1992;61:941–955. doi: 10.1016/S0006-3495(92)81901-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruppersberg JP, Frank R, Pongs O, Stocker M. Cloned neuronal IK(A) channels reopen during recovery from inactivation. Nature. 1991;353:657–660. doi: 10.1038/353657a0. [DOI] [PubMed] [Google Scholar]
- Ruta V, Chen J, MacKinnon R. Calibrated measurement of gating-charge arginine displacement in the KvAP voltage-dependent K+ channel. Cell. 2005;123:463–475. doi: 10.1016/j.cell.2005.08.041. [DOI] [PubMed] [Google Scholar]
- Salinas M, De Weille J, Guillemare E, Lazdunski M, Hugnot J-P. Modes of regulation of Shab K+ channel activity by the Kv8.1 subunit. J Biol Chem. 1997;272:8774–8780. doi: 10.1074/jbc.272.13.8774. [DOI] [PubMed] [Google Scholar]
- Scannevin RH, Wang K, Jow F, Megules J, Kopsco DC, Edris W, Carroll KC, Lu Q, Xu W, Xu Z, Katz AH, Olland S, Lin L, Taylor M, Stahl M, Malakian K, Somers W, Mosyak L, Bowlby MR, Chanda P, Rhodes KJ. Two N-terminal domains of Kv4 K+ channels regulate binding to and modulation by KChIP1. Neuron. 2004;41:587–598. doi: 10.1016/s0896-6273(04)00049-2. [DOI] [PubMed] [Google Scholar]
- Schmidt D, Cross SR, MacKinnon R. A gating model for the archeal voltage-dependent K+ channel KvAP in DPhPC and POPE:POPG decane lipid bilayers. J Mol Biol. 2009;390:902–912. doi: 10.1016/j.jmb.2009.05.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoppa NE, Westbrook GL. Regulation of synaptic timing in the olfactory bulb by an A-type potassium current. Nat Neurosci. 1999;2:1106–1113. doi: 10.1038/16033. [DOI] [PubMed] [Google Scholar]
- Serodio P, Kentros C, Rudy B. Identification of molecular components of A-type channels activating at subthreshold potentials. J Neurophysiol. 1994;72:1516–1529. doi: 10.1152/jn.1994.72.4.1516. [DOI] [PubMed] [Google Scholar]
- Serodio P, Vega-Saenz de Miera E, Rudy B. Cloning of a novel component of A-type K+ channels operating at subthreshold potentials with unique expression in heart and brain. J Neurophysiol. 1996;75:2174–2179. doi: 10.1152/jn.1996.75.5.2174. [DOI] [PubMed] [Google Scholar]
- Shahidullah M, Covarrubias M. The link between ion permeation and inactivation gating of Kv4 potassium channels. Biophys J. 2003;84:928–941. doi: 10.1016/S0006-3495(03)74910-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata R, Nakahira K, Shibasaki K, Wakazono Y, Imoto K, Ikenaka K. A-type K+ current mediated by the Kv4 channel regulates the generation of action potential in developing cerebellar granule cells. J Neurosci. 2000;20:4145–4155. doi: 10.1523/JNEUROSCI.20-11-04145.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin KS, Maertens C, Proenza C, Rothberg BS, Yellen G. Inactivation in HCN channels results from reclosure of the activation gate: desensitization to voltage. Neuron. 2004;41:737–744. doi: 10.1016/s0896-6273(04)00083-2. [DOI] [PubMed] [Google Scholar]
- Shirokov R, Ferreira G, Yi J, Ríos E. Inactivation of gating currents of L-type calcium channels. Specific role of the α2δ subunit. J Gen Physiol. 1998;111:807–823. doi: 10.1085/jgp.111.6.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerritt MR, Campbell DL. Role of S4 positively charged residues in the regulation of Kv4.3 inactivation and recovery. Am J Physiol Cell Physiol. 2007;293:C906–914. doi: 10.1152/ajpcell.00167.2007. [DOI] [PubMed] [Google Scholar]
- Skerritt MR, Campbell DL. Non-native R1 substitution in the S4 domain uniquely alters Kv4.3 channel gating. PLoS One. 2008;3:e3773. doi: 10.1371/journal.pone.0003773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skerritt MR, Campbell DL. Contribution of electrostatic and structural properties of Kv4.3 S4 arginine residues to the regulation of channel gating. Biochim Biophys Acta. 2009;1788:458–469. doi: 10.1016/j.bbamem.2008.09.012. [DOI] [PubMed] [Google Scholar]
