

Abstract
Mycobacterium tuberculosis arabinogalactan (AG) is an essential cell wall component. It provides a molecular framework serving to connect peptidoglycan to the outer mycolic acid layer. The biosynthesis of the arabinan domains of AG and lipoarabinomannan (LAM) occurs via a combination of membrane bound arabinofuranosyltransferases, all of which utilize decaprenol-1-monophosphorabinose as a substrate. The source of arabinose ultimately destined for deposition into cell wall AG or LAM originates exclusively from phosphoribosyl-1-pyrophosphate (pRpp), a central metabolite which is also required for other essential metabolic processes, such as de novo purine and pyrimidine biosyntheses. In M. tuberculosis, a single pRpp synthetase enzyme (Mt-PrsA) is solely responsible for the generation of pRpp, by catalyzing the transfer of pyrophosphate from ATP to the C1 hydroxyl position of ribose-5-phosphate. Here, we report a detailed biochemical and biophysical study of Mt-PrsA, which exhibits the most rapid enzyme kinetics reported for a pRpp synthetase.
Keywords: arabinan, cell wall, Mycobacteria, phosphoribosylpyrophosphate, polysaccharides
Introduction
As we enter into the second decade of the twenty-first century, tuberculosis (TB) continues to cause more deaths and human suffering than any other infectious disease (Dye 2006). The number of new TB cases is increasing at an alarming pace, as does the prevalence of drug-resistant stains of the causative agent, Mycobacterium tuberculosis, in the form of multidrug-resistant TB (MDR-TB) and extensively drug-resistant TB (XDR-TB; Madariaga et al. 2008). The need for new drugs and vaccines to combat this major burden to human health is overwhelming.
The unique hydrophobic cell wall of M. tuberculosis forms a primary defensive barrier to the onslaught of toxic insult during the host immune response. The current chemotherapeutic regime used to treat TB includes the administration of isoniazid and ethambutol, both of which target crucial steps in the biosynthesis and the assembly of the M. tuberculosis cell wall (Banerjee et al. 1994; Belanger et al. 1996). However, whilst these cell wall inhibitors are excellent at effectively killing actively dividing cells, certain subpopulations of bacilli, labeled as “persisters”, exhibit transient resistance to many of the drugs used to treat TB (Wayne and Sohaskey 2001). The precise molecular mechanism of this latent infection caused by persistent bacilli is poorly understood. However, in the pursuit of identifying suitable cellular targets and the development of new anti-TB chemotherapeutic agents, we must address the phenomenon of persistence to truly eradicate the disease. Recently, it has been shown that certain processes of metabolic pathways involved in mycobacterial cellular energy metabolism, such as respiratory ATP biosynthesis, represent excellent physiological targets for the effective killing of latent populations of TB bacilli (Koul et al. 2007, 2008; Rao et al. 2008; Bald and Koul 2010). Phospho-α-d-ribosyl-1-pyrophosphate (pRpp) is a central metabolite that links the pentose phosphate pathway to the de novo and the salvage biosynthetic pathways of purine and pyrimidine production, NAD and NADP cofactor biosynthesis, as well as histidine and tryptophan biosynthesis (Hove-Jensen 1988). It is therefore considered that pRpp is required at all times during the life cycle of both prokaryotic and eukaryotic cells (Hove-Jensen 1989; Tozzi et al. 2006). In addition to these central metabolic processes, members belonging to the Corynebacteriacae, such as mycobacteria, have evolved an almost unique biochemical pathway, utilizing pRpp as a high-energy biosynthetic precursor for cell wall arabinan biosynthesis (Wojtkiewicz et al. 1988; Scherman et al. 1995, 1996; Figure 1). The gene product of Rv3806c encodes for a membrane bound pRpp:decaprenol-1-monophosphate 5-phosphoribosyltransferase (DPPR synthase) which catalyzes the formation of DPPR and pyrophosphate from pRpp and decaprenol-1-monophosphate (DP; Huang et al. 2005). DPPR then undergoes C5-dephosphorylation to decaprenol-1-monophosphoribose (DPR) and epimerization about the C2–OH position of the ribosyl moiety catalyzed by a two-step heteromeric oxidation/reduction process (Mikusova et al. 2005). In M. tuberculosis, Rv3790 encodes for an FAD-containing oxidoreductase (DprE1) which oxidizes the ribosyl C2–OH group to the keto sugar decaprenol-1-monophosphoryl-2-keto-β-erythro-pentofuranose (DPX) which is subsequently reduced by DprE2 (encoded by Rv3791) forming decaprenol-1-monophosphoarabinose (DPA; Mikusova et al. 2005), which is the sole high-energy lipid-linked substrate for the GT-C family arabinofuranosyltransferases (AraTs) which are involved in cell wall arabinan polymerization (Alderwick et al. 2007).
Fig. 1.

Proposed metabolic pathway depicting the importance of Mt-PrsA for the generation of pRpp in M. tuberculosis. R5P is generated via the catabolism of glucose through the pentose phosphate pathway, a process ubiquitous throughout nature. Like all pRpp synthetase enzymes, Mt-PrsA catalyses the transfer of pyrophosphate from ATP to the C1–OH group of R5P forming pRpp and AMP. In a process unique to members belonging to the Corynebacteriaceae, such as M. tuberculosis, pRpp is utilized as the sole metabolic intermediate in the formation of DPA, which is a crucial substrate required for mycobacterial cell wall assembly. DP, decaprenol-1-monophosphate; DPA, decaprenol-1-monophosphoarabinose; DPR, decaprenol-1-monophosphoribose; DPPR, decaprenol-1-monophosphoribose-5-phosphate; DPX, decaprenol-1-monophosphoryl-2-keto-β-erythro-pentofuranose; pRpp, phosphoribosyl-1-pyrophosphate; R5P, ribose -5-phosphate.
