

Abstract
In this study, fast synaptic transmission at vertebrate CNS connections mediated by several different nicotinic ACh receptors (nAChRs) was investigated with paired recordings from pre- and postsynaptic neurons. Analysis of the response kinetics at the axo-axonic connections between the Mauthner (M-) axon and cranial relay neurons (CRN) indicates up to three main components are present and can be characterized by fast, ∼1.5 ms, intermediate, ∼6 ms and slow, ∼15 ms, decay time constants. Further analysis indicates most responses have multiexponential decays and each response falls into one of six classes dependent on the weight and combination of kinetic components. Pharmacological results suggest that up to three nAChRs, α7*, α3β2* and α3β4*, mediate the postsynaptic responses and correspond to the fast, intermediate and slow decay components, respectively. The fast decay component is blocked by ∼35 nm methyllycaconitine (MLA), 100 nmα-bungarotoxin (α-Btx) or 150 nmα-conotoxin (α-Ctx) ArIB. The intermediate decay component is blocked by 2 μm dihydro-beta-erythroidine (DHβE) or 200 nmα-Ctx GIC. The slow decay component is blocked by 10 μmα-Ctx AuIB, but not by 7.25 μm DHβE. Intriguingly, the mEPSPs (minis) at connections with evoked EPSPs best fitted by multiple exponentials, were not composite; rather, there were multiple populations of minis, each with single exponential decay times corresponding to those of the different evoked EPSP components. This indicates that the different receptors are topographically segregated at the connection between the M-axon and CRN axon. These results suggest that, as with glutamate, fast nicotinic synaptic transmission in the CNS can be mediated by multiple receptors in the same postsynaptic neuron. The coexistence of EPSPs of different durations may have implications for network function and plasticity.
Non-technical summary
Usually nicotinic receptors in the central nervous system only influence the strength of a signal between neurons. At a few critical connections, for instance some of those involved in the flight response, nicotinic receptors not only modulate the signal, they actually determine whether a signal is conveyed or not. We show at one of the few such connections accessible for study, up to three different nicotinic receptor subtypes mediate the signal. The subtypes appear to be clustered in separate locations. Depending on the number and combination of the subtypes present the signal can range from short to long duration and from low to high amplitude. This provides a critical connection with a built-in plasticity and may enable it to adapt to a changing environment.
Introduction
Nicotinic transmission is widespread in both the central and autonomic nervous systems and most studies have focused on the modulatory role of nicotinic ACh receptors (nAChRs), largely because there are relatively few examples of their postsynaptic function (McGehee et al. 1995; Jones et al. 1999; Berg & Conroy, 2002; McIntosh et al. 2005; Wonnacott et al. 2006; Dani & Bertrand, 2007). Unfortunately, models of fast nicotinic transmission in the CNS have not been accessible for paired recordings (Roerig et al. 1997; Frazier et al. 1998; Alkondon et al. 1998; Nong et al. 1999; Bradaia & Trouslard, 2002; Hatton & Yang, 2002; Guo et al. 2005; Thinschmidt et al. 2005). A notable exception is the nicotinic axo-axonic connection between the Mauthner (M-) axon and cranial relay neuron (CRN) of the goldfish, which is readily accessible for paired recordings and pharmacological manipulations. As this is an in vivo model, correlations between behaviour and physiology are also feasible (Weiss et al. 2006).
Key insights into the physiology of α7 and non-α7 nACh receptors have come from paired recordings in dissected chick ciliary ganglion of the autonomic system. There, fast synaptic transmission (Zhang et al. 1996; Ullian et al. 1997) is mediated by α7 nAChRs, concentrated on spines and largely excluded from postsynaptic densities (PSDs), and by α3* nAChRs, where * indicates the possible presence of additional subunits (Lukas et al. 1999), located within somatic PSDs (Jacob & Berg, 1983; Jacob et al. 1984; Loring et al. 1985; Horch & Sargent, 1995; Williams et al. 1998; Shoop et al. 1999). The means by which α7 nAChRs are activated is debated. Possible contributions to the α7 nAChR response include (1) acetylcholine release at sites not apposed to PSDs, that is, ectopic release on spines (Shoop et al. 1999, 2001; Coggan et al. 2005; Sargent, 2009), (2) multiquantal release at the relatively rare spinous active zones and/or (3) diffusion of transmitter released somatically to PSDs on spines (Nguyen & Sargent, 2002). These observations raise the question of the functional organization of fast nicotinic synapses in the vertebrate CNS.
To study fast nicotinic transmission we have used an in vivo CNS model system, the connection in the goldfish hindbrain between the M-axon and the postsynaptic CRNs. The M-cell is responsible for a stereotyped escape behaviour in the goldfish, the C-start (Zottoli, 1977). CRNs relay the command from the M-axon to motorneurons controlling jaw, opercular and ocular muscles (Diamond, 1971; Hackett & Faber, 1983a; Hackett & Buchheim, 1984) and to interneurons that mediate feedback inhibition of the M-cells (Hackett & Faber, 1983b). Based on morphology, at least two classes of CRNs have been reported. In one class, the axon projects medially from the cell body, crosses over the nearest M-axon and midline before projecting only rostrally to innervate the trigeminal nucleus (Hackett & Buchheim, 1984). In another class, CRN axons bifurcate and course both rostrally and caudally after crossing the midline (Titmus & Faber, 1987). By inspection of the CRN homologues in the zebrafish (Kimmel et al. 1985), the T-reticular interneurons, coupled with the morphological evidence in the goldfish, it is likely that different types of CRNs serve different motoneuron targets. The homologue of the M-axon–CRN connection in the hatchetfish is the nicotinic connection between the Mauthner and giant fibres, which controls pectoral fins in addition to the aforementioned targets (Auerbach & Bennett, 1969).
We have used the M-axon–CRN connection to investigate fast nAChR-mediated transmission in the CNS and the properties imparted by distinct nAChR subtypes. We found that the postsynaptic receptor complement is diverse and can include combinations of α7* and non-α7 nAChRs, which produce different decay kinetics in the EPSPs among CRN types. Furthermore, electrophysiological data indicate these receptor subtypes are localized to separate clusters, possibly within one contact or on the separate distinct contacts that comprise a connection. This insight lays the foundation for studying the functional contributions of the different receptor subtypes and pre- and postsynaptic factors influencing the strength and dynamics of the EPSP.
Methods
Ethical approval
All experiments were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) protocols approved at Albert Einstein College of Medicine, consistent with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals and compliant with all federal, state and local regulations.
Surgery
One hundred adult male or female goldfish (Carassius auratis, length 7.5–10 cm, measured without the tail) were obtained from EECHO Systems (North Kansas City, MO, USA), Hunting Creek Fisheries (Thurmond, MD, USA) and Billy Bland Fisheries (Taylor, AR, USA), and were maintained in conditioned water at 17–18°C (Szabo et al. 2006). Goldfish were anaesthetized using 60 mg l−1 3-aminobenzoic acid ethyl ester (MS222) in conditioned water and subsequently mounted in an experimental chamber in which animals were respirated through the mouth to provide a continuous flow of aerated conditioned water over the gills. This water contained 60 mg l−1 MS222 and was cooled to 6°C. The spinal cord was exposed for antidromic stimulation of the M-cell and goldfish were immobilized with d-tubocurarine chloride (Abbot Laboratories, Chicago, IL, USA) injected intramuscularly (1 mg (gm body weight)−1) after the anaesthesia took effect. The cranium was opened dorsally at the location of the fourth ventricle and the cerebellum and the facial lobes were gently retracted rostrally and retained with paper spatulas fashioned from Kimwipes (Mississauga, Ontario, Canada). Subsequently, the vagal lobes were retracted laterally and retained to reveal the section of the brainstem containing the CRN cell bodies and to enable placement of electrodes in the M-axon and CRN as illustrated in Fig. 1 (Faber & Korn, 1978; Waldeck et al. 2000). At the end of the experiment, the goldfish used for this project were killed by decapitation in accordance with IACUC protocols.
Figure 1. Characteristics of the excitatory axo-axonic connection between the Mauthner (M-) and cranial relay neuron (CRN) axons.
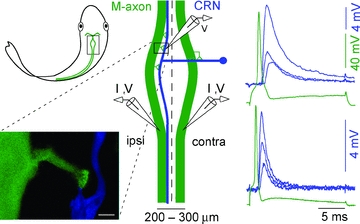
Location of M-axon–CRN connection in goldfish hindbrain is highlighted with a rectangle (upper left). For clarity only one CRN is illustrated and as indicated, a CRN receives input from both M-axons. Expanded diagram (centre) depicts the typical contact locations (triangles) between a presynaptic M-axon (green) and a postsynaptic CRN axon (blue). M-axons 50–100 μm in diameter run within 75–400 μm of each other and parallel to the midline (dashed line). The lower left panel shows a confocal image of a M-axon (green)–CRN (blue) axo-axonic contact. The calibration bar is 20 μm. Right, current pulses (I) injected into either the ipsilateral or contralateral M-axon generate a presynaptc action potential followed by EPSPs that depress with increasing stimulus number (1, 2 and 10) at 1 Hz. The EPSP decay kinetics differ among CRNs and can range from slow (upper right) to fast (lower right).
Electrophysiology
Paired recordings from the M-axon and CRN axon were made in current clamp mode using sharp electrodes and an Axoprobe (Axon Instruments, Foster City, CA, USA), a two-electrode amplifier with cross talk neutralization. The presynaptic electrodes were filled with 3 m KCl buffered with 10 mm Hepes, pH 7.2 (resistance 8–11 MΩ). The postsynaptic electrodes were filled with 3 m KCl, 75 mmN-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide (QX-314) and 2.5% neurobiotin (resistance 14–18 MΩ). QX-314 eliminated contamination by regenerative sodium-dependent responses during collection of EPSPs that were otherwise suprathreshold at low stimulus frequency. The presynaptic M-axon, a myelinated axon 70–100 μm in diameter situated 100–200 μm from the midline and at most 100–150 μm below the hindbrain surface, was penetrated after being located with the aid of a stereomicroscope. The CRN axons, which run alongside and within 50 μm of the M-axon in the hindbrain, were found by systematically probing the hindbrain while stimulating the M-axon antidromically with an electrode on the exposed caudal spinal column. Once a postsynaptic CRN was penetrated, the M-axon was stimulated with transmembrane current pulses (15–30 nA, 18 ms duration). The identity of a CRN was confirmed by the rapid depression of its response to repeated M-axon stimulation, evidence of inputs from both M-axons, the EPSP waveform and monosynaptic latency (Hackett & Faber, 1983a; Hackett et al. 1989; Waldeck et al. 2000).
Miniature EPSPs (mEPSPs) were collected continuously at a 60 μs per point sampling rate, first without depolarization and subsequently with a 10–20 nA depolarization of the M-axon. Asynchronous mEPSPs rarely followed the large stimulus-evoked responses. Without depolarization spontaneous mEPSPs frequency was 1.8 ± 1.3 events s−1 (n= 6).
Perfusion of drugs
The brain was superfused with normal fish saline (in mm: 124.0 NaCl, 5.1 KCl, 2.8 NaH2PO4.H2O), 0.9 MgSO4, 1.6 CaCl2.2H2O, 5.6 glucose and 20.0 Hepes, pH 7.2), and the perfusate was drained continuously via a shunt posterior and lateral to the hindbrain. Two protocols were employed for delivery of antagonists to the hindbrain. α-Conotoxins (α-Ctx) GIC, AuIB and ArIB were injected via a septum into the fish saline superfusion stream, which was maintained at 6°C and flowed at a rate of 30 μl min−1. A bolus of α-Ctx (20–50 μl) was injected over 1 min. A 0.5 ml volume between the injection site and the point at which the superfusion was delivered to the brain surface enabled adequate mixing of antagonist before the delivery point to provide consistent results. This arrangement enabled a reasonably rapid delivery and exchange of drugs, with minimum consumption of antagonist and minimum mechanical disruption at the brain surface. A higher perfusion flow rate (150–250 μl min−1) protocol, which provided even greater stability and enabled longer recording times, was used to deliver: α-Btx (Tocris, Ellisville, MO, USA), DHβE and MLA. Unless otherwise specified, chemicals and reagents were purchased from Sigma (St Louis, MO, USA). α-Ctxs were provided by J. M. McIntosh and synthesized as previously described: ArIB (Whiteaker et al. 2007), AuIB[V11L;V16D] (Luo et al. 1998) and GIC (McIntosh et al. 2002).