- Solc CK, Aldrich RW. Gating of single non-Shaker A-type potassium channels in larval Drosophila neurons. J Gen Physiol. 1990;96:135–165. doi: 10.1085/jgp.96.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler-Llavina GJ, Chang TH, Swartz KJ. Functional interactions at the interface between voltage-sensing and pore domains in the Shaker Kv channel. Neuron. 2006;52:623–634. doi: 10.1016/j.neuron.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Song WJ, Tkatch T, Baranauskas G, Ichinohe N, Kitai ST, Surmeier DJ. Somatodendritic depolarization-activated potassium currents in rat neostriatal cholinergic interneurons are predominantly of the A type and attributable to coexpression of Kv4.2 and Kv4.1 subunits. J Neurosci. 1998;18:3124–3137. doi: 10.1523/JNEUROSCI.18-09-03124.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stotz SC, Hamid J, Spaetgens RL, Jarvis SE, Zamponi GW. Fast inactivation of voltage-dependent calcium channels. A hinged-lid mechanism? J Biol Chem. 2000;275:24575–24582. doi: 10.1074/jbc.M000399200. [DOI] [PubMed] [Google Scholar]
- Sun Y, Olson R, Horning M, Armstrong N, Mayer M, Gouaux E. Mechanism of glutamate receptor desensitization. Nature. 2002;417:245–253. doi: 10.1038/417245a. [DOI] [PubMed] [Google Scholar]
- Swartz KJ. Sensing voltage across lipid membranes. Nature. 2008;456:891–897. doi: 10.1038/nature07620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadross MR, Yue DT. Systematic mapping of the state dependence of voltage- and Ca-dependent inactivation using simple open channel measurements. J Gen Physiol. 2010;135:217–227. doi: 10.1085/jgp.200910309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadross MR, Johny MB, Yue DT. Molecular endpoints of Ca2+/calmodulin- and voltage-dependent inactivation of Cav1.3 channels. J Gen Physiol. 2010;135:197–215. doi: 10.1085/jgp.200910308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera K, Nilius B. Biophysics and structure-function relationship of T-type Ca2+ channels. Cell Calcium. 2006;40:97–114. doi: 10.1016/j.ceca.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Tombola F, Pathak MM, Isacoff EY. How far will you go to sense voltage? Neuron. 2005;48:719–725. doi: 10.1016/j.neuron.2005.11.024. [DOI] [PubMed] [Google Scholar]
- Vandenberg CA, Bezanilla F. Single-channel, macroscopic, and gating currents from sodium channels in the squid giant axon. Biophys J. 1991;60:1499–1510. doi: 10.1016/S0006-3495(91)82185-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev PM, Scheuer T, Catterall WA. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science. 1988;241:1658–1661. doi: 10.1126/science.241.4873.1658. [DOI] [PubMed] [Google Scholar]
- Villalba-Galea CA, Sandtner W, Starace DM, Bezanilla F. S4-based voltage sensors have three major conformations. Proc Natl Acad Sci U S A. 2008;105:17600–17607. doi: 10.1073/pnas.0807387105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yan Y, Liu Q, Huang Y, Shen Y, Chen L, Chen Y, Yang Q, Hao Q, Wang K, Chai J. Structural basis for modulation of Kv4 K+ channels by auxiliary KChIP subunits. Nat Neurosci. 2007;10:32–39. doi: 10.1038/nn1822. [DOI] [PubMed] [Google Scholar]
- Wang S, Bondarenko VE, Qu YJ, Bett GC, Morales MJ, Rasmusson RL, Strauss HC. Time- and voltage-dependent components of Kv4.3 inactivation. Biophys J. 2005;89:3026–3041. doi: 10.1529/biophysj.105.059378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster SM, Del Camino D, Dekker JP, Yellen G. Intracellular gate opening in Shaker K+ channels defined by high-affinity metal bridges. Nature. 2004;428:864–868. doi: 10.1038/nature02468. [DOI] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc Natl Acad Sci U S A. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G. The moving parts of voltage-gated ion channels. Q Rev Biophys. 1998;31:239–295. doi: 10.1017/s0033583598003448. [DOI] [PubMed] [Google Scholar]
- Yifrach O, MacKinnon R. Energetics of pore opening in a voltage-gated K+ channel. Cell. 2002;111:231–239. doi: 10.1016/s0092-8674(02)01013-9. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 1990;250:568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Shaker potassium channel gating. III: Evaluation of kinetic models for activation. J Gen Physiol. 1994;103:321–362. doi: 10.1085/jgp.103.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Morais-Cabral JH, Mann S, MacKinnon R. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 2001a;411:657–661. doi: 10.1038/35079500. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 Å resolution. Nature. 2001b;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- Zhou W, Qian Y, Kunjilwar K, Pfaffinger PJ, Choe S. Structural insights into the functional interaction of KChIP1 with Shal-type K+ channels. Neuron. 2004;41:573–586. doi: 10.1016/s0896-6273(04)00045-5. [DOI] [PubMed] [Google Scholar]