Mycobacterial arabinan is a highly branched polysaccharide consisting of arabinose units arranged into specific repeating motifs, and it predominantly features in the heteropolysaccharide arabinogalactan (AG), which is a crucial component of the mycobacterial cell wall (Daffé et al. 1990; Besra et al. 1995). Three arabinan domains are covalently linked via an Araf-α(1 → 5)-Galf glycosidic bond to the 8th, 10th and 12th positions of a single linear polysaccharide composed of ∼30 alternating β(1 → 5) and β(1 → 6) Galf residues forming the galactan domain of AG (Alderwick et al. 2005). AG is a structural macromolecule and serves to function as a molecular scaffold linking peptidoglycan to the mycolic acids, forming a highly impermeable and hydrophobic layer surrounding the mycobacterial cell (McNeil et al. 1991). Arabinan is also present in the form of lipoarabinomannan (LAM), a highly immunogenic lipoglycan, which is involved in modulating the host immune response (Briken et al. 2004). In terms of chemical composition, arabinan represents ∼18% of mycobacterial cell wall biomass (Ortalo-Magne et al. 1995). Therefore, during cell elongation and division of the mycobacterial bacilli, it is necessary for there to be a readily available supply of pRpp for the efficient incorporation of arabinose into newly synthesized cell wall material. During this process, the metabolic demand for pRpp is likely to increase dramatically and the elevated levels required for effective arabinan biosynthesis must be maintained. The enzyme pRpp synthetase (ATP:α-d-ribose-5-phosphate (R5P) pyrophosphotransferase; EC2.7.6.1) belongs to a family of enzymes that catalyze the transfer of the β,γ-diphosphate moiety from ATP to the C1–OH group of α-d-R5P and the gene(s) encoding for this enzyme(s) is present in all living organisms (Hove-Jensen 1985; Carter et al. 1997; Krath and Hove-Jensen 1999). It is predicted that the M. tuberculosis H37Rv genome codes for only one pRpp synthetase (Rv1017c), which is annotated as Mt-PrsA (Cole and Barrell 1998).
In this study, we report a detailed biochemical characterization of recombinant Mt-PrsA and provide evidence which confirms the essentiality of this gene for the maintenance of cell growth, division and integrity. Mt-PrsA displays an absolute requirement for inorganic phosphate for enzyme activation and will accept either Mg2+ or Mn2+ as divalent cations, which serve to coordinate the phosphate moieties of ATP during enzyme catalysis. Using a continuous enzyme coupled spectrophotometric assay, we report a detailed kinetic characterization of Mt-PrsA, which displays the highest specific activity measured for any pRpp synthetase studied to date. By investigating the oligomeric state of Mt-PrsA in solution and measuring the ligand-binding properties of its substrates, we provide evidence suggesting that the mechanism by which ADP inhibits Mt-PrsA activity, results from the binding of ADP to an allosteric site, and as a consequence, induces the stabilization of an inactive, hexameric oligomeric species.
Results
Evidence suggesting that Mt-prsA is an essential gene for the maintenance of cellular integrity
Mt-prsA is predicted to be an essential gene as determined by high-density transposon mutagenesis (Sassetti et al. 2003). This is not unsurprising, considering the apparent physiological importance of Mt-PrsA in the provision of pRpp for central metabolic pathways and the formation of the cell wall biosynthetic precursor DPA (Wolucka et al. 1994; Scherman et al. 1995). We have previously used Corynebacterium glutamicum as an excellent model system for the study of complex cell wall biosynthetic processes, and we have reported the phenotypic characterization of several cell wall mutants, which would otherwise be essential in mycobacterial systems (Gande et al. 2004; Alderwick et al. 2005; Alderwick, Dover, et al. 2006; Alderwick, Seidel, et al. 2006; Seidel et al. 2007; Birch et al. 2008). A bioinformatics analysis of several Corynebacteriaceae genomes revealed that NCgl0945 (Cg-prsA) was the likely C. glutamicum ortholog of Mt-prsA (Rv1017c). These corresponding gene products share 71% identity (83% similarity), and the gene locus is conserved within the Corynebacteriaceae indicating that this region is syntenic. We attempted to inactivate Cg-prsA in C. glutamicum, by using the disruption vector pK18mob-Cg-prsA-int, carrying 304 bp of Cg-prsA. In a control experiment, we used the previously reported pK18mob-Cg-ppm1-int construct, which affords the successful inactivation of the gene encoding for the polyprenyl monophosphomannose synthase (ppm1) of C. glutamicum, which is a gene of similar size to that of Cg-prsA (Gibson et al. 2003). Both inactivation vectors were used in parallel experiments for the transformation of wild-type (WT) C. glutamicum to kanamycin resistance (KanR). Whilst we were able to easily generate C. glutamicum KanR colonies from the pK18mob-Cg-ppm1-int construct, we were required to conduct several rounds of electroporation using pK18mob-Cg-prsA-int before obtaining three separate C. glutamicum KanR clones. This indicates that the recombination events appeared to have taken place at unexpected and, as yet, undefined loci. From this analysis, we concluded that Cg-prsA is an essential gene and its function is necessary for the growth and integrity of C. glutamicum, even when grown on rich Brain Heart Infusion (BHI) medium.