Neurobiotin injection and immunohistological protocol
In some experiments, after gathering electrophysiological data the morphology of the M-axon–CRN connection was marked by injecting Neurobiotin (Vector Laboratories, Burlingame, CA, USA). The marker was injected iontophoretically into the CRN for at least 30 min using 90 nA depolarizing current injections (400 ms at 1 Hz). Subsequently, the fish was perfused intracardially with phosphate buffer and 4% paraformaldehyde in PBS, pH 7.4. The dissected brain was refrigerated in fresh 4% paraformaldehyde for 24 h, transferred to cryoprotectant (30% sucrose in 0.1 m phosphate buffer) and stored at 4°C. Using a CM3050S cryostat (Leica Microsystems, Nussloch, Germany), 60 μm horizontal slices were made and retrieved in a 24-well plate containing 0.1 m phosphate buffer and refrigerated until further processing. Slices were washed in 0.5% Triton in 0.1 m phosphate buffer and treated for 1 h in a blocking solution containing 10% normal goat serum (Jackson ImmunoResearch, West Grove, PA, USA). Brain sections were incubated overnight in primary antibodies to zonula occludins II (rabbit anti-ZO-2) (Molecular Probes, Eugene, OR, USA) at a 1:125 dilution in 0.5% Triton and 5% normal goat serum in PBS at 4°C. ZO-2 antibodies were used because they have been shown to selectively stain the M-axon (Flores et al. 2008). To image the CRN injected with Neurobiotin, sections were incubated for 2 h at room temperature in streptavidin AlexaFluor 488 (Molecular Probes) at a 1:250 dilution. At the same time secondary antibodies conjugated to AlexaFluor goat anti-rabbit (Molecular Probes) at a 1:250 dilution were added to visualize the M-axon. AlexaFluor conjugated α-bungarotoxin was used to mark the putative α7* acetylcholine receptors located at the M-axon–CRN connection.
Serial sections were analysed and images were recorded with an upright Olympus BX61WI and Zeiss LSM 5 Duo V2 confocal microscopes of the Morphology and Image Analysis Facility of the Kennedy Center at Albert Einstein College of Medicine. FluoView and Imaris software were used to create projection images.
Data analysis
Custom software (Trace Analyzer 2 and 3) was used to collect data and analyse EPSP amplitudes. The sampling rate for EPSPs and mEPSPs were 10–25 μs point−1 and 60 μs point−1, respectively. Artifacts caused by current injection on the tails of some EPSPs were removed from the traces. Decay times were analysed using Igor (Wavemetrics, Lake Oswego, OR, USA) and Detectivent (Ankri et al. 1994). To improve the signal-to-noise ratio 5–20 EPSP traces were averaged for decay time analysis. Only data taken under steady-state conditions were averaged, and only one frequency was used during a given experiment, and therefore skewing the relative amplitudes of the different components as a function of changing depression was avoided. Decay time constants of EPSPs were determined by fitting the decaying phase initially with a single exponential and then increasing to the optimum number of exponentials as determined by the F test and P value ≤ 0.05. The robustness of each fit was explored by varying the initial conditions up to one order of magnitude from time constants determined by the initial fit and by increasing the width of the fitting window in increments by 20 ms.
Only the EPSP decays best fitted by a combination of four time constants, τ1–τ4 (class 6), exhibited sensitivity to the width of the fit windows and the initial conditions. For the cases in which one component had been isolated or determined as a difference peak by using antagonists, the exponential fit of the composite peak that contained the time constant that most closely matched that of the isolated single component was selected. If such an empirical constraint was not available, the average values for τ1–τ4 were used (Table 1).
Table 1.
Decay kinetics of EPSPs
| n | Rel. Wt | τ1 (ms) Rel. Wt | τ2 (ms) Rel. Wt | τ3 (ms) Rel. Wt | τ4 (ms) Rel. Wt | |
|---|---|---|---|---|---|---|
| Class 1 α7* | ||||||
| τ < 2.20 | 48 | 1.4 ± 0.3 | 14.4 ± 9.2 | |||
| 88.6±3.6% | 11.4±3.6% | |||||
| All class 1 | 75 | 54.7% | 1.7 ± 0.4 | 15.7 ± 10.0 | ||
| 87.9±4.3% | 12.1±4.3% | |||||
| Class 2 α3β2*/α3β4* | ||||||
| All class 2 | 6 | 4.4% | 5.3 ± 1.3 | 14.7 ± 4.2 | ||
| 42.7±24.1% | 57.3±24.1% | |||||
| Class 3 α3β4* | ||||||
| All class 3 | 4 | 2.9% | 9.0 ± 1.6 | |||
| 100.0% | ||||||
| Class 4 α7*/α3β2* | ||||||
| 2 kinetic components | 5 | 1.7 ± 0.3 | 6.2 ± 1.6 | |||
| 52.2±24.9% | 47.8±24.9% | |||||
| 3 kinetic components | 7 | 1.4 ± 0.2 | 22.0 ± 4.9 | 4.3 ± 1.0 | ||
| 72.5±15.7% | 10.4±4.8% | 17.1±12.6% | ||||
| All class 4 | 12 | 8.8% | 1.5 ± 0.3 | 22.0 ± 4.9 | 5.1 ± 1.6 | |
| 64.1±21.7% | 6.1±6.4% | 29.9±23.7% | ||||
| Class 5 α7*/α3β4* | ||||||
| 2 kinetic components | 23 | 1.8 ± 0.4 | 13.2.0 ± 4.2** | 13.2 ± 4.2** | ||
| 51.9±23.9% | 6.4±3.0%** | 41.7±26.8%** | ||||
| 3 kinetic components | 2 | 1.8 ± 0.4 | 15.7 ± 10.0 | 13.2 ± 2.2 | ||
| 76.9±2.9% | 8.5±3.2% | 14.6±6.1% | ||||
| All class 5 | 25 | 18.2% | 1.8 ± 0.4 | 13.4 ± 4.6 | 13.2.0 ± 4.1 | |
| 53.8±23.9% | 6.6±3.0% | 39.6±26.8% | ||||
| Class 6 α7*/α3β2*/α3β4* | ||||||
| 3 kinetic components | 11 | 1.7 ± 0.5 | 16.3 ± 4.1*** | 6.2 ± 1.3 | 16.3 ± 4.1*** | |
| 23.9±10.7% | 2.9±1.3%*** | 40.0±1.4% | 33.2±16.5%*** | |||
| 4 kinetic components | 4 | 1.6 ± 0.6 | 22.3 ± 13.3 | 5.0 ± 2.1 | 10.5 ± 1.3 | |
| 26.2±10.9% | 5.1±0.7% | 37.0±17.3% | 31.6±16.6 | |||
| All class 6 | 15 | 10.9% | 1.6 ± 0.5 | 17.9 ± 7.6 | 5.9 ± 1.5 | 14.8 ± 4.4 |
| 24.5±10.4% | 3.5±1.5% | 39.2±14.0% | 32.8±16.0% | |||
| All classes | 137 | |||||
| Classes blocked/isolated | ||||||
| Class 1 | 9 | 1.6 ± 0.2 | 13.4 ± 8.0 | |||
| Class 2 | 4 | 6.0 ± 1.7 | ||||
| Class 2, 3 & 6 | 6 | 13.2 ± 3.8 | ||||
Within tau group and across classes, t test P > 0.05, except τ4 of class 3, which is significantly different from τ4 for all other associated classes, t test P < 0.05; ** and *** indicate that the t2/t4 component has been partitioned between τ2 and τ4 class 5 (**) and class 6 (***).
To ensure that the mEPSPs were detected without bias, measurements were automated. Detectivent software was used to detect randomly occurring mEPSPs and analyse mEPSP decay time constants. Initially mEPSPs were treated as monoexponential. The resulting decay time constant distributions ranged from mono- to trimodal and could be fitted with sums of Gaussians. These decay times were confirmed using Igor software. Origin 7 (Northampton, MA, USA) was used to construct the decay time distributions. Averaging of mEPSPs was used to improve the signal-to-noise ratio (Zhang et al. 1996). For averaging, between 20 to 50 mEPSPs from each population defined by a mode and within 3 standard deviations of each Gaussian mean were selected. mEPSPs were aligned by their rising edges. Events with amplitudes of less than three times the standard deviation of the noise or with tails or onsets overlapping another mEPSP were not used in the averaged data.
Basic logical alignment search tool (BLAST) and vector alignment search tool (VAST) available at the National Center for Biological Information were used to compare sequence and structural identity of α3, α7, β2 and β4 nAChRs across species.
Results
Data from CRNs in the goldfish (Hackett and Faber, 1983a) and their homologues in hatchetfish (Model et al. 1975; Gilat et al. 1986; Barry & Bennett, 1990) and zebrafish (Kimmel et al. 1985) indicate each CRN receives inputs from both M-axons via axo-axonic contacts. The CRN axons are ∼20 μm in diameter and are myelinated (Hackett & Buchheim, 1984). As shown schematically in Fig. 1, there are a limited number of axo-axonic contacts between short collaterals issued by both the Mauthner and CRN axons (Hackett & Buchheim, 1984; Waldeck et al. 2000). For clarity only, one CRN on one side of the midline is illustrated. Because of their axonal location, the contacts comprising this connection represent the dominant input to the CRN. Both pre- and postsynaptic elements are accessible for intracellular physiology, and the large visible M-axon enables manipulations designed to elucidate presynaptic factors influencing synaptic transmission. Since the connections are axo-axonic, it is possible to detect and analyse mEPSPs largely uncontaminated by non-M-axon inputs (Barry & Bennett, 1990; Waldeck et al. 2000).
In the electrophysiological context, the M-axon that is immediately adjacent to the rostral–caudal or parallel branch of the CRN is termed the ipsilateral M-axon. Since the M-axons decussate, the CRN and Mauthner cell bodies are on the same side of the hindbrain. Typical contact locations, one between the crossing CRN branch and contralateral M-axon and several between the parallel CRN branch and ipsilateral M-axon, are indicated in Fig. 1, centre panel. Contacts from the ipsilateral M-axon are most often observed within ∼200 μm of crossing branch of the CRN axon. A confocal image shows a typical junction between the M-axon and CRN axon collaterals (Fig. 1, lower left). In the goldfish (Hackett et al. 1989) and in the hatchetfish (Model et al. 1975), each contact contains multiple active zones. Transmission between the M-axon and a CRN is blocked by d-tubocurarine and is believed to be cholinergic based on this finding and on the observation of α-Btx staining in the hatchetfish (Day et al. 1983).
Paired recordings were obtained with the pre- and postsynaptic electrodes placed within the confines of the vagal lobe, and we estimated that the CRN electrode was typically within 200 μm of the contact zone (Fig. 1). EPSPs were triggered by a spike, which was evoked by a brief depolarizing current pulse in the recorded M-axon. M-axon spike height and duration were monitored throughout the experiment. We estimated that the CRN axon length constant and effective membrane time constant were ∼2 mm and ∼400 μs, respectively, on the basis of paired simultaneous recordings from single CRN axons, while applying 7 nA through one electrode in two preparations. This time constant was faster than the decay time of any EPSP component described in the present work. For EPSP components with decay time constants >2 ms, it was unlikely that either the size or kinetics of the spontaneous and evoked EPSPs were appreciably degraded by CRN cable properties. Although some degradation may have occurred with EPSPs components having decay time constants of ∼1.5 ms, it was still possible to correlate kinetics and pharmacology.
EPSP kinetics
Data collected from 137 M-axon–CRN axon pairs exhibited a wide range of the evoked EPSP decay kinetics. This was quantified by inspection of the distribution of decay time constants, obtained by assuming monoexponential decay (Fig. 2). On this basis, the decay time constant ranged from ∼1 ms to 12 ms, i.e. over one order of magnitude. As shown in Fig. 2, at least one clear population of EPSPs could be distinguished on this basis, with τmean= 2.1 ± 0.5 ms. Subsequently, we asked if each EPSP decay could be better fitted as a multiexponential, using the F test to establish the optimum number of decay components. On this basis, the EPSPs fell into six different classes characterized by some combination of up to four time constants, τ1–τ4. The following combinations exist: class 1: τ1 and τ2; class 2: τ3 and τ4; class 3: τ4 only; class 4: τ1, τ2 and τ3; class 5: τ1, τ2 and τ4; and class 6: τ1, τ2, τ3 and τ4. Among the exemplars from each class (Fig. 2) only the class 3 EPSP is well fitted by one exponential using the F test criterion. The average values and weight of the decay time constants and the relative prevalence of class type are listed in Table 1. The EPSPs with values of τ1–τ4 blocked or isolated by antagonists are also listed in Table 1.
Figure 2. Decay time histogram of M-axon–CRN pairs.
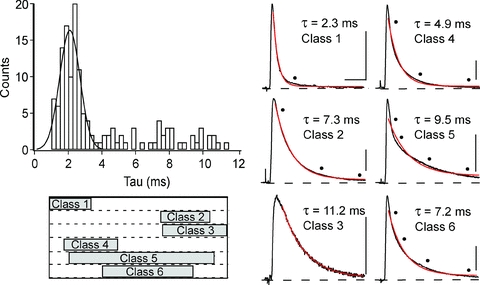
The decay phase of 137 EPSPs were fitted with one exponential. One distinct mode representing class 1 EPSPs is present in the histogram with a mean at 2.1 ms. Exemplars of the single exponential fits of classes 1–6 EPSPs, illustrate that these EPSPs will best be fitted by more than one exponential, except for the class 3 EPSP, which is well fitted with one exponential. Dots mark the points of divergence between the trace and the fit. EPSP classes 1, 4, 5 and 6 represent the majority of all EPSPs recorded. The width of the decay time windows associated with all EPSP classes, 1–6, are indicated by bars below the histogram. Horizontal calibration bar, 10 ms, applies to all classes. Vertical calibration bars, 2 mV for class 3 and 4 mV for all other classes.
As mentioned, only one distinct mode is present in the histogram of decay time values (Fig. 2). A total of 48 EPSPs in the first mode of the histogram, which are termed class 1 EPSPs, are well separated from the right shoulder of this mode and are best fitted by two time constants, τ 1: 1.4 ± 0.3 ms and τ2: 14.4 ± 9.2 ms with relative weights of 88.6 ± 3.6% and 11.4 ± 3.6%, respectively (Table 1). We chose this subset, 48 of the 75 EPSPs comprising this distinct mode, to avoid the overlap region between class 1, 4 and 5 EPSPs. The dominant time constant, τ1, is the fastest decaying of the four time constants observed. In 8.9% of the cases, class 4 EPSPs, there was a third, intermediate component, with τ3 ≈ 5–6 ms, in combination with τ1 and τ2. In 18.2% of the responses, class 5 EPSPs, the contribution of a slowly decaying component, τ4 ≈ 14 ms, is observed in combination with τ1 and τ2. Note that although τ4 and τ2 have similar values, they are associated with different receptor types as will be clarified by the pharmacological data. Finally, in 10.9% of the responses, class 6 EPSPs, both the intermediate and slowly decaying components are present along with τ1 and τ2 components. Thus, the majority of the EPSPs have τ1 and τ2 components, in isolation or in combination with the other taus, and we focused first on the characterization of this primary response.
Fast kinetic decay: EPSP class 1
The majority of CRNs, 85%, were investigated by stimulating the ipsilateral M-axon, thus probing connections with multiple contacts. Including these, as well as responses to input from contralateral M-axons, EPSPs from class 1 were observed 53.7% of the time. As nicotinic antagonists can block transmission at the M-axon–CRN connection and its homologue in the hatchetfish (Model et al. 1972; Day et al. 1983), we asked if the responses could be attributed to the nAChRs.
In the example of Fig. 3A the decay of a class 1 EPSP was fitted by a double exponential with time constants of 1.7 and 19.3 ms for τ1 and τ2, respectively, and the EPSP was completely blocked by 200 nm MLA. Overall, we found that EPSPs of this class are blocked by ∼35 nm MLA, as shown in the dose–response curve representing the results from 10 M-axon–CRN pairs at nine concentrations of MLA, n= 2 for 10 nm MLA (Fig. 3B). The inset is an example of the block at 35 nM MLA. As discussed below, this suggests that class 1 EPSPs are mediated by α7* nAChRs (Alkondon & Albuquerque, 1993). Access of antagonists to the M-axon–CRN connection in the in vivo preparation may be more limited than to nicotinic connections that have been investigated in slice and culture preparations in other species or to nAChRs expressed in oocytes. As 35 nm MLA can take more than 1 h to completely block the response, concentrations of 100–200 nm MLA were routinely used to reduce the time to block to ∼30 min.
Figure 3. MLA and α-Ctx ArIB block class 1 EPSPs.
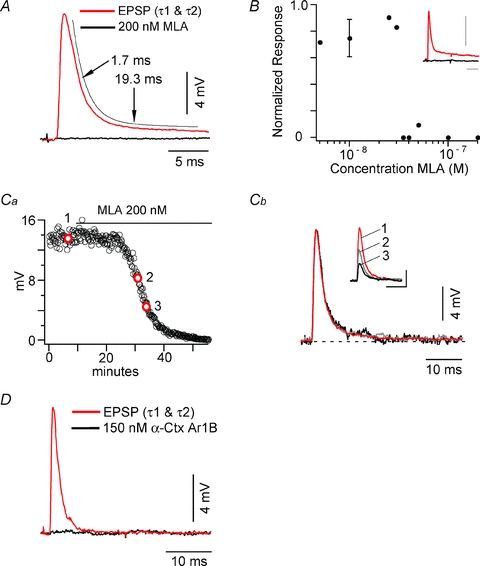
A, class 1 EPSP obtained before (red) and after (black) superfusion with 200 nm MLA. The decay of the control EPSP is fitted as the sum of two exponentials (offset thin black line), with τ1 = 1.7 ms and τ2 = 19.3 ms. B, dose–response curve for MLA. Inset shows block of class 1 EPSP with 35 nm MLA. Calibration bars are 10 ms and 4 mV. C, time course of MLA block. Ca, plot of EPSP amplitude vs. time in the same experiment as in A (frequency: 0.08 Hz). Red circles 1, 2 and 3 refer to time points at which responses in Cb were obtained. Cb, EPSPs from time points before MLA (1 – red, control), after ∼40% block (2 – grey) and after ∼70% block (3 – black) are scaled to the control EPSP and superimposed, showing both exponentials, τ1 and τ2, are equally sensitive to the antagonist. Inset shows the unscaled EPSPs. Calibration bars pertain to the unscaled data. D, class 1 EPSP obtained before (red) and after (black) superfusion with 150 nmα-Ctx ArIB [V11L;V16D].
To confirm that the two decay time constants characterizing class 1 EPSPs can both be attributed to α7* nAChRs, we compared the waveforms of the EPSPs at different stages of the block. Figure 3Ca illustrates the time course of antagonism by 200 nm MLA; the EPSP was reduced by ∼40% and 70% from control (point 1) at points 2 and 3, respectively. In Fig. 3Cb the corresponding EPSPs are normalized to match the size of the control, and the superimposed responses demonstrate that all three EPSPs have the same waveform. Class 1 EPSPs were also blocked by 50–100 nmα-Btx, an α7 nAChR antagonist (Zhang et al. 1994). Finally, to further establish that goldfish class 1 EPSPs are mediated by α7* nAChRs, we used 150 nmα-Ctx ArIB[V11L;V16D], a highly specific α7 nAChR antagonist (Whiteaker et al. 2007), to block class 1 EPSPs (Fig. 3D).
Intermediate and slow kinetic decay: EPSP classes 2 and 3
Two classes associated with two additional time constants, τ3 and τ4, were identified from decay time analysis (Fig. 2, and Table 1). The EPSPs in class 2 are best fitted by intermediate, τ3, and slow, τ4, time constants, while those in class 3 are fitted by only the slow time constant, τ4. In class 2 the average values and relative weights for τ3 and τ4 are 5.3 ± 1.3 ms, 42.7 ± 24.1% and 14.7 ± 4.2 ms, 57.3 ± 24.1%, respectively, and τ4 has a value of 9.0 ± 1.6 for EPSPs in class 3 (Table 1). Although EPSP classes 2 and 3 accounted for only 7% of all pairs studied, the components, τ3 and/or τ4, are present in five of the six classes observed. Overall these components are found in 45% of the EPSPs.
As all time constants identified in this study are represented in EPSP classes 1–3, primarily these classes were used for the pharmacological studies. As just discussed, class 1 EPSPs, associated with τ1 and τ2, are mediated by α7* nAChRs. We next asked if non-α7 nAChRs mediated the EPSP components associated with the intermediate and slow time constants, τ3 and τ4. In class 2 EPSPs the weight of each of the components, τ3 and τ4, is large enough to be readily detected and only one time constant, τ4, is associated with class 3 EPSPs, making these classes advantageous for the pharmacological experiments.
In contrast to class 1 EPSPs, pharmacological evidence suggested that two receptor subtypes mediate a class 2 EPSP. In the example of Fig. 4A, the control EPSP decay was fitted as the sum of two exponentials, τ3 = 4.9 ms and τ4 = 12.8 ms. This EPSP was partially blocked by 1.5 μm MLA and the residual response decayed monoexponentially with a time constant, τ4, of 13.5 ms, similar to that extracted from the fit of the composite control. To minimize access time, 1.5–2 μm MLA routinely was used to block the signal associated with τ3 time constant. The residual response, i.e. that associated with τ4, is insensitive to MLA at concentrations as high as ∼20 μm. In Fig. 4A the difference signal, i.e. the MLA-sensitive component, had a decay time constant of 4.5 ms, a value within the τ3 range (Table 1).
Figure 4. Pharmacological dissection of class 2 and class 3 EPSPs.

For A–C, superimposed records of averaged class 2 EPSPs in control (black) and after perfusion with antagonist for ∼30 min (green) and the difference trace (blue). Control is fitted (offset thin black line) with the sum of two exponentials, τ3 and τ4. Monoexponential fits of the decays for the difference (τ3) and antagonist-insensitive (τ4) peaks are marked with offset thin black lines. A, 1.5 μm MLA, Control: τ3 = 4.9 and τ4 = 12.8 ms, Difference: τ3 = 4.5 ms, Antagonist insensitive: τ4 = 13.5 ms. B, 2 μm DHβE, Control: τ3 = 7.5 and τ4 = 11.3 ms, Difference: τ3 = 7.5 ms, Antagonist insensitive: τ4 = 13.2 ms. C, 200 nmα-Ctx GIC, Control: τ3 = 6.4 and τ4 = 16.7 ms, Difference: τ3 = 6.7 ms, Antagonist insensitive: τ4 = 20.2 ms. D, superimposed records of averaged of class 3 EPSP in control (black), after perfusion with 10 μmα-Ctx AuIB ∼30 min (green) and after washing out the antagonist for ∼15 min (grey). Control: τ4 = 14.4 ms, Mostly blocked: τ4 = 20.8 ms, after wash: τ4 = 24.9 ms.
The τ3 component was also blocked by 2 μm DHβE (Fig. 4B) and by 200 nmα-Ctx GIC (Fig. 4C). Taken together, these data suggest that the τ3 response is mediated by α3β2* nAChRs. In other systems, α3β2* nAChRs were blocked by > 1 μm MLA (Drasdo et al. 1992; Astles et al. 2002) or by 1–1.6 μm DHβE (Chavez-Noriega et al. 1997; Faria et al. 2003). α-Ctx GIC is a peptide that blocks nAChRs in the sequence α3β2≫α4β2 > α3β4. At 100 nm, α-Ctx GIC completely blocks human α3β2 expressed in oocytes (McIntosh et al. 2002), and it also blocks α6β2* nAChRs (J. M. McIntosh, unpublished observation); since 200 nm MLA can block α6β2* nAChRs in rat (Mogg et al. 2002), the contribution of α6β2* nAChRs has not yet been ruled out in class 2 EPSPs.
In Fig. 4A–C, the EPSP component remaining in the presence of MLA, DHβE or α-Ctx GIC has a decay time constant ranging from 13.2–20.2 ms, i.e. τ4. Because of the relatively slow decay associated with this component, we asked if it could be mediated by α3β4* nAChRs. Relative to the decay kinetics of α7 and α3β2 nAChRs expressed in oocytes, those of α3β4 AChRs are slow (Chavez-Noriega et al. 1997). The τ4 component was blocked completely by 10 μmα-Ctx AuIB (Fig. 4D). At 10 μmα-Ctx AuIB antagonized responses of rat α3β4 nAChRs expressed in oocytes (Luo et al. 1998), of rat major pelvic ganglion neurons (Park et al. 2006), and of chick ciliary ganglion neurons (Nai et al. 2003) by 80–95%. Additionally, a slow decay of a nAChR EPSP is consistent with the presence of a heteromer containing β4 subunits (Figl & Cohen, 2000).
MLA is primarily used as an α7 nAChR antagonist. However, it partially blocks (∼57 to 95%) α3β4 nAChRs in a number of species at concentrations approximately three orders of magnitude greater than that used for α7 nAChRs (Fucile et al. 1998; Lopez et al. 1998; Astles et al. 2002; Bryant et al. 2002). In our experiments with the goldfish ∼20 μm MLA has no effect on the τ4 component. However, the identification of the τ4 component as an α3β4* nAChR is consistent with the observation that 7.25 μm DHβE does not block the τ4 component, given that human α3β4 nAChR expressed in oocytes has a Kb, the equilibrium dissociation constant for the antagonist-receptor complex, of 13.77 μm (Chavez-Noriega et al. 1997). High MLA concentrations, severalfold greater than the IC50 values, reduce access time of exogenous antagonists to the cleft and have enabled the block of putative α7* and α3β2* components within 30 to 60 min. As such, the other β4-containing candidates, α2β4 and α4β4, are less likely matches, as their Kb values for DHβE are 3.61 and 0.01 μm, respectively (Chavez-Noriega et al. 1997). Taken together these results are consistent with α3β4* nAChRs giving rise to the τ4 component.
As already noted, EPSPs associated with only a τ4 component have been observed and designated as class 3 EPSPs (Table 1). While they have been observed only four times, pharmacological and kinetic dissection of composite EPSPs allows us to infer the involvement of the τ4 component at another 34% of the connections studied, i.e. class 2, 5 and 6 connections (Table 1).
Relative weights of τ2 and τ4
An inspection of Table 1 reveals that the τ2 and τ4 time constants are similar and are the slowest time constants associated with the M-axon–CRN connection. The only distinct mode of the decay time constant distribution (Fig. 2) is associated with EPSPs that decay biexponentially with time constants, τ1 and τ2, with relative weights of 87.9 ± 4.3 and 12.1 ± 4.3%, respectively, (n= 75). Our antagonist data indicate that the kinetic and relative weight profiles of these EPSPs, which have been assigned to class 1, are associated with α7* nAChRs. For the case in which the long decay time constant is not associated with the fastest decay time constant, τ1, it is designated as τ4 and not τ2. Antagonist data suggest that α3β4* nAChRs produce the τ4 kinetic profile of class 3 EPSPs. The designation of τ2 and τ4 is straightforward in each of these cases.