Overexpression, purification and pRpp synthetase activity of recombinant Mt-PrsA
Overexpression of Mt-PrsA in Escherichia coli C41 cells and the subsequent purification by IMAC resulted in a preparation of Mt-PrsA that was stable in solution at a concentration of 2 mg/mL. A coomassie-stained 12% SDS–PAGE analysis of Mt-PrsA revealed a band that migrated to a position of 35 kDa, which is in accordance with the expected molecular weight of Mt-PrsA, and was estimated to be at least 98% pure. To determine if recombinant Mt-PrsA was active, we performed a radiochemical assay to measure the pyrophosphate transfer from ATP to the C1–OH group of [14C]-uniformly labeled R5P (Boss et al. 1984). Starting with [14C]-U-d-glucose and replicating the first three enzymic reactions of the pentose phosphate pathway in vitro using commercially available enzymes (Roche and Sigma), we prepared a source of purified [14C]-U-α-d-R5P as a substrate for subsequent enzyme assays. In a time course assay, recombinant Mt-PrsA exhibited rapid pRpp synthetase activity which was dependent upon the presence of either of the divalent cations Mg2+ and Mn2+, with slight preference for the latter (Figure 2A). During the assay, p[14C]Rpp is susceptible to product degradation due to hydrolysis of the labile C1–OH pyrophosphate bond, which accounts for the relatively small increase in band density (relating to p[14C]Rpp) over the course of the assay. Addition of EDTA to the reaction mix inhibited the formation of p[14C]Rpp (data not shown) and the inclusion of CaCl2, in replacement of MgCl2 or MnCl2, also resulted in the loss of Mt-PrsA activity (Figure 2A). Mt-PrsA was found to be most active at a pH of 6.5, but also showed activity across a broad range of pH levels (4.5–9.5) and was only found to be inactive at high pH (10.5; data not shown). In accordance with previous studies of pRpp synthetases (Prs) from other organisms, Mt-PrsA was strongly inhibited by the presence of ADP, which has been shown to be an allosteric modulator of this class of enzymes (Figure 2B) (Switzer and Sogin 1973; Gibson et al. 1982; Arnvig et al. 1990; Eriksen et al. 2000; Willemoes et al. 2000).
Fig. 2.

Time course assay showing the Mt-PrsA catalyzed formation of p[14C]Rpp from [14C]R5P and ATP. Assays contained Mt-PrsA, 50 mM Pi, 2 mM ATP supplemented with either 2 mM Mg2+, Mn2+ or Ca2+ as illustrated in (A), and in the presence of ADP at a final concentration of 2 mM in (B). Assays were incubated at 37°C, and aliquots were removed from the assay mix at 0, 10, 30 and 60 s and the reactions halted by quenching with the addition of formic acid. Samples were applied to a glass-backed PEI-cellulose TLC plate and developed in 0.85 M KH2PO4 (pH 3.4). The plate was inspected for radioactive spots relating to p[14C]Rpp and [14C]R5P by autoradiography through the exposure of TLCs to X-ray film (Kodak X-Omat).
Mt-PrsA-dependent metabolic channeling of R5P into mycobacterial cell wall biosynthetic precursors
Apart from being a key metabolite in the de novo and salvage pathways of purine and pyrimidine biosynthesis, as well as histidine, tryptophan, NAD and NADP metabolism, pRpp serves as a high-energy intermediate in the formation of the mycobacterial cell wall precursor DPR (Figure 1; Scherman et al. 1995). We investigated the conversion of [14C]-U-d-R5P ([14C]-R5P) into the cell wall AraT substrate DPA and its precursor, DPR. Assays were conducted using membranes prepared from M. smegmatis which provided a source of DPPR synthase (Huang et al. 2005), which was necessary to investigate incorporation of [14C]-R5P into the lipid-linked organic extractable material. When assays were performed with just [14C]-R5P and membranes, no radioactive product was detectable in the organic phase, as depicted in Figure 3 (lane 1). In this instance, all radioactivities remained in the aqueous phase of the assay work-up (data not shown). In addition, M. smegmatis P60 (a preparation of cell wall material rich in proteins associated with cell wall processes) exhibited no pRpp synthetase activity (Figure 3, lane 2). When recombinant Mt-PrsA is included in the assay, the formation of DPPR and DPR can be clearly observed (Figure 3, lane 3). It is likely that DPPR undergoes enzyme catalyzed 5'-dephosphorylation, resulting in the formation of DPR. In M. tuberculosis, Rv3807c is an excellent candidate for this enzyme since it codes for a putative membrane bound PAP2 phosphatase directly adjacent to the DPPR synthase (Rv3806c; Figure 1; Cole and Barrell 1998). Mycobacterium smegmatis has a direct ortholog of this phosphatase encoded by MSMEG6402, which explains the apparent conversion of DPPR to DPR observed in this assay. The combined addition of sodium fluoride and sodium tungstate (known phosphatase inhibitors; Stankiewicz and Gresser 1988) to the assay mix, results in a diminished level of DPR formation and a corresponding increase in a band migrating to a lower position on the TLC identified as being DPPR (Figure 3, lane 4). When Mt-PrsA is used in combination with M. smegmatis P60, two additional bands can be observed migrating in close proximity to DPR, which correspond to the subsequent conversion of DPR to DPX and DPA, by the endogenous P60 activity of DprE1 and DrpE2, respectively (Figure 3, lane 5; Mikusova et al. 2005). The inclusion of sodium fluoride and sodium tungstate dramatically reduced the conversion of DPPR to DPA, suggesting that the two-step epimerization of the C2–OH group of ribose by DprE1 and DprE2 occurs after 5' dephosphorylation (Figure 3, lane 6). The inclusion of ADP to the reaction mix completely ablated the formation of any organic extractable material, clearly indicating that the ADP inhibition of Mt-PrsA results in the total loss of DPPR formation (Figure 3, lane 7).
Fig. 3.