However, the situation is more complex if τ1, τ2 and τ4 are all present at a given connection. Two EPSP classes contain these decay time constants, class 5 (τ1, τ2 and τ4) and class 6 (τ1, τ2, τ3 and τ4) (Table 1). In some cases, kinetic analysis yields different values for τ2 and τ4, which are determined to not be redundant by use of the F test. In these analyses τ2, associated with the α7* nAChRs, and τ4, associated with α3β4* nAChRs, are readily assigned. The long decay time constant that most closely matches the τ1/τ2 relative weight ratio for class 1 EPSPs as indicated in Table 1 is given the designation τ2 and the other long decay time constant is designated as τ4.
However, if τ2 and τ4 have values such that a kinetic analysis only assigned one value to the slowly decaying phase (>8 ms), τ2/τ4, we apportioned the weight of this slowly decaying phase between τ2 and τ4. In order to estimate the relative contribution of τ2- and τ4-associated nAChRs to a composite EPSP, we used the observation that class 1 EPSPs, believed to be mediated by α7* nAChRs have a ratio of weights, τ2w/τ1w, of 0.124 (Table 1, first 48 class 1 EPSPs). The weight of the τ4 component, Wτ4, can be estimated from the combined weight of τ2 and τ4 (Wτ2/τ4) using the formula:
where Wτ1 is the weight of the τ1 component.
Combination of fast with intermediate and/or slow decay: EPSP classes 4–6
Kinetic analysis suggests that additional receptor combinations mediate a significant fraction of the EPSPs, namely class 4 (α7* and α3β2*) and class 5 (α7* and α3β4*), and class 6, which not only contains all kinetic components (α7*, α3β2* and α3β4*), but has also been investigated with antagonists and is described below.
Three populations of receptors have been identified so far. Class 1 EPSPs with two decay time constants, τ1 and τ2, have been attributed to only one receptor type, the α7* nAChR. Class 2 EPSPs have decay time constants of τ3 and τ4 and are most probably mediated by α3β2* and α3β4* nAChRs, respectively. Kinetic analysis indicates that class 6 responses (Fig. 5A and B) exhibit τ1/τ2, τ3 and τ4 components, suggesting that EPSPs of this class are mediated by a combination of three different nAChRs, α7*, α3β2* and α3β4*. In the first illustrated example (Fig. 5A), the control EPSP decay was fitted as the sum of four exponentials, τ1 = 1.4 ms, τ2 = 9.8 ms, τ3 = 5.9 ms and τ4 = 16.4 ms. The α7* EPSP component, blocked by 750 nm MLA and illustrated as a difference peak, has a decay phase best fitted by two exponentials, τ1 = 1.4 and τ2 = 9.8 ms, consistent with the presence of α7* nAChRs. The EPSP components that were insensitive to 750 nm MLA in this experiment were fitted by two exponentials, τ3 = 6.2 and τ4 = 16.5 ms. These values for τ3 and τ4 are consistent with the presence of α3β2* and α3β4* nAChRs, respectively. Since 200 nm MLA can block α6β2* nAChRs in rat (Mogg et al. 2002), the contribution of α6β2* nAChRs has yet to be ruled out in class 6 EPSPs.
Figure 5. Curve fit of class 6 EPSPs and effect of two concentrations of MLA.
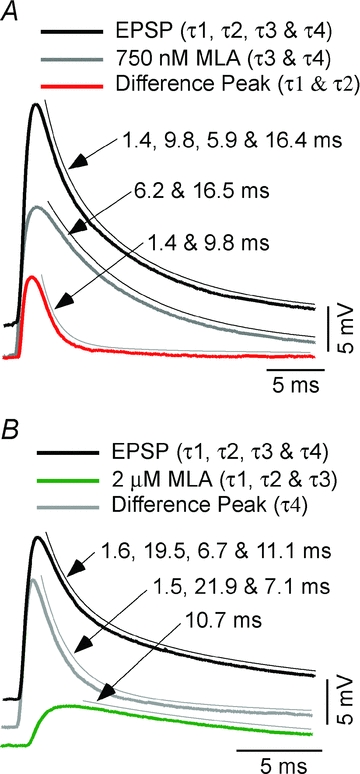
A, EPSP of class 6 CRN (thick black) is fitted as the sum of four decay exponentials τ1 = 1.4, τ2 = 9.8 ms, τ3 = 5.9 ms and τ4 = 16.4 ms with weights of 7.8, 1.0, 12.3 and 5.3 mV, respectively. The EPSP is partially blocked by 750 nm MLA, leaving a response (grey) fitted by two exponentials, τ3 = 6.2 and τ4 = 16.5 ms with weights of 11.7 and 5.0 mV, respectively. The difference trace (red), representing the component blocked by MLA, is fitted with a double exponential, τ1 = 1.4 and τ2 = 9.8 ms with relative weights of 7.9 and 1.0 mV. B, EPSP of class 6 CRN (thick black) is fitted as the sum of four decay exponentials τ1 = 1.6, τ2 = 19.5 ms, τ3 = 6.7 ms and τ4 = 11.1 ms with weights of 11.4, 2.6, 2.2 and 4.6 mV, respectively. The EPSP is partially blocked by 2 μm MLA, leaving an antagonist-insensitive component (green), fitted by a single exponential, τ4 = 10.7 ms with a weight of 4.5 mV. The difference trace (grey) is fitted by three exponentials, τ1 = 1.5, τ2 = 21.9 and τ3 = 7.1 ms with weights of 11.9, 2.3 and 2.1 mV, respectively, representing the components blocked by MLA. Stimulus and calibration artifacts are removed from the tail of each class 6 EPSP to facilitate curve fitting. All fits (thin black lines) are offset for clarity.
The multiexponential fit of the decay in the second illustrated class 6 EPSP example (Fig. 5B) has four decay times, with τ1–τ4, being 1.6, 19.5, 6.7 and 11.1 ms, respectively. In this case we found that 2 μm MLA blocked the τ1, τ2 and τ3 components and isolated the τ4 EPSP component with a decay time of 10.7 ms. The fit of the decay of the MLA-sensitive EPSP components yielded values of τ1, τ2 and τ3 equal to 1.5, 21.9 and 7.1 ms, respectively. Thus, the values of τ1–τ4 derived from the fit of the decay of the control EPSP correspond quite well to the estimates obtained separately from analysis of MLA-sensitive and -insensitive EPSPs components. Based on these kinetic profiles and the MLA sensitivities of goldfish nAChRs established in this study, we suggest that these results indicate α7* and α3β2* nAChRs were blocked by 2 μm MLA and that only an α3β4* nAChR-mediated response remained. Table 1 provides the decay time constants and weights of the components comprising the six classes we have identified. The time constants as blocked or isolated by antagonists are: τ1 = 1.6 ± 0.2, n= 9, τ2 = 13.4 ± 8.0, n= 9, τ3 = 6.0 ± 1.7 ms, n= 4, τ4 = 13.2 ± 3.8 ms, n= 6. Although transmission was mediated by α7* nAChRs alone at the majority of the connections studied, multiple nAChR types mediated transmission at a large fraction of connections.
In both class 6 cases the absolute weight of the residual components, noted in the figure legend (Fig. 5A–B), matches the weight of those components determined by analysis of the decay kinetics of the unblocked EPSP. This suggests that the nAChRs are located only postsynaptically. A presynaptic locus might alter release probability and the weight of the components between the control and antagonized conditions.
Decay kinetics of mEPSPs
The M-axon–CRN parallel branch connection comprises a few contacts with multiple active zones (Hackett et al. 1989) and as indicated gives rise to the majority of these multiple exponential EPSPs. This raises several questions about the structure of the connection. Is the transmitter released at a contact detected by only one or by several receptor types? If several receptor types mediate the signal at one contact, are distinct receptor types or a mixture of receptor types associated with each postsynaptic density (PSD) in apposition to an active zone?
We postulated that aspects of the arrangement of the nAChRs at the M-axon–CRN connection could be inferred from the kinetics of the mEPSPs if the sampled mEPSPs were due to asynchronous release from only one M-axon. While there are multiple soma–dendritic inputs to the hatchetfish giant-fibre cell body, a CRN analogue (Barry & Bennett, 1990), the only known axonal inputs are from the two M-axons. Thus, we took advantage of this fact by injecting a steady subthreshold depolarizing current directly into one M-axon, so that the frequency of mEPSPs attributed to that M-axon could be enhanced.
Most often no mEPSP activity was observed in the absence of current injection. When observed, the basal frequency of mEPSPs averaged 1.8 ± 1.3 events s−1 (n= 6) (Fig. 6A). The mEPSP frequency increased dramatically as the depolarizing current in the M-axon was increased from 0 to 20 nA. Figure 6A is an example from a connection characterized as having a class 1 EPSP. A comparison of the number of mEPSP events recorded in the presence or absence of depolarizing current is illustrated in the amplitude histogram of Fig. 6B, and it shows that the depolarized M-axon is the main source of the mEPSPs. The events ratio (depolarized to control) in Fig. 6B is 12.2 and overall was 10.2 ± 5.4 (n= 6). The decay time constants of an averaged mEPSP for this connection (Fig. 6C), τ1 = 1.0 ms and τ2 = 9.2 ms, coincide with those of the class 1 EPSPs (Table 1).
Figure 6. M-axon is the main source of mEPSPs in CRN axons.
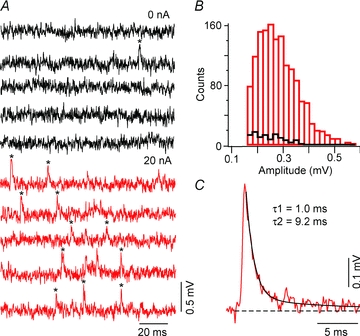
A, black and red traces are continuous recordings from a class 1 CRN with 0 nA (black) and 20 nA (red) depolarizing current injected steadily into the presynaptic M-axon. mEPSPs that meet criteria for use in averages are marked with an asterisk. B, amplitude histogram for mEPSPs recorded at 0 nA (black) and 20 nA (red) M-axon depolarization. C, averaged mEPSP of class 1 CRN is fitted by two exponentials, τ1 = 1.0 ms and τ2 = 9.2 ms.
Different nAChRs distributed in separated clusters
We next asked if the waveforms of mEPSPs could allow us to determine whether receptor populations were isolated or mixed at connections with composite EPSPs. For example, the presence of two populations of mEPSPs that decayed with different time constants, τ3 and τ4, at a class 2 connection, putatively associated with α3β2* and α3β4* nAChRs, would imply separate receptor clusters. However, detection of only one mEPSP population with a biexponential decay, τ3 and τ4, would imply that α3β2* and α3β4* nAChRs were intermingled within a receptor cluster (Sargent, 2009). We approached this problem by constructing mEPSP decay time distributions, averaging mEPSPs taken from each mode of the distribution and analyzing decay kinetics of these averaged mEPSPs.
By depolarizing one M-axon, populations of mEPSPs were collected from connections at which the EPSPs were of class 1, 2 or 6. When the evoked responses were mediated by only one nAChR, as with connections mediated by class 1 EPSPs, the decay time distributions of the associated mEPSPs were uni-modal. This is depicted in Fig. 7A, where the mean decay time constant was 1.2 ms. In this particular case, a sufficient number of these mEPSPs were averaged to adequately improve the signal-to-noise ratio and the second slower exponential associated with α7* nAChR EPSP decay, τ2, emerged (Fig. 7A, right). That is, this averaged class 1 mEPSP could be fitted by two exponentials, τ1 = 1.0 and τ2 = 10.1 ms. The weight of the second exponential was 13.8% of the total amplitude, consistent with the weighting for the evoked signal being 12.1 ± 4.3% (n= 75). Overall, including those cases in which only τ1 was resolved, the mEPSP decay associated with class 1 EPSPs was τ1 = 1.1 ± 0.4, n= 6.
Figure 7. CRN mEPSPs are not composites.
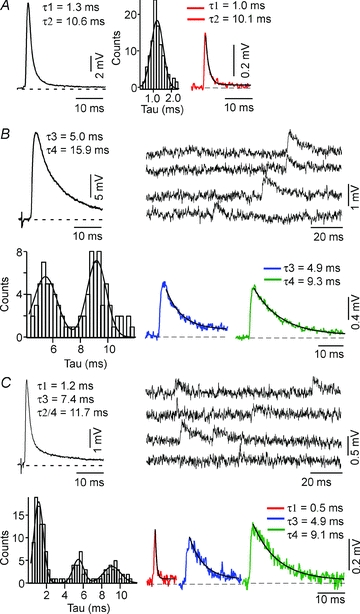
A, B and C, comparison of decay kinetics of evoked and mEPSPs, for class 1, class 2 and class 6 connections, respectively. In each example, averaged evoked EPSPs (left panel, A and upper left panels, B and C) have multiple exponential decays as indicated by τ values. The corresponding mEPSP decay time histograms (centre, A and lower left panels, B and C) are fitted as the sums of one, two or three Gaussians, respectively. In right panel, A, and lower right panels, B and C, are averaged mEPSPs for traces selected from each mode. In the upper right panels, B and C, are continuous recordings with mEPSPs of varying decay kinetics. A, class 1 EPSP decay is fitted by two exponentials, with τ1 = 1.3 and τ2 = 10.6 ms. The histogram of mEPSP decay times is fitted as one Gaussian. The decay times of the corresponding averaged mEPSP are τ1 = 1.0 and τ2 = 10.1 ms. B, class 2 EPSP decay is fitted with two exponentials τ3 = 5.0 and τ4 = 15.9 ms. The histogram of mEPSP decay times is fitted as the sum of two Gaussians. The decay times of the corresponding averaged mEPSPs are τ3 = 4.9 and τ4 = 9.3 ms. C, class 6 EPSP decay is fitted by three exponentials τ1 = 1.2, τ3 = 7.4 and τ2/τ4 = 11.7 ms. The histogram of mEPSP decay times is fitted as the sum of three Gaussians. The decay times of the corresponding averaged mEPSPs are τ1 = 0.5, τ3 = 4.9 and τ4 = 9.1 ms.
At class 2 and class 6 connections, at which evoked responses were composites, mEPSP decay time distributions were bi-modal (Fig. 7B) or tri-modal (Fig. 7C), respectively. These distributions were also obtained by treating single events as having monoexponential decays. Therefore, additional decay time constants may have been missed at this point in the analysis. To test for composite mEPSPs and improve the signal-to-noise ratio, responses in each mode of a distribution were averaged. As shown in Fig. 7B and C, these averaged mEPSPs all decayed monoexponentially, indicating that in class 2 and class 6 EPSPs the different nAChR subtypes mediating these responses were in separate clusters. Additionally, averaged mEPSP decay time constants of the class 2 connection (τ3 = 5.2 ± 0.4, τ4 = 10.3 ± 1.5, n= 3) were consistent with the taus attributed to the α3β2* and α3β4* nAChR-mediated EPSPs and those obtained for a class 6 connection (τ1 = 0.6 ± 0.3, τ3 = 5.8 ± 0.9, τ4 = 10.6 ± 2.0, n= 3, P < 0.05 for τ1 vs.τ3 or τ4 and for τ3 vs.τ4) were consistent with those attributed to the α7*, α3β2* and α3β4* nAChR-mediated EPSPs. Note that at the class 6 connection of Fig. 7C the signal-to-noise ratio of the averaged α7* mEPSPs was such that the slow decay time constant could not be extracted from the noise. Therefore only the fast time constant is indicated. The lack of mEPSPs fitted by multiple exponentials associated with different nAChR types is in contrast to those seen in chick ciliary ganglion neurons (Chen et al. 2001; Sargent, 2009).
To confirm the validity of averaging minis by their rising edge, the class 6 mini data were also aligned by their peaks for averaging and the decays of the averaged mEPSPs were subsequently fitted with one and two exponentials. In all cases, these averaged minis were again best fitted by one exponential as per F test criteria. Additionally, at one class 6 connection where the M-axon-CRN pair was held long enough to observe the influence of 100 nm MLA, τ1 (n= 13) and τ3 (n= 10) asynchronous minis before the block reduced to only τ3 (n= 7) minis after the block (data not shown).
Up to three contact sites were detected between the parallel branch of the CRN and the M-axon (Fig. 8A), and these were separated by more than 50 μm. In contrast, only one contact was ever observed on the crossing branch of the CRN. The finding that mEPSPs are not composite, but rather are generated by activation of homogeneous receptor populations, implies that the different receptor types are functionally isolated from each other. This functional organization could be achieved in two ways: either each of several contacts between a M-axon and a CRN involves a single receptor type, or the different receptors are segregated into separate clusters within a contact having multiple active zones (Hackett et al. 1989). The latter is the likely structure for the contralateral M-axon–CRN connection where only one contact has been observed and would require sufficient separation between neighbouring active zones, and their apposed postsynaptic sites, to preclude activation by spillover with low-frequency activation (Hartzell et al. 1975; Faber et al. 1985; Faber & Korn, 1988).
Figure 8. M-axon and CRN anatomy and contact sites.
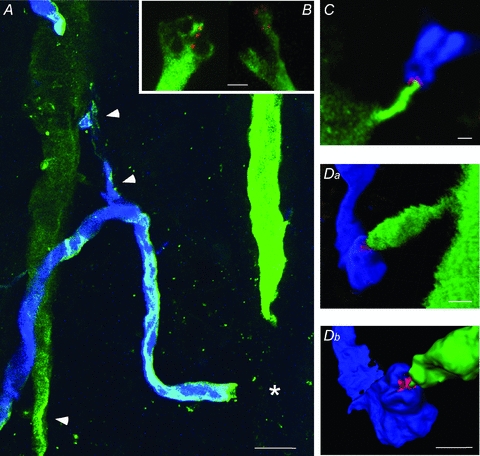
A, B, C and D are confocal images from four separate preparations depicting the M-axon (green), CRN (blue) and α7* nAChRs (red). A, three putative contact locations, marked by arrowheads, between the ipsilateral M-axon and CRN. The approximate location of the contact site between the CRN and contralateral M-axon (not captured in this slice), is marked by an asterisk (*). Calibration bar equals 50 μm. B, two examples of M-axon collaterals exhibiting structures that could support the segregation of different nAChR types. C, high magnification of the contact associated with the crossing branch of a CRN with class 1 EPSPs (location near asterisk in A, but different preparation). Overlap of CRN (blue) and α7* nAChRs (red) depicted by rose colour. Da, Z-stack image of a contact associated with the parallel branch of the CRN (location near rostral contact and arrowhead in A, but different preparation). Db, a thresholded rendering of same contact seen in Da magnified and tilted to highlight α7* nAChRs and CRN structure. B–D, calibration bars equal 5 μm.
Overall, we observed M-axon collateral structures that would allow for the separation of receptor clusters (e.g. Fig. 8B). In these experiments, receptors located at these sites were identified with α-Btx. An example from a different experiment of a contact site between an M-axon and a crossing CRN axon that exhibited class 1 EPSPs is shown in Fig. 8C. In this case, the EPSP was mediated by α7* nAChRs only. In another experiment, an α7* nAChR contact of an ipsilateral M-axon–CRN connection exhibiting class 1 EPSPs was found ∼200 μm rostral to the intersection of the contralateral M-axon and the crossing branch of the CRN (Fig. 8Da). An enlarged three-dimensional rendering of this contact site (Fig. 8Db) shows the α7* nAChR embedded in the postsynaptic CRN axon. The nAChRs appear to be embedded in the CRN terminal and not associated with the M-axon, consistent with what we observed as we stepped through the confocal stacks of such connections. Additionally, transmission electron microscopy studies showed a postsynaptic locus for α-Btx staining of receptors at the homologous connection in the hatchetfish (Day et al. 1983).
Discussion
The major findings of this study are that (1) multiple subtypes of nAChRs can mediate the EPSPs at the connection between an M-axon and a CRN, with correspondingly distinct kinetics and (2) when different nAChRs are present they appear to be segregated into separate clusters. These conclusions are based upon comparison of the kinetic properties of spike-triggered EPSPs with those of mEPSPs attributed to release of single quanta, as well as the pharmacological profiles of the EPSPs. Through the use of antagonists we identified three pharmacologically distinguishable nAChR populations associated with the EPSPs. Population 1 nAChRs, putatively α7*, were sensitive to MLA, α-Btx and α-Ctx ArIB[V11L;V16D], population 2 nAChRs, putatively α3β2*, were sensitive to MLA, DHβE, α-Ctx GIC and population 3 nAChRs, putatively α3β4*, were sensitive to α-Ctx AuIB.
Filtering
The morphological relationships described here support the argument that EPSPs and mEPSPs recorded in a CRN accurately reflect the decay kinetics of the nAChRs and are not subject to significant filtering by axonal cable properties, even in the case of the fast α7* nAChR responses (Jack et al. 1975). The CRN axon, which is myelinated, had a membrane time constant of ∼400 μs and a length constant, λ, of ∼2 mm (Funch et al. 1984), based on paired recordings from the same axon in two experiments. This decay time constant was less than that of any of the EPSP components. Thus, unless the CRN electrode was electrotonically quite far from the synaptic loci, which would be inconsistent with the morphology, the measured decay kinetics accurately reflect the time course of deactivation for putative α3β2* and α3β4* nAChR channels. The τ1 component kinetics (α7* nAChRs) of ∼1.7 ms may be degraded, but only moderately.
Based on the contact locations observed with confocal microscopy, the maximum distance between the CRN recording site and the contact would be 400–500 μm, or ∼0.25λ. In confirmation, we measured the decrement and filtering of two types of responses recorded simultaneously with two electrodes that were in the same CRN axon and separated by 350 to 500 μm: (i) an EPSP evoked by caudal spinal cord stimulation, was reduced in amplitude by about 20% rostrally, but its kinetics were unchanged, and (ii) the full width half maximum of a passively conducted action potential evoked by current injected caudally increased only slightly, from 1.1 to 1.2 ms. The action potential propagated passively because QX-314 diffusing from the rostral electrode blocked voltage-dependent sodium current locally. Finally, the frequency of minis with intermediate, ∼5 ms, and long, ∼10 ms, decay time constants did not change after treatment with 200 nm MLA to block α7* nAChR receptors, while those with the fast decay time constant disappeared. This observation indicates that the minis associated with tau values > 2.5 ms were not filtered α7* mEPSPs generated at sites distant from the recording electrode.
Specificity of pharmacological dissection
The pharmacological profile for α7, α3β2 and α3β4 nAChRs in the goldfish may be different to that for other species. Much of the pharmacology for these receptors has been studied in oocyte expression systems and in rat, mouse and chick. BLAST and VAST analyses were used to find the common structural and sequence features between the teleost (goldfish and/or zebrafish) and the species in which the specificity of antagonists had been demonstrated. Comparison of sequence and structural homology across species with each of the α7, α3, β2 and β4 nAChR subunit goldfish or zebrafish gene sequences shows a high degree of identity. This holds true particularly in the case of the sequence fragments and residues of α7, α3 and β2 nAChR subunits known to be critical for binding the pertinent ligands used in this study.
Due to potential species differences, we cannot definitively assign specific receptor subtypes to a population type. Nevertheless, the nAChR populations in the goldfish M-axon–CRN connection are consistent with α7* (population 1), α3β2* (population 2) and α3β4* (population 3) by analogy to mammalian and chick nAChRs. These three populations contribute to six classes of EPSPs that have different decay time constants as follows: class 1 (τ1, τ2, population 1 nAChRs), class 2 (τ3, τ4, populations 2 and 3 nAChRs), class 3 (τ4, population 3 nAChRs), class 4 (τ1, τ2, τ3, populations 1 and 2 nAChRs), class 5 (τ1, τ2, τ4, populations 1 and 3 nAChRs) and class 6 (τ1, τ2, τ3, τ4, populations 1, 2 and 3 nAChRs). The pharmacological sensitivities of the different EPSP components and their kinetics are consistent with known properties of the different nAChRs in other species.
Class 1 EPSPs are completely blocked by 35–50 nm MLA. For comparison, α7 nAChRs in rat dorsal root ganglion neurons (Genzen et al. 2001), rat dorsal motor vagal neurons (Sahibzada et al. 2002), rat striatal slices (Kaiser & Wonnacott, 2000) and rat superior cervical ganglion neurons (Cuevas et al. 2000) were blocked by 10 nm, 10 nm, 50 nm and 200 nm MLA, respectively. Class 1 EPSPs were also blocked by 50–100 nmα-Btx, the concentrations typically used to block putative α7* nAChRs (Day et al. 1983; Zhang et al. 1996; Ullian et al. 1997; Chang & Berg, 1999; Sahibzada et al. 2002). At the M-axon–CRN connection, the α-Btx and MLA blocks were not reversible after up to 45 min of washing. A block of putative α7* nAChRs with 50 nmα-Btx was similarly found to be irreversible in chick ciliary ganglion (Ullian et al. 1997).
Both α-Btx and MLA block α9* nAChRs (Elgoyhen et al. 2001; Baker et al. 2004) and α-Btx also blocks α1* nAChRs. MLA also blocks α6β2* nAChRs (Ki 33 nm), (Mogg et al. 2002). Therefore, to further establish that goldfish class 1 EPSPs are probably mediated by α7* nAChRs we utilized 150 nmα-Ctx ArIB to block a class 1 EPSP (Fig. 3D). α-Ctx ArIB is a new α7 nAChR antagonist that does not block α1*, α2*, α3*, α4*, α6* or α9* nAChRs (Whiteaker et al. 2007).
The pharmacological profiles of α7* nAChRs in the goldfish may be different to those in mammalian and avian homologues and require longer access times, particularly in an in vivo preparation. However, comparison across species of sequence and structural homology of the α7 nAChR ligand binding domain (Brejc et al. 2001) with that of the zebrafish, indicates a high level of identity with human, rat, mouse and chick (Table 2). The zebrafish and goldfish are both members of the teleost infraclass and are expected to have similar antagonist sensitivities. Indeed, they show 100% sequence identity in the nAChR binding domain.
Table 2.
Percentage sequence identity
| Binding domain α7 Zebrafish | 14 Antagonist interaction sites α7 Zebrafish | 4 MLA specific sites α7 Zebrafish | Binding domain α3 Goldfish | Binding domain β2 Goldfish | Binding domain β4 Goldfish | |
|---|---|---|---|---|---|---|
| Human | 83.0 | 78.6 | 50 | 79.8 | 77.8 | 75.6 |
| Rat | 82.5 | 78.6 | 50 | 77.9 | 77.3 | 74.6 |
| Mouse | 82.5 | 78.6 | 50 | 77.9 | 77.3 | 74.6 |
| Chick | 82.5 | 100 | 100 | 78.4 | 78.4 | 79.9 |
| Zebrafish | 100 | 100 | 100 | 99.0 | 96.4 | 100 |
| Pufferfish | ___ | ___ | ___ | ___ | ___ | 91.4 |
Cysteines: α7 at 190, 191 (Hansen et al. 2005); α3 at 192, 193 (Cauley et al. 1990; Hieber et al. 1990a); β2 at 130, 144 (Heiber et al. 1990b); β4 at 130, 144 (XP 696993, NCBI database).