Analysis of Mt-PrsA-dependent [14C]-R5P incorporation into the M. smegmatis AraT substrate DPA via p[14C]Rpp. The basic assays mixture contained (amongst other components described in the Materials and methods section) membranes prepared from M. smegmatis, 100,000 cpm [14C]-R5P, 50 µg of DP and 2 mM ATP. Individual assays were carried out at 37°C and included the addition of no further additive; lane 1, M. smegmatis P60; lane 2, Mt-PrsA; lane 3, Mt-PrsA and NaWO4 and NaF both at a final concentration of 2 mM; lane 4, Mt-PrsA and M. smegmatis P60; lane 5, Mt-PrsA, M. smegmatis P60 and 2 mM of both NaWO4 and NaF; lane 6 and finally Mt-PrsA, M. smegmatis P60 and 2 mM ADP in lane 7. After an incubation at 37°C for 30 min, assays were quenched and products extracted from the assay mix by organic solvent extraction as described in the Materials and methods section. Samples relating to each assay were loaded onto a silica gel TLC plates, developed in CHCl3/CH3OH/CH3COONH4/NH4OH/H2O (180:140:9:9:23 v/v/v/v/v) and bands migrating to the position of DPA, DPR, DPX and DPPR were visualized by autoradiography, exposure of TLCs to X-ray film (Kodak X-Omat) and compared to previously isolated known standards (Mikusova et al. 2005; Alderwick et al. 2006a).
In a separate experiment, assays were carried out in the presence of a neoglycolipid acceptor [Araf-α(1 → 5)-Araf-O-(CH2)CH3], which is specific for measuring the activity of the cell wall biosynthetic arabinofuranosyltransferases EmbA, EmbB, EmbC and AftB (Lee et al. 1997; Seidel et al. 2007), all of which reside in the membrane due to the polytopic nature of these GT-C family glycosyltransferases. AraT activity was measured after product isolation by organic extraction and analysis by TLC and autoradiography. AraT assays carried out in the absence of Mt-PrsA afforded no AraT-dependent product formation (Figure 4, lane 1). Upon addition of Mt-PrsA, two closely migrating bands could be observed with the top bad attributed to α(1 → 5)-Araf transfer to the acceptor by either EmbA, EmbB, EmbC or a combination and the lower band attributed to β(1 → 2)-Araf transfer resulting from AftB activity (Figure 4, lane 2), as described previously (Lee et al. 1997). Addition of the EmbA, EmbB and EmbC inhibitor ethambutol (EMB), resulted in the loss of the upper band with only the lower band remaining, this being due to the noninhibitory effect of EMB on the terminal β(1 → 2) capping enzyme, AftB (Seidel et al. 2007; Figure 4, lane 3). As a consequence of allosteric inhibition of Mt-PrsA and the resultant loss of in vitro Mt-PrsA-dependent pRpp formation, the inclusion of ADP in the assay mixture led to the inhibition of any AraT-dependent product formation (Figure 4, lane 4).
Fig. 4.

AraT assays carried out via the Mt-PrsA-dependent conversion of [14C]-R5P to p[14C]Rpp and subsequent chase into a neoglycolipid acceptor. The absolute dependence of the cell wall AraT enzymes on the availability of pRpp was investigated using the neoglycolipid acceptor Ara2. The basic assay contained (amongst other components described in the Materials and methods section) 0.5 mg of membranes and P60 from M. smegmatis, 2 mM α-d-Araf-(1 → 5)-α-d-Araf-O-(CH2)7CH3, 100,000 cpm [14C]-R5P, 50 µg of DP and 2 mM ATP. Assays were initiated by the addition of [14C]-R5P and incubated for 1 h at 37°C. Individual assays carried out contained the addition of no Mt-PrsA, lane 1; 30 µg of Mt-PrsA, lane 2; 30 µg of Mt-PrsA and 100 µg/mL of EMB, lane 3 and 30 µg of Mt-PrsA and 2 mM ADP, lane 4. [14C]-Araf-linked products were extracted from the assay mix as described in the Materials and methods section and subsequently applied to a silica gel TLC plates, developed in CHCl3:CH2OH:H2O:NH4OH (65:25:3.6:0.5, v/v/v/v) and visualized by autoradiography from exposure of TLCs to X-ray film (Kodak X-Omat; Lee et al. 1997; Seidel et al. 2007).
Kinetic characterization of Mt-PrsA substrates and inhibitors using a continuous spectrophotometric assay
Due to the rapid enzymic activity of recombinant Mt-PrsA using the radioactive assay described previously, we sought to link Mt-PrsA catalyzed AMP product release to a three-enzyme coupled assay system, thus enabling us to measure NADH oxidation at 340 nm in a continuous spectrophotometric assay, which has been described similarly elsewhere (Braven et al. 1984). This system (described in the Materials and methods section) enabled us to record detailed measurements of the steady-state enzyme kinetics of Mt-PrsA. Plotting the steady-state reaction velocity (ν) calculated from Eq. (1), as a function of increasing concentrations of R5P at the saturating levels of ATP, results in a hyperbolic curve displaying the Michelis–Menton kinetics (Figure 5A). Nonlinear regression analysis using Eq. (2) derived a Km of 8.2 µM (±2.72 SE) for R5P and a Vmax of 530 µmol−1 min−1 mg−1 and a specificity constant (Kcat/Km) of 7.43 × 106 M−1 s−1. As previously described using the radiochemical assay, Mt-PrsA requires the presence of Pi at a concentration of 50 mM for full enzyme activation, which is illustrated in Figure 5A. However, at reduced levels of Pi (5 mM), the subsequent addition of R5P results in partial substrate inhibition, an effect that has been observed elsewhere [Willemoes et al. 2000; data fitted to Eq. (3)], with little or no activity recorded in the absence of Pi (Figure 5A). This is likely due to Mt-PrsA being incompletely activated by Pi at low [Pi], meaning that at low R5P concentrations, the relatively high-affinity catalytic site for R5P is occupied and the normal Micheli–Menton kinetics occur until R5P reaches a concentration that results in the occupancy of a second low-affinity inhibitory site which would otherwise be occupied by Pi at higher [Pi]. Kinetic analysis of Mt-PrsA with increasing ATP concentrations at saturating levels of R5P results in a sigmoidal response showing positive cooperativity (Figure 5B). Data obtained from measuring ν against increasing concentrations of ATP at various Pi concentrations was fitted to Eq. (4). At activating levels of [Pi] (50 mM), the S0.5 and the Vmax for ATP were calculated as being 62.65 µM (±12.04 SE) and 601 µmol−1 min−1 mg−1 (±66.39 SE), respectively. An apparent Hill coefficient (n) of 1.68 indicates a strong positive cooperativity during increasing [ATP]. However, when fitted to Eq. (4) using nonlinear regression analysis, this effect diminishes at low [Pi] resulting in a ν vs. [ATP] response with altered kinetic constants (Figure 5B). It has been reported elsewhere that Prs enzymes from other organisms are allosterically inhibited by the presence of ADP (Switzer and Sogin 1973; Gibson et al. 1982; Arnvig et al. 1990; Eriksen et al. 2000; Willemoes et al. 2000). We measured the effect of ADP on ν as a function of increasing [R5P] and [ATP]. At saturating concentrations of ATP, the addition of ADP resulted in noncompetitive inhibition of Mt-PrsA with a calculated Ki of 320 µM (±53.51 SE) against increasing [R5P] [data fitted to Eq. (5); Figure 6A]. When we measured the effect of ADP on the kinetic response of Mt-PrsA to increasing [ATP] and subsequent fitting of the data to Eq. (6) (Figure 6B), we calculated a Ki of 522 µM (±45.78 SE).