MLA binding to the acetylcholine binding protein, a surrogate for the nAChR binding domain, includes interactions with 14 residues of the α7 nAChR subunit interfaces, four of which are specific to MLA (Hansen et al. 2005). At these 14 interaction sites, the zebrafish subunits share 100% and 78.6% identity with residues of chick and mammalian subunits, respectively (Supplemental Fig. S1). Overall, in the ligand binding domain, the zebrafish shares 82.5–83% gene sequence identity with chick, rat, mouse and human (Table 2). These homologies suggest that zebrafish and goldfish α7 nAChRs have similar MLA sensitivities to those of human, rat and mouse, but particularly chick.
Only one decay time constant has been associated with putative α7 nAChR responses to synaptic current in the chick ciliary ganglion preparation, 1.84 ± 0.14 ms (Zhang et al. 1996) and 1.04 ± 0.35 ms (Ullian et al. 1997), and our observed τ1 value of class 1 EPSPs is in good agreement with these reports. However, two decay time constants have been reported for α7 nAChR-mediated synaptic current in hippocampal interneurons in rat brain slices (Frazier et al. 1998). In our study the nAChRs that underlie the two time constants, τ1 and τ2, of class 1 EPSPs are both sensitive to α7 nAChR antagonists. In that kinetic elements of class 1 EPSPs do not exhibit a differential sensitivity to antagonists (Fig. 3Ca and b), it is unlikely that two different receptor types give rise to τ1 and τ2 (Frazier et al. 1998).
Three possible explanations for the source of the second kinetic element, τ2, are (1) the lack of desensitization of homomeric α7 nAChRs, (2) the presence of heteromeric α7* nAChRs and (3) a small contribution from α3β4* nAChRs not detected by kinetic analysis. The biexponential decay of class 1 EPSPs could reflect multiple open states or burst properties (Nai et al. 2003) and the absence of desensitization at agonist concentrations and profiles generated in the synaptic context, enabling the presence of a slowly decaying current similar to the τ2 element, (Frazier et al. 1998; Papke & Thinschmidt, 1998; Papke et al. 2000; Mike et al. 2000; Shoop et al. 2001; Placzek et al. 2005). In a few cases α7 nAChRs have been reported to occur as a heteromers in association with β2 subunits (Yu & Role, 1998; Shao & Yakel, 2000). It is possible that in our preparation a similar situation might account for the two observed time constants. Lastly, a small contribution from α3β4* nAChRs may be misidentified as a τ2 element in class 1 EPSPs analysed only by decay kinetics, but this cannot account for the susceptibility of the τ2 element to antagonists specific for α7 nAChRs.
The τ3 component is blocked by an α3β2 nAChR-specific antagonist, α-Ctx GIC (McIntosh et al. 2002), and has decay kinetics similar to synaptically evoked monoexponential EPSCs (∼5–7 ms) (Ullian et al. 1997; Chen et al. 2001) in the chick ciliary ganglion. The ciliary α3* EPSCs are mediated in part by α3 and β2 subunits (Conroy & Berg, 1995), are blocked by α-Ctx MII (Ullian et al. 1997; Chen et al. 2001), an antagonist that targets α3/β2 nAChRs interfaces (Cartier et al. 1996), and are associated with a 40 pS nAChR channel having a mean open time of ∼2.8 ms under 0.5 μm nicotine activation (Nai et al. 2003).
Comparison of sequence homology across species within the nAChR ligand binding domain (Brejc et al. 2001) for the goldfish α3 (Cauley et al. 1990; Hieber et al. 1990a) and goldfish β2 (Hieber et al. 1990b) nAChR subunit gene sequences reveals 77–80% identity, as shown in Table 2. Note that there is a very high identity, 96–99%, between goldfish and zebrafish. The sequence fragments and residues crucial for binding α-Ctx GIC, one of the antagonists used in the present pharmacological investigation of the τ3 component, are inferred by comparison with α-Ctx MII and outlined in vertical registers in Supplemental Fig. S2A and B. The goldfish α3 and β2 nAChR sequences contain the same residues shown to be critical for binding α-Ctx MII in the α3β2 human nAChR, namely K185 and I188 for the α3 subunit and T59 for the β2 subunit (Harvey et al. 1997; Chi et al. 2005). α-Ctx MII and α-Ctx GIC are expected to have similar interactions with α3β2 nAChRs, based on structural analysis (Chi et al. 2004). Additionally, the high level of sequence and structural homology across species suggest that MLA and α-Ctx GIC are likely to interact with the goldfish α3β2 nAChRs as they do in other species (Table 2).
The τ4 EPSP component is blocked by an α3β4-specific antagonist, α-Ctx AuIB (Luo et al. 1998). Interestingly a ciliary α3* channel lacking the β2, but containing the β4, subunit had a slightly longer mean open time, ∼3.2 ms, than its α3β2* counterpart (Nai et al. 2003). Also, α3β4 nAChRs expressed in oocytes (Boorman et al. 2003) have complex channel kinetics with a mean open time of 9.4 ms, consistent with the decay kinetics of the τ4 EPSP and mEPSP (Table 1).
The sequence identity of the teleost β4 subunit when compared with that of human, rodents and chick (Supplemental Fig. S2C) is ∼74–80% (Table 2). α-Ctx AuIB (10 μm) blocks α3β4 nAChRs expressed in oocytes heterologously and in chick and rat endogenously (Luo et al. 1998; Nai et al. 2003; Park et al. 2006). The sites that convey this specificity are not yet known. Nevertheless, the zebrafish 54–63 segment in the β4 nAChR subunit that is part of the complementary face of the antagonist binding pocket (Brejc et al. 2001; Hansen et al. 2005) is similar across species, suggesting that this toxin may also have specificity in the goldfish.
Physiological relevance of multiple nAChR types
The M-axon action potential triggers an escape response, and the classic role for the CRNs or their analogues is to activate supraspinal motorneuron pools that control the opercular, ocular, jaw and pectoral fin muscles (Auerbach & Bennett, 1969; Diamond, 1971; Model et al. 1972; Hackett & Faber, 1983a; Hackett & Buchheim, 1984). During an escape, CRN activation leads to jaw closure, making the fish shape more streamlined (Barry and Bennett, 1990). Why are three different nAChR populations present at a connection that has a high safety factor and that is associated with a stereotyped behaviour, especially since the escape response is optimized by factors limiting M-cell activation to a single spike? Indeed it has been assumed that CRNs also do not fire repetitively. Nevertheless, the multiplicity of receptor types may enable a wide variety of important subcellular (Sabatini et al. 2001; Berg et al. 2006) and circuit-level adaptive properties (Korn & Faber, 2005). These include (1) increasing the safety factor of the connection; (2) temporal tuning of the CRN depolarization window to enhance spatial and temporal integration, for example, of delayed inputs to the CRN dendritic arbor from other reticulospinal neurons; (3) differential timing of the excitation of the different CRN classes; and (4) differential modulation of channel biophysical properties and activation of downstream pathways unique to each receptor type.
Augmenting the EPSP by activating multiple receptor components enables a fast rise time via α7* nAChRs and an increased probability that threshold will be attained with the added contributions from the α3β2* and α3β4* receptors that prolong the EPSPs (Zhang et al. 1996). Both of these attributes would be consistent with the need to quickly and reliably relay the signal from the M-axon, initiating a startle response.
Behavioural observations suggest that in certain cases the escape may consist of two successive fast body bends. The input underlying the second bend could come from either the M-axon or its homologues known to participate in some escapes (Korn & Faber, 2005); in either case, the mixed signal CRNs (classes 2, 4, 5 or 6) would be more likely to integrate these inputs, especially asynchronous excitation from the homologues.
Second messengers (Tan et al. 1998; Du & Role, 2001; Fischer et al. 2005) interact with the intercellular region of particular nAChR subunits and can differentially alter the biophysical characteristics of the receptor. The presence of several receptor subtypes enables the cell to regulate multiple downstream processes simultaneously. Specific members of the PSD95 family of PDZ-containing proteins are associated with particular nAChR subtypes (Conroy et al. 2003; Baer et al. 2007) and mediate such processes (Parker et al. 2004; Berg et al. 2006).
Glutamatergic synapses also are characterized by the co-activation of multiple receptor types that mediate fast synaptic transmission with different kinetics, notably AMPA and NMDA receptors. Activating NMDA receptors, which have slower kinetics and a higher Ca2+ permeability, has been implicated in activity-dependent plasticities of synaptic transmission. While a similar arrangement might pertain for nAChRs, it is notable that the Ca2+-permeable channel, α7*, has the fastest kinetics.
We have demonstrated the presence of six EPSP classes coupled with at least two CRNs types (Titmus & Faber, 1987; Hackett & Buchheim, 1984), indicating a significantly greater diversity in CRNs than previously appreciated in the goldfish (Kimmel et al. 1985; Barry & Bennett, 1990). This has raised questions as to the physiological role of this diversity. Instead of the limited stereotyped role of CRNs in the escape response, CRNs may be associated with many more behaviours. Indeed, the possibility of varied inputs to the giant fibre cell body, a CRN homologue in the hatchet fish, suggests the involvement of the giant fibre in non-Mauthner initiated C-starts (Barry & Bennett, 1990). This system lends itself to investigating pre- and postsynaptic transmission mechanisms, and given that it can be studied in vivo, provides a compelling model for exploring cellular correlates of a wide variety of behaviours.
Acknowledgments
This work was supported by NIH grants MH53631 and GM48677 to J.M.M., NS21848 to D.S.F. and C.L.G. was supported by T32DK075-13-17. We thank Drs Heike Neumeister, Alberto Pereda, Thomas Preuss and Peter Sargent for critical reading of earlier versions of this manuscript. We thank Dr Kostantin Dobrenis and Pamela Cabahug of the Morphology Core and Kevin Fisher of the Imaging Core of the Kennedy Center and Kathy Grove for their technical expertise.
Glossary
Abbreviations
- α-Btx
α-bungarotoxin
- α-Ctx
α-conotoxin
- BLAST
basic logical alignment search tool
- CRN
cranial relay neuron
- DHβE
dihydro-beta-erythroidine
- M-
Mauthner
- MLA
methyllycaconitine
- nAChR
nicotinic ACh receptors
- m
mini
- PSD
post synaptic density
- PSD95
postsynaptic density-95
- QX-314
N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide
- VAST
vector alignment search tool
Author contributions
C.L.G., T.M.S., J.M.M. and D.S.F. designed the research; C.L.G., T.M.S., S.C.D. and R.F.W. performed the experiments; J.M.M. contributed analytical tools; C.L.G., T.M.S., J.M.M., S.C.D., R.F.W. and D.S.F analysed and interpreted data, C.L.G. wrote the paper with critical input from T.M.S., J.M.M. and D.S.F. All authors were involved in the design or analysis of experiments and in the critical reading and revision of the manuscript. All have approved the final version of the paper. The experiments were done at the Department of Neuroscience, Albert Einstein College of Medicine.
Supplementary material
Supplemental Figure 1.
Supplemental Figure 2.