Fig. 5.

Enzyme activity of Mt-PrsA and kinetic characterization of R5P (A) and ATP (B) using a continuous enzyme-coupled spectrophotometric assay. (A) A plot of velocity (ν) of Mt-PrsA to increasing [R5P] in the presence of 50 mM Pi (filled circles), 5 mM Pi (filled squares) and no Pi (filled triangle). Data for 50 mM Pi fitted to Eq. (2), and data obtained for 5 mM Pi and no Pi were fitted to Eq. (3). (B) A plot of velocity (ν) of Mt-PrsA to increasing [ATP] in the presence of 50 mM Pi (filled circles), 5 mM Pi (filled squares) and no Pi (filled triangle), all of which were fitted to Eq. (4) using nonlinear regression analysis. All data points plotted (calculated mean) represent experiments performed in triplicate using three independent preparations of recombinant Mt-PrsA. Calculated standard errors (±SE) for kinetic constants are reported in the manuscript where appropriate.
Fig. 6.

Inhibition of Mt-PrsA activity by the addition of ADP when titrated with R5P (A) and ATP (B) using a continuous enzyme-coupled spectrophotometric assay. (A) A plot of velocity (ν) of Mt-PrsA response to increasing [R5P] in the presence of no ADP (filled circles), 0.1 mM ADP (filled squares) and 2 mM ADP (filled triangle). Data were fitted to Eq. (5) using nonlinear regression analysis. (B) A plot of velocity (ν) of Mt-PrsA response to increasing [ATP] in the presence of no ADP (filled circles), 0.1 mM ADP (filled squares) and 2 mM ADP (filled triangles). Data were fitted to Eq. (6) using nonlinear regression analysis. All data points plotted (calculated mean) represent experiments performed in triplicate using three independent preparations of recombinant Mt-PrsA. Calculated standard errors (±SE) for kinetic constants are reported in the manuscript where appropriate.
Biophysical characterization of Mt-PrsA: oligomeric state and ligand-binding studies
Mt-PrsA homologs from other organisms have been reported to aggregate in solution in various homo-oligomeric states, ranging from dimer to octamer (Schubert et al. 1975; Arnvig et al. 1990; Li et al. 2007). Using analytical ultracentrifugation, we performed a set of sedimentation velocity experiments on Mt-PrsA in the absence or the presence of R5P, ATP and ADP at saturating concentrations (Figure 7). Analysis of each experiment by interpretation of the molar mass distribution c(M) indicates that Mt-PrsA in solution alone reaches a dynamic equilibrium between two molecular species with peaks centered over ∼100,000 and ∼205,000 Da, which relate to Mt-PrsA in a trimeric and hexameric aggregation state, respectively (Figure 7, black line). The inclusion of 5 mM R5P showed no apparent change in the equilibrium or oligomeric state of Mt-PrsA in solution, and the c(M) vs. Mw distribution aligned well with that of Mt-PrsA in the absence of ligand (Figure 7, red line). The addition of 5 mM ATP to Mt-PrsA resulted in an altered c(M) distribution with a peak centered over ∼70,000 Da, equating to a Mt-PrsA homodimer, and concomitant reduction in the intensity of the hexameric species, when compared with Mt-PrsA alone (Figure 7, blue line). Interestingly, when we performed a similar experiment in combination with 5 mM ADP, the ratio of c(M) distribution shifted toward an increase in the hexameric state with a corresponding reduction in trimeric species (Figure 7, green line). Mt-PrsA contains within its primary amino acid sequence, three naturally occurring tryptophan residues at position Trp 5, Trp 74 and Trp 188. We used intrinsic tryptophan fluorescence (ITF) spectroscopy to probe the binding properties of R5P, ATP and ADP for Mt-PrsA. We performed a fluorescence emission (Femission) scan of Mt-PrsA in solution by recording (Femission) output between 300 and 400 nm, upon excitation at λ292nm. Mt-PrsA gave a maximum fluorescence emission 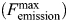 at a wavelength of 335 nm, thus providing a basal Femission coordinate for the collection of subsequent ITF data. The change in fluorescence emission (ΔFemission) was calculated by subtracting the Femission (recorded 5 min after each ligand titration) away from
at a wavelength of 335 nm, thus providing a basal Femission coordinate for the collection of subsequent ITF data. The change in fluorescence emission (ΔFemission) was calculated by subtracting the Femission (recorded 5 min after each ligand titration) away from 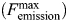 , and the data were then plotted against [L]. Nonlinear regression analysis of ΔFemission vs. [R5P] using Eq. (7) shows a hyperbolic isothermal ligand-binding curve for R5P, with a calculated Kd of 24.8 µM (±6.77 SE; Figure 8A). We sought to investigate what effect ATP might have on the Kd for R5P; however, the inclusion of both ATP and R5P would obviously result in enzyme catalysis and the formation of pRpp and AMP. Due to the relatively high specific activity of Mt-PrsA, the addition of R5P and ATP at concentrations required for the effective measurement of ligand-binding constants by ITF would result in a reaction that would go to completion within the 5 min time-frame required to establish binding equilibrium. Therefore, due to the stability of the imidodiphophate moity and lack of enzyme-catalyzed phosphate transfer, we used the ATP analog 5'-adenylyl-β,γ-imidodiphosphate (AMPPNP) as an ATP ligand substitute for binding assays, when carried out in conjunction with R5P. Mt-PrsA was titrated with increasing [AMPPNP] using identical increments carried out during ATP ligand-binding experiments. Subsequent nonlinear regression analysis and comparison of the ΔFemission vs. [AMPPNP] and ΔFemission vs. [ATP] plots (data not shown) resulted in a strong correlation (R2 = 0.986) as well as extremely similar dissociation constants of 28.29 µM (±0.54 SE) and 31.02 µM (±0.62 SE) for ATP and AMPPNP, respectively. With the similarly calculated Hill coefficients (n) of ∼4, the almost identical binding properties of ATP and AMPPNP for Mt-PrsA allowed us to confidently substitute AMPPNP for ATP during binding assays carried out in combination with R5P. In this regard, when R5P was titrated against Mt-PrsA in the presence of 5 mM AMPPNP, the apparent Kd for R5P decreased to 21.36 µM (±4.84 SE). This data indicates that the Mt-PrsA–AMPPNP complex exhibits a slightly higher affinity for R5P when compared with Apo–Mt-PrsA (Figure 8A). In the reverse experiment where ΔFemission vs. [AMPPNP] was measured in the presence or the absence of 5 mM R5P (Figure 8B), the affinity of Mt-PrsA–R5P for AMPPNP increased slightly, as is evident from the reduction in Kd to 28.22 µM (±0.54 SE) and an increase in the Hill coefficient (n) to 5 by a value of 1. In light of the previous AUC experiment, which suggests that the addition of ADP induces a shift toward Mt-PrsA hexamerization, we performed an ITF experiment by titrating Mt-PrsA with increasing [ADP] (Figure 9A). Unsurprisingly, the ΔFemission vs. [ADP] plot displays a sigmoidal cooperative-binding response with an apparent Kd of 512.6 µM (±39.57 SE) and a Hill coefficient of 2. Since ATP and ADP bind to Mt-PrsA at separate sites and induce very different aggregation states, the apparent ∼2-fold difference in the Fmax recorded for ATP and ADP is most likely due to the differing effects each of these ligands have on the chemical environments surrounding the tryptophan residues providing the source of Femission in Mt-PrsA (Eriksen et al. 2000). The inclusion of ADP at two different concentrations (0.1 and 2.0 mM) in separate ITF [ATP] ligand-binding assays causes a significant increase in the apparent Kd of ATP for its Mt-PrsA-binding site (
, and the data were then plotted against [L]. Nonlinear regression analysis of ΔFemission vs. [R5P] using Eq. (7) shows a hyperbolic isothermal ligand-binding curve for R5P, with a calculated Kd of 24.8 µM (±6.77 SE; Figure 8A). We sought to investigate what effect ATP might have on the Kd for R5P; however, the inclusion of both ATP and R5P would obviously result in enzyme catalysis and the formation of pRpp and AMP. Due to the relatively high specific activity of Mt-PrsA, the addition of R5P and ATP at concentrations required for the effective measurement of ligand-binding constants by ITF would result in a reaction that would go to completion within the 5 min time-frame required to establish binding equilibrium. Therefore, due to the stability of the imidodiphophate moity and lack of enzyme-catalyzed phosphate transfer, we used the ATP analog 5'-adenylyl-β,γ-imidodiphosphate (AMPPNP) as an ATP ligand substitute for binding assays, when carried out in conjunction with R5P. Mt-PrsA was titrated with increasing [AMPPNP] using identical increments carried out during ATP ligand-binding experiments. Subsequent nonlinear regression analysis and comparison of the ΔFemission vs. [AMPPNP] and ΔFemission vs. [ATP] plots (data not shown) resulted in a strong correlation (R2 = 0.986) as well as extremely similar dissociation constants of 28.29 µM (±0.54 SE) and 31.02 µM (±0.62 SE) for ATP and AMPPNP, respectively. With the similarly calculated Hill coefficients (n) of ∼4, the almost identical binding properties of ATP and AMPPNP for Mt-PrsA allowed us to confidently substitute AMPPNP for ATP during binding assays carried out in combination with R5P. In this regard, when R5P was titrated against Mt-PrsA in the presence of 5 mM AMPPNP, the apparent Kd for R5P decreased to 21.36 µM (±4.84 SE). This data indicates that the Mt-PrsA–AMPPNP complex exhibits a slightly higher affinity for R5P when compared with Apo–Mt-PrsA (Figure 8A). In the reverse experiment where ΔFemission vs. [AMPPNP] was measured in the presence or the absence of 5 mM R5P (Figure 8B), the affinity of Mt-PrsA–R5P for AMPPNP increased slightly, as is evident from the reduction in Kd to 28.22 µM (±0.54 SE) and an increase in the Hill coefficient (n) to 5 by a value of 1. In light of the previous AUC experiment, which suggests that the addition of ADP induces a shift toward Mt-PrsA hexamerization, we performed an ITF experiment by titrating Mt-PrsA with increasing [ADP] (Figure 9A). Unsurprisingly, the ΔFemission vs. [ADP] plot displays a sigmoidal cooperative-binding response with an apparent Kd of 512.6 µM (±39.57 SE) and a Hill coefficient of 2. Since ATP and ADP bind to Mt-PrsA at separate sites and induce very different aggregation states, the apparent ∼2-fold difference in the Fmax recorded for ATP and ADP is most likely due to the differing effects each of these ligands have on the chemical environments surrounding the tryptophan residues providing the source of Femission in Mt-PrsA (Eriksen et al. 2000). The inclusion of ADP at two different concentrations (0.1 and 2.0 mM) in separate ITF [ATP] ligand-binding assays causes a significant increase in the apparent Kd of ATP for its Mt-PrsA-binding site (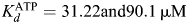 at a concentration of 0.1 and 2.0 mM ADP, respectively), clearly illustrating allosteric inhibition of Mt-PrsA–ATP complex formation (Figure 9B). The change in the slope of the curve as a function of [ATP] in separate ITF experiments carried out at increasing ADP concentrations, clearly illustrates a reduction in the homotropic cooperative binding of ATP for Mt-PrsA, which was confirmed by calculating the resultant Hill coefficients (Figure 9B). The ligand-binding properties of ADP for Mt-PrsA, in the presence of ATP, compare well with the inhibition constant calculated for ADP [Ki = 522 µM (±45.78 SE)] obtained from the kinetic analysis of ν vs. [ATP] at saturating [R5P] (Figure 6B).