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Alkondon M, Albuquerque EX. Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. I. Pharmacological and functional evidence for distinct structural subtypes. J Pharmacol Exp Ther. 1993;265:1455–1473. [PubMed] [Google Scholar]
- Alkondon M, Pereira EFR, Albuquerque EX. α-Bungartotoxin- and methyllycaconitine-sensitive nicotinic receptors mediate fast synaptic transmission in interneurons of rat hippocampal slices. Brain Res. 1998;810:257–263. doi: 10.1016/s0006-8993(98)00880-4. [DOI] [PubMed] [Google Scholar]
- Ankri N, Legendre P, Faber DS, Korn H. Automatic detection of spontaneous synaptic responses in central neurons. J Neurosci Methods. 1994;52:87–100. doi: 10.1016/0165-0270(94)90060-4. [DOI] [PubMed] [Google Scholar]
- Astles PC, Baker SR, Boot JR, Broad LM, Dell CP, Keenan M. Recent progress in the development of subtype selective nicotinic acetylcholine receptor ligands. Curr Drug Targets CNS Neurol Disord. 2002;1:337–348. doi: 10.2174/1568007023339256. [DOI] [PubMed] [Google Scholar]
- Auerbach AA, Bennett MV. Chemically mediated transmission at a giant fiber synapse in the central nervous system of a vertebrate. J Gen Physiol. 1969;53:183–210. doi: 10.1085/jgp.53.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer K, Burli T, Huh KH, Wiesner A, Erb-Vogtli S, Gockeritz-Dujmovic D, et al. PICK1 interacts with α7 neuronal nicotinic acetylcholine receptors and controls their clustering. Mol Cell Neurosci. 2007;35:339–355. doi: 10.1016/j.mcn.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker ER, Zwart R, Sher E, Millar NS. Pharmacological properties of α9 α10 nicotinic acetylcholine receptors revealed by heterologous expression of subunit chimeras. Mol Pharmacol. 2004;65:453–460. doi: 10.1124/mol.65.2.453. [DOI] [PubMed] [Google Scholar]
- Barry MA, Bennett MV. Projections of giant fibers, a class of reticular interneurons, in the brain of the silver hatchetfish. Brain Behav Evol. 1990;36:391–400. doi: 10.1159/000115321. [DOI] [PubMed] [Google Scholar]
- Berg DK, Conroy WG. Nicotinic α7 receptors: synaptic options and downstream signaling in neurons. J Neurobiol. 2002;53:512–523. doi: 10.1002/neu.10116. [DOI] [PubMed] [Google Scholar]
- Berg DK, Conroy WG, Liu Z, Zago WM. Nicotinic signal transduction machinery. J Mol Neurosci. 2006;30:149–152. doi: 10.1385/JMN:30:1:149. [DOI] [PubMed] [Google Scholar]
- Boorman JP, Beato M, Groot-Kormelink PJ, Broadbent SD, Sivilotti LG. The effects of β3 subunit incorporation on the pharmacology and single channel properties of oocyte-expressed human α3β4 neuronal nicotinic receptors. J Biol Chem. 2003;278:44033–44040. doi: 10.1074/jbc.M211719200. [DOI] [PubMed] [Google Scholar]
- Bradaia A, Trouslard J. Fast synaptic transmission mediated by α-bungarotoxin-sensitive nicotinic acetylcholine receptors in lamina X neurones of neonatal rat spinal cord. J Physiol. 2002;544:727–739. doi: 10.1113/jphysiol.2002.028894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, Van Der Oost J, Smit AB, Sixma TK. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- Bryant DL, Free RB, Thomasy SM, Lapinsky DJ, Ismail KA, McKay SB, et al. Structure-activity studies with ring E analogues of methyllycaconitine on bovine adrenal α3β4* nicotinic receptors. Neurosci Res. 2002;42:57–63. doi: 10.1016/s0168-0102(01)00304-2. [DOI] [PubMed] [Google Scholar]
- Cartier GE, Yoshikami D, Gray WR, Luo S, Olivera BM, McIntosh JM. A new α-conotoxin which targets α3β2 nicotinic acetylcholine receptors. J Biol Chem. 1996;271:7522–7528. doi: 10.1074/jbc.271.13.7522. [DOI] [PubMed] [Google Scholar]
- Cauley K, Agranoff BW, Goldman D. Multiple nicotinic acetylcholine receptor genes are expressed in goldfish retina and tectum. J Neurosci. 1990;10:670–683. doi: 10.1523/JNEUROSCI.10-02-00670.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KT, Berg DK. Nicotinic acetylcholine receptors containing α7 subunits are required for reliable synaptic transmission in situ. J Neurosci. 1999;19:3701–3710. doi: 10.1523/JNEUROSCI.19-10-03701.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez-Noriega LE, Crona JH, Washburn MS, Urrutia A, Elliott KJ, Johnson EC. Pharmacological characterization of recombinant human neuronal nicotinic acetylcholine receptors h α2β2, h α2β4, h α3β2, h α3β4, h α4β2, h α4β4 and h α7 expressed in Xenopus oocytes. J Pharmacol Exp Ther. 1997;280:346–356. [PubMed] [Google Scholar]
- Chen M, Pugh PC, Margiotta JF. Nicotinic synapses formed between chick ciliary ganglion neurons in culture resemble those present on the neurons in vivo. J Neurobiol. 2001;47:265–279. doi: 10.1002/neu.1034. [DOI] [PubMed] [Google Scholar]
- Chi SW, Kim DH, Olivera BM, McIntosh JM, Han KH. Solution conformation of α-conotoxin GIC, a novel potent antagonist of α3β2 nicotinic acetylcholine receptors. Biochem J. 2004;380:347–352. doi: 10.1042/BJ20031792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Lee SH, Kim DH, Kim JS, Olivera BM, McIntosh JM, Han KH. Solution structure of α-conotoxin PIA, a novel antagonist of α6 subunit containing nicotinic acetylcholine receptors. Biochem Biophys Res Commun. 2005;338:1990–1997. doi: 10.1016/j.bbrc.2005.10.176. [DOI] [PubMed] [Google Scholar]
- Coggan JS, Bartol TM, Esquenazi E, Stiles JR, Lamont S, Martone ME, et al. Evidence for ectopic neurotransmission at a neuronal synapse. Science. 2005;309:446–451. doi: 10.1126/science.1108239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conroy WG, Berg DK. Neurons can maintain multiple classes of nicotinic acetylcholine receptors distinguished by different subunit compositions. J Biol Chem. 1995;270:4424–4431. doi: 10.1074/jbc.270.9.4424. [DOI] [PubMed] [Google Scholar]
- Conroy WG, Liu Z, Nai Q, Coggan JS, Berg DK. PDZ-containing proteins provide a functional postsynaptic scaffold for nicotinic receptors in neurons. Neuron. 2003;38:759–771. doi: 10.1016/s0896-6273(03)00324-6. [DOI] [PubMed] [Google Scholar]
- Couturier S, Erkman L, Valera S, Rungger D, Bertrand S, Boulter J, Ballivet M, Bertrand D. α5, α3, and non-α3. Three clustered avian genes encoding neuronal nicotinic acetylcholine receptor-related subunits. J Biol Chem. 1990b;265:17560–17567. [PubMed] [Google Scholar]
- Cuevas J, Roth AL, Berg DK. Two distinct classes of functional 7-containing nicotinic receptor on rat superior cervical ganglion neurons. J Physiol. 2000;525:735–746. doi: 10.1111/j.1469-7793.2000.t01-1-00735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol. 2007;47:699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- Day JW, Hall DH, Hall LM, Bennett MV. α-Bungarotoxin labeling and acetylcholinesterase localization at the Mauthner fiber giant synapse in the hatchetfish. J Neurosci. 1983;3:272–279. doi: 10.1523/JNEUROSCI.03-02-00272.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond J. The Mauthner cell. In: Hoar WS, Randall DJ, editors. Fish Physiology. New York: Academic Press; 1971. pp. 265–346. [Google Scholar]
- Drasdo A, Caulfield M, Bertrand D, Bertrand S, Wonnacott S. Methyl lycaconitine: A novel nicotinic antagonist. Mol Cell Neurosci. 1992;3:237–243. doi: 10.1016/1044-7431(92)90043-2. [DOI] [PubMed] [Google Scholar]
- Du C, Role LW. Differential modulation of nicotinic acetylcholine receptor subtypes and synaptic transmission in chick sympathetic ganglia by PGE2. J Neurophysiol. 2001;85:2498–2508. doi: 10.1152/jn.2001.85.6.2498. [DOI] [PubMed] [Google Scholar]
- Elgoyhen AB, Vetter DE, Katz E, Rothlin CV, Heinemann SF, Boulter J. α10: a determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proc Natl Acad Sci U S A. 2001;98:3501–3506. doi: 10.1073/pnas.051622798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber DS, Funch PG, Korn H. Evidence that receptors mediating central synaptic potentials extend beyond the postsynaptic density. Proc Natl Acad Sci U S A. 1985;32:3504–3508. doi: 10.1073/pnas.82.10.3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber DS, Korn H. Electrophysiology of the Mauthner cell: basic properties, synaptic mechanisms, and associated networks. In: Faber DS, Korn H, editors. Neurobiology of the Mauthner Cell. New York: Raven; 1978. pp. 47–131. [Google Scholar]
- Faber DS, Korn H. Synergism at central synapses due to lateral diffusion of transmitter. Proc Natl Acad Sci U S A. 1988;85:8708–8712. doi: 10.1073/pnas.85.22.8708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria M, Oliveira L, Timoteo MA, Lobo MG, Correia-De-Sa P. Blockade of neuronal facilitatory nicotinic receptors containing α3β2 subunits contribute to tetanic fade in the rat isolated diaphragm. Synapse. 2003;49:77–88. doi: 10.1002/syn.10211. [DOI] [PubMed] [Google Scholar]
- Figl A, Cohen BN. The subunit dominates the relaxation kinetics of heteromeric neuronal nicotinic receptors. J Physiol. 2000;524:685–699. doi: 10.1111/j.1469-7793.2000.00685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer H, Liu DM, Lee A, Harries JC, Adams DJ. Selective modulation of neuronal nicotinic acetylcholine receptor channel subunits by Go-protein subunits. J Neurosci. 2005;25:3571–3577. doi: 10.1523/JNEUROSCI.4971-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores CE, Ene S, Pereda AE. An immunochemical marker for goldfish Mauthner cells. J Neurosci Methods. 2008;175:64–69. doi: 10.1016/j.jneumeth.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier CJ, Buhler AV, Weiner JL, Dunwiddie TV. Synaptic potentials mediated via α-bungarotoxin-sensitive nicotinic acetylcholine receptors in rat hippocampal interneurons. J Neurosci. 1998;18:8228–8235. doi: 10.1523/JNEUROSCI.18-20-08228.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fucile S, Matter JM, Erkman L, Ragozzino D, Barabino B, Grassi F, et al. The neuronal α6 subunit forms functional heteromeric acetylcholine receptors in human transfected cells. Eur J Neurosci. 1998;10:172–178. doi: 10.1046/j.1460-9568.1998.00001.x. [DOI] [PubMed] [Google Scholar]
- Funch PG, Wood MR, Faber DS. Localization of active sites along the myelinated goldfish Mauthner axon: morphological and pharmacological evidence for saltatory conduction. J Neurosci. 1984;4:2397–2409. doi: 10.1523/JNEUROSCI.04-09-02397.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genzen JR, Van Cleve W, McGehee DS. Dorsal root ganglion neurons express multiple nicotinic acetylcholine receptor subtypes. J Neurophysiol. 2001;86:1773–1782. doi: 10.1152/jn.2001.86.4.1773. [DOI] [PubMed] [Google Scholar]
- Gilat E, Hall DH, Bennett MV. The giant fiber and pectoral fin adductor motoneuron system in the hatchetfish. Brain Res. 1986;365:96–104. doi: 10.1016/0006-8993(86)90726-2. [DOI] [PubMed] [Google Scholar]
- Guo JZ, Liu Y, Sorenson EM, Chiappinelli VA. Synaptically released and exogenous ACh activates different nicotinic receptors to enhance evoked glutamatergic transmission in the lateral geniculate nucleus. J Neurophysiol. 2005;94:2549–2560. doi: 10.1152/jn.00339.2005. [DOI] [PubMed] [Google Scholar]
- Hackett JT, Buchheim A. Ultrastructural correlates of electrical-chemical synaptic transmission in goldfish cranial motor nuclei. J Comp Neurol. 1984;224:425–436. doi: 10.1002/cne.902240310. [DOI] [PubMed] [Google Scholar]
- Hackett JT, Cochran SL, Greenfield LJ., Jr Quantal transmission at Mauthner axon target synapses in the goldfish brainstem. Neuroscience. 1989;32:49–64. doi: 10.1016/0306-4522(89)90107-3. [DOI] [PubMed] [Google Scholar]
- Hackett JT, Faber DS. Mauthner axon networks mediating supraspinal components of the startle response in the goldfish. Neuroscience. 1983a;8:317–331. doi: 10.1016/0306-4522(83)90069-6. [DOI] [PubMed] [Google Scholar]
- Hackett JT, Faber DS. Relay neurons mediate collateral inhibition of the goldfish Mauthner cell. Brain Res. 1983b;264:302–306. doi: 10.1016/0006-8993(83)90829-6. [DOI] [PubMed] [Google Scholar]
- Hansen SB, Sulzenbacher G, Huxford T, Marchot P, Taylor P, Bourne Y. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 2005;24:3635–3646. doi: 10.1038/sj.emboj.7600828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartzell HC, Kuffler SW, Yoshikami D. Post-synaptic potentiation: interaction between quanta of acetylcholine at the skeletal neuromuscular synapse. J Physiol. 1975;251:427–463. doi: 10.1113/jphysiol.1975.sp011102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SC, McIntosh JM, Cartier GE, Maddox FN, Luetje CW. Determinants of specificity for α-conotoxin MII on α3β2 neuronal nicotinic receptors. Mol Pharmacol. 1997;51:336–342. doi: 10.1124/mol.51.2.336. [DOI] [PubMed] [Google Scholar]
- Hatton GI, Yang QZ. Synaptic potentials mediated by α7 nicotinic acetylcholine receptors in supraoptic nucleus. J Neurosci. 2002;22:29–37. doi: 10.1523/JNEUROSCI.22-01-00029.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieber V, Bouchey J, Agranoff BW, Goldman D. Nucleotide and deduced amino acid sequence of the goldfish neural nicotinic acetylcholine receptor α-3 subunit. Nucleic Acids Res. 1990a;18:5293. doi: 10.1093/nar/18.17.5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieber V, Bouchey J, Agranoff BW, Goldman D. Nucleotide and deduced amino acid sequence of the goldfish neural nicotinic acetylcholine receptor β-2 subunit. Nucleic Acids Res. 1990b;18:5307. doi: 10.1093/nar/18.17.5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horch HL, Sargent PB. Perisynaptic surface distribution of multiple classes of nicotinic acetylcholine receptors on neurons in the chicken ciliary ganglion. J Neurosci. 1995;15:7778–7795. doi: 10.1523/JNEUROSCI.15-12-07778.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack JB, Noble D, Tsien RW. Electric Current Flow in Excitable Cells. London: Oxford University Press; 1975. [Google Scholar]
- Jacob MH, Berg DK. The ultrastructural localization of α-bungarotoxin binding sites in relation to synapses on chick ciliary ganglion neurons. J Neurosci. 1983;3:260–271. doi: 10.1523/JNEUROSCI.03-02-00260.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob MH, Berg DK, Lindstrom JM. Shared antigenic determinant between the Electrophorus acetylcholine receptor and a synaptic component on chicken ciliary ganglion neurons. Proc Natl Acad Sci U S A. 1984;81:3223–3227. doi: 10.1073/pnas.81.10.3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Sudweeks S, Yakel JL. Nicotinic receptors in the brain: correlating physiology with function. Trends Neurosci. 1999;22:555–561. doi: 10.1016/s0166-2236(99)01471-x. [DOI] [PubMed] [Google Scholar]
- Kaiser S, Wonnacott S. α-bungarotoxin-sensitive nicotinic receptors indirectly modulate [3H]dopamine release in rat striatal slices via glutamate release. Mol Pharmacol. 2000;58:312–318. doi: 10.1124/mol.58.2.312. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Metcalfe WK, Schabtach E. T reticular interneurons: a class of serially repeating cells in the zebrafish hindbrain. J Comp Neurol. 1985;233:365–376. doi: 10.1002/cne.902330306. [DOI] [PubMed] [Google Scholar]
- Korn H, Faber DS. The Mauthner cell half a century later: a neurobiological model for decision-making? Neuron. 2005;47:13–28. doi: 10.1016/j.neuron.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Lopez MG, Montiel C, Herrero CJ, Garcia-Palomero E, Mayorgas I, Hernandez-Guijo JM, et al. Unmasking the functions of the chromaffin cell α7 nicotinic receptor by using short pulses of acetylcholine and selective blockers. Proc Natl Acad Sci U S A. 1998;95:14184–14189. doi: 10.1073/pnas.95.24.14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loring RH, Dahm LM, Zigmond RE. Localization of α-bungarotoxin binding sites in the ciliary ganglion of the embryonic chick: an autoradiographic study at the light and electron microscopic level. Neuroscience. 1985;14:645–660. doi: 10.1016/0306-4522(85)90316-1. [DOI] [PubMed] [Google Scholar]
- Lukas RJ, Changeux JP, Le Novere N, Albuquerque EX, Balfour DJ, Berg DK, et al. International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev. 1999;51:397–401. [PubMed] [Google Scholar]
- Luo S, Kulak JM, Cartier GE, Jacobsen RB, Yoshikami D, Olivera BM, McIntosh JM. α-conotoxin AuIB selectively blocks α3β4 nicotinic acetylcholine receptors and nicotine-evoked norepinephrine release. J Neurosci. 1998;18:8571–8579. doi: 10.1523/JNEUROSCI.18-21-08571.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- McIntosh JM, Dowell C, Watkins M, Garrett JE, Yoshikami D, Olivera BM. α-conotoxin GIC from Conus geographus, a novel peptide antagonist of nicotinic acetylcholine receptors. J Biol Chem. 2002;277:33610–33615. doi: 10.1074/jbc.M205102200. [DOI] [PubMed] [Google Scholar]
- McIntosh JM, Plazas PV, Watkins M, Gomez-Casati ME, Olivera BM, Elgoyhen AB. A novel α-conotoxin, PeIA, cloned from Conus pergrandis, discriminates between rat α9α10 and α7 nicotinic cholinergic receptors. J Biol Chem. 2005;280:30107–30112. doi: 10.1074/jbc.M504102200. [DOI] [PubMed] [Google Scholar]
- Mike A, Castro G, Albuquerque EX. Choline and acetylcholine have similar kinetic properties of activation and desensitization on the α7 nicotinic receptors in rat hippocampal neurons. Brain Res. 2000;882:155–168. doi: 10.1016/s0006-8993(00)02863-8. [DOI] [PubMed] [Google Scholar]
- Model PG, Highstein SM, Bennett MV. Depletion of vesicles and fatigue of transmission at a vertebrate central synapse. Brain Res. 1975;98:209–228. doi: 10.1016/0006-8993(75)90002-5. [DOI] [PubMed] [Google Scholar]
- Model PG, Spira ME, Bennett MV. Synaptic inputs to the cell bodies of the giant fibers of the hatchetfish. Brain Res. 1972;45:288–295. doi: 10.1016/0006-8993(72)90240-5. [DOI] [PubMed] [Google Scholar]
- Mogg AJ, Whiteaker P, McIntosh JM, Marks M, Collins AC, Wonnacott S. Methyllycaconitine is a potent antagonist of α-conotoxin-MII-sensitive presynaptic nicotinic acetylcholine receptors in rat striatum. J Pharmacol Exp Ther. 2002;302:197–204. doi: 10.1124/jpet.302.1.197. [DOI] [PubMed] [Google Scholar]
- Nai Q, McIntosh JM, Margiotta JF. Relating neuronal nicotinic acetylcholine receptor subtypes defined by subunit composition and channel function. Mol Pharmacol. 2003;63:311–324. doi: 10.1124/mol.63.2.311. [DOI] [PubMed] [Google Scholar]
- Nguyen D, Sargent PB. Synaptic vesicle recycling at two classes of release sites in giant nerve terminals of the embryonic chicken ciliary ganglion. J Comp Neurol. 2002;448:128–137. doi: 10.1002/cne.10237. [DOI] [PubMed] [Google Scholar]
- Nong Y, Sorenson EM, Chiappinelli VA. Fast excitatory nicotinic transmission in the chick lateral spiriform nucleus. J Neurosci. 1999;19:7804–7811. doi: 10.1523/JNEUROSCI.19-18-07804.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke RL, Meyer E, Nutter T, Uteshev VV. α7 receptor-selective agonists and modes of α7 receptor activation. Eur J Pharmacol. 2000;393:179–195. doi: 10.1016/s0014-2999(00)00009-1. [DOI] [PubMed] [Google Scholar]
- Papke RL, Thinschmidt JS. The correction of α7 nicotinic acetylcholine receptor concentration-response relationships in Xenopus oocytes. Neurosci Lett. 1998;256:163–166. doi: 10.1016/s0304-3940(98)00786-1. [DOI] [PubMed] [Google Scholar]
- Park KS, Cha SK, Kim MJ, Kim DR, Jeong SW, Lee JW, Kong ID. An α3β4 subunit combination acts as a major functional nicotinic acetylcholine receptor in male rat pelvic ganglion neurons. Pflugers Arch. 2006;452:775–783. doi: 10.1007/s00424-006-0086-1. [DOI] [PubMed] [Google Scholar]
- Parker MJ, Zhao S, Bredt DS, Sanes JR, Feng G. PSD93 regulates synaptic stability at neuronal cholinergic synapses. J Neurosci. 2004;24:378–388. doi: 10.1523/JNEUROSCI.3865-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Placzek AN, Grassi F, Meyer EM, Papke RL. An α7 nicotinic acetylcholine receptor gain-of-function mutant that retains pharmacological fidelity. Mol Pharmacol. 2005;68:1863–1876. doi: 10.1124/mol.105.016402. [DOI] [PubMed] [Google Scholar]
- Roerig B, Nelson DA, Katz LC. Fast synaptic signaling by nicotinic acetylcholine and serotonin 5-HT3 receptors in developing visual cortex. J Neurosci. 1997;17:8353–8362. doi: 10.1523/JNEUROSCI.17-21-08353.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Maravall M, Svoboda K. Ca2+ signaling in dendritic spines. Curr Opin Neurobiol. 2001;11:349–356. doi: 10.1016/s0959-4388(00)00218-x. [DOI] [PubMed] [Google Scholar]
- Sahibzada N, Ferreira M, Jr, Williams B, Wasserman A, Vicini S, Gillis RA. Nicotinic ACh receptor subtypes on gastrointestinally projecting neurones in the dorsal motor vagal nucleus of the rat. J Physiol. 2002;545:1007–1016. doi: 10.1113/jphysiol.2002.021337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent P. Nicotinic receptors concentrated in the subsynaptic membrane so not contribute significantly to synaptic currents at an embryonic synapse in the chicken ciliary ganglion. J Neurosci. 2009;29:3749–3759. doi: 10.1523/JNEUROSCI.5404-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Yakel JL. Single channel properties of neuronal nicotinic ACh receptors in stratum radiatum interneurons of rat hippocampal slices. J Physiol. 2000;527:507–513. doi: 10.1111/j.1469-7793.2000.00507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoop RD, Chang KT, Ellisman MH, Berg DK. Synaptically driven calcium transients via nicotinic receptors on somatic spines. J Neurosci. 2001;21:771–781. doi: 10.1523/JNEUROSCI.21-03-00771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoop RD, Martone ME, Yamada N, Ellisman MH, Berg DK. Neuronal acetylcholine receptors with α7 subunits are concentrated on somatic spines for synaptic signaling in embryonic chick ciliary ganglia. J Neurosci. 1999;19:692–704. doi: 10.1523/JNEUROSCI.19-02-00692.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo TM, Weiss SA, Faber DS, Preuss T. Representation of auditory signals in the M-cell: role of electrical synapses. J Neurophysiol. 2006;95:2617–2629. doi: 10.1152/jn.01287.2005. [DOI] [PubMed] [Google Scholar]
- Tan W, Du C, Siegelbaum SA, Role LW. Modulation of nicotinic AChR channels by prostaglandin E2 in chick sympathetic ganglion neurons. J Neurophysiol. 1998;79:870–878. doi: 10.1152/jn.1998.79.2.870. [DOI] [PubMed] [Google Scholar]
- Thinschmidt JS, Frazier CJ, King MA, Meyer EM, Papke RL. Medial septal/diagonal band cells express multiple functional nicotinic receptor subtypes that are correlated with firing frequency. Neurosci Lett. 2005;389:163–168. doi: 10.1016/j.neulet.2005.07.038. [DOI] [PubMed] [Google Scholar]
- Titmus MJ, Faber DS. 20th Anniversary Commemorative Symposium. San Jaun, Puerto Rico: University of Puerto Rico, Institute of Neurobiology; 1987. Alteration of identified output synapses spared by axotomy. [PubMed] [Google Scholar]
- Ullian EM, McIntosh JM, Sargent PB. Rapid synaptic transmission in the avian ciliary ganglion is mediated by two distinct classes of nicotinic receptors. J Neurosci. 1997;17:7210–7219. doi: 10.1523/JNEUROSCI.17-19-07210.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldeck RF, Pereda A, Faber DS. Properties and plasticity of paired-pulse depression at a central synapse. J Neurosci. 2000;20:5312–5320. doi: 10.1523/JNEUROSCI.20-14-05312.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss SA, Zottoli SJ, Do SC, Faber DS, Preuss T. Correlation of C-start behaviors with neural activity recorded from the hindbrain in free-swimming goldfish (Carassius auratus) J Exp Biol. 2006;209:4788–4801. doi: 10.1242/jeb.02582. [DOI] [PubMed] [Google Scholar]
- Whiteaker P, Christensen S, Yoshikami D, Dowell C, Watkins M, Gulyas J, et al. Discovery, synthesis, and structure activity of a highly selective α7 nicotinic acetylcholine receptor antagonist. Biochemistry. 2007;46:6628–6638. doi: 10.1021/bi7004202. [DOI] [PubMed] [Google Scholar]
- Williams BM, Temburni MK, Levey MS, Bertrand S, Bertrand D, Jacob MH. The long internal loop of the α3 subunit targets nAChRs to subdomains within individual synapses on neurons in vivo. Nat Neurosci. 1998;1:557–562. doi: 10.1038/2792. [DOI] [PubMed] [Google Scholar]
- Wonnacott S, Barik J, Dickinson J, Jones IW. Nicotinic receptors modulate transmitter cross talk in the CNS: nicotinic modulation of transmitters. J Mol Neurosci. 2006;30:137–140. doi: 10.1385/JMN:30:1:137. [DOI] [PubMed] [Google Scholar]
- Yu CR, Role LW. Functional contribution of the α7 subunit to multiple subtypes of nicotinic receptors in embryonic chick sympathetic neurones. J Physiol. 1998;509:651–665. doi: 10.1111/j.1469-7793.1998.651bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZW, Coggan JS, Berg DK. Synaptic currents generated by neuronal acetylcholine receptors sensitive to α-bungarotoxin. Neuron. 1996;17:1231–1240. doi: 10.1016/s0896-6273(00)80253-6. [DOI] [PubMed] [Google Scholar]
- Zhang ZW, Vijayaraghavan S, Berg DK. Neuronal acetylcholine receptors that bind α-bungarotoxin with high affinity function as ligand-gated ion channels. Neuron. 1994;12:167–177. doi: 10.1016/0896-6273(94)90161-9. [DOI] [PubMed] [Google Scholar]
- Zottoli SJ. Correlation of the startle reflex and Mauthner cell auditory responses in unrestrained goldfish. J Exp Biol. 1977;66:243–254. doi: 10.1242/jeb.66.1.243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.