at a concentration of 0.1 and 2.0 mM ADP, respectively), clearly illustrating allosteric inhibition of Mt-PrsA–ATP complex formation (Figure 9B). The change in the slope of the curve as a function of [ATP] in separate ITF experiments carried out at increasing ADP concentrations, clearly illustrates a reduction in the homotropic cooperative binding of ATP for Mt-PrsA, which was confirmed by calculating the resultant Hill coefficients (Figure 9B). The ligand-binding properties of ADP for Mt-PrsA, in the presence of ATP, compare well with the inhibition constant calculated for ADP [Ki = 522 µM (±45.78 SE)] obtained from the kinetic analysis of ν vs. [ATP] at saturating [R5P] (Figure 6B).
Fig. 7.

Mt-PrsA self-assembly analysis using analytical ultracentrifugation. Sedimentation velocity experiments were conducted to investigate the oligomeric state of Mt-PrsA in solution in the presence of no additional ligands (black line), 5 mM R5P (red line), 5 mM ATP (blue line) and 5 mM ADP (green line). Individual plots represent the molar mass distribution c(M) vs. the apparent molecular weight (Da). Arrows indicate the calculated oligomeric state of Mt-PrsA in each sedimentation velocity experiment.
Fig. 8.

Binding kinetics of R5P (A) and the ATP analog (AMPPNP) (B) to Mt-PrsA using ITF spectroscopy. ITF spectroscopy was used to determine the binding properties of Mt-PrsA for its substrates R5P and ATP. Ligand-binding assays were carried out as described in the Materials and methods section. (A) A plot of ΔFemission vs. [R5P] in the presence of no AMPPNP (filled squares) and 5 mM AMPPNP (filled circles) which was fitted to Eq. (7). (B) A plot of ΔFemission vs. [AMPPNP] in the presence of no R5P (filled squares) and 5 mM R5P (filled circles) which was fitted to Eq. (9).
Fig. 9.

Binding kinetics of ADP and ATP to Mt-PrsA using ITF spectroscopy. ITF spectroscopy was used to determine the binding properties of Mt-PrsA for it's allosteric inhibitor ADP in competition with ATP. Ligand-binding assays were carried out as described in the Materials and methods section. (A) A plot of ΔFemission vs. [ADP] (filled circles) which was fitted to Eq. (8). (B) A plot of ΔFemission vs. [ATP] in the presence of no ADP (filled squares), 0.1 mM ADP (filled triangles) and 2.0 mM ADP (filled inverted triangles) which was fitted to Eq. (10).
Structural model of Mt-PrsA
An alignment of Mt-PrsA with its related Corynebacterineae PrsA orthologs as well as other bacterial Prs homologs from Bacillus subtilis (Bs_PrsA), E. coli (Ec_PrsA) and Streptomyces coelicolor (Sc_PrsA), and the three human Prs isoforms (Human_PrsA1–3) clearly illustrate the high level of primary sequence conservation between all variants (Figure 10). Mt-PrsA shares very high sequence identity with its Corynebacterineae counterparts (92% Mycobacterium leprae, 87% M. smegmatis and 71% C. glutamicum) and a moderate level of sequence identity with Mt-PrsA homologs in S. coleicolor (61%), E. coli (43%) and B. subtilis (43%). Interestingly, the three human Prs isoforms also share 40% sequence identity with Mt-PrsA. Both of the human type 1 and B. subtilis pRpp synthetase X-ray crystal structures have been solved (Eriksen et al. 2000; Li et al. 2007). Each of these structures was solved in the Apo form and in complex with AMP, which is located in the ATP-binding pocket of the active site. Interestingly, the Bs-PrsA structure was solved in complex with an additional molecule of ADP, thus revealing its position and mode of action in the allosteric regulatory site of the enzyme (Eriksen et al. 2000). Since Mt-PrsA shares highest sequence homology with Bs-PrsA, we constructed a homology model of Mt-PrsA using the structure of Bs-Prs in complex with AMP and ADP as a template (Eriksen et al. 2000; Figure 11). Using crystallographic symmetry data from the Bs-Prs structure, we arranged the Mt-PrsA homology model into its hexameric form (Figure 11A). Each monomer is composed of two subunits, which is related by a 3-fold axis perpendicular to an additional 2-fold planar rotational symmetry. Many of the residues involved in binding at the interface between monomers are well conserved, with notable differences highlighted by the position of red spheres in Figure 11A. With only a few exceptions, the sequence alignment of Mt-PrsA and Bs-Prs indicates that most of the residues involved in enzyme catalysis and allosteric regulation are conserved which, in turn, are suitably positioned in the Mt-PrsA homology model compared with Bs-Prs. A noteworthy difference in the Mt-PrsA catalytic site is the presence of Asn45, which is arranged to form contacts with the ADP ligand (Figure 11B). In the Bs-Prs structure, the equivalent residue is Asp42. In addition, further comparison of the two structures suggests that the Mt-PrsA homology model retains all of the important residues required for the regulatory site, apart from Gly88 (Ala85 in Bs-Prs), which participates in the formation of contacts to ADP with a neighboring Ser89 (Figure 11B). An interesting observation in the model of Mt-PrsA is the presence of His109 in the “flexible loop” formed by Mt-PrsA residues Lys108–Arg112. Ala106 is the equivalent residue in Bs-Prs, and it appears to play little if no part in binding or coordination of the ADP ligand. This flexible loop region, which contains many conserved residues (Figure 10), has been identified as displaying some of the largest variation in backbone conformation, and the conformational changes that occur upon substrate binding are considered to be important for catalysis (Eriksen et al. 2000). The presence of a His residue in the Mt-PrsA flexible loop region might allow for multiple hydrogen bonding opportunities between His109 and the ATP substrate in the catalytic site (Figure 11B). Gly111 is another conserved residue located in the flexible loop of mycobacterial PrsA enzymes, which in the majority of other bacterial homologs is typically replaced by a Ser that makes H-bond interactions with the α-phosphate of ADP in the allosteric site (Figure 11C). Both the Human-PrsA1 and the Bs-PrsA homodimers are stabilized by a salt bridge at the N-terminal interface formed between two conserved residues (Asn114 and Asp139, and Asn120 and Asp145, respectively) (Eriksen et al. 2000; Li et al. 2007). From the sequence alignment and the Mt-PrsA homology model, it is worth noting that the equivalent Asn residue is replaced by an aspartic acid (Asp122) which would sit in close proximity to Asp147. Therefore, it is reasonable to assume that unlike Human-PrsA1 and Bs-PrsA, a salt bridge does not occur at the Mt-PrsA dimer interface. Interestingly, the Human-PrsA1 Asn114 (Asp122 in Mt-PrsA) is often mutated to a Ser in the cells of patients suffering from gout (Becker et al. 1995). It has been reported that the molecular mechanism behind the 50% increase in N114S mutant enzyme activity is caused by a disruption of a salt bridge, which would otherwise stabilizes the dimer interface (Liu et al. 2009).
Fig. 10.

Sequence alignment of various prokaryotic pRpp synthetases. Protein sequences from M. tuberculosis (Mt_PrsA), M. smegmatis (Ms_PrsA), M. leprae (Ml_PrsA), C. glutamicum (Cg_PrsA), S. coleicolor (Sc_PrsA), B. subtilis (Bs_PrsA), E. coli (Ec-PrsA) and the three PrsA isoforms (Human_PrsA1–3) were aligned using CLUSTALW and annotated with ESPRIPT. Triangles represent residues involved in catalysis. Filled circles indicate conserved and open circles highlight nonconserved residues involved in allosteric regulation. His109 and Gly111 are highlighted with stars, indicating two residues only found in the flexible loop region of Corynebacterianeae PrsA homologs. The diamonds indentify the location of the residues involved in the formation of a salt bridge in Bs-PrsA and Human-PrsA1.
Fig. 11.

Homology model of Mt-PrsA. (A) Mt-PrsA arranged into its hexameric quaternary conformation. Carbon atoms of the ADP ligands located at the catalytic and regulatory sites of the Mt-PrsA hexamer are colored in orange and green, respectively. Red spheres located at K10, D169, G194, D205-V208 and F322 indicate regions of the Mt-PrsA dimmer that include insertions or deletions with respect to the Bs-Prs structure. The triangle in the middle represents 3-fold symmetry, where as the arrow parallel to the plane indicates 2-fold rotational symmetry. Catalytic (B) and regulatory (C) sites of the Mt-PrsA homology model. The yellow and the blue spheres indicate A85/G88 and S109/G111 substitution between the Bs-Prs and Mt-PrsA structures, respectively.
Discussion
The presence of d-arabinose in living organisms is rarely observed. Whilst the utilization of d-arabinopyranose seems to be confined to glycoconjugate formation in trypanosomatid parasites (Turnock and Ferguson 2007), arabinose in the furanose configuration (Araf) is exclusively used by bacteria in the formation of cell surface polysaccharides and the biosynthesis of glycolipid antigens. Members belonging to the Corynebacterineae, such as M. tuberculosis and C. glutamicum, contain large quantities of Araf in the form of arabinan, which in turn, is covalently attached to galactan and mannan, thus forming AG and LAM, respectively. Whilst LAM exerts its biological activity by subverting the host immune response, the primary function of AG is to provide a highly branched molecular scaffold, linking the murein sacculus to the hydrophobic mycolate layer. Since arabinan is an essential component of the TB cell wall and is targeted by treatment with EMB, other enzymes involved in its formation might present themselves as potential putative drug targets. In this regard, DPPR synthetase (Rv3806c) is a novel enzyme catalyzing the transfer of pRpp to decaprenol-1-monphosphate forming DPPR (Figure 1), represents the first committed step toward the biosynthesis of DPA (an exclusive cell wall AraT substrate) and, as a consequence of its genetic disruption in C. glutamicum (a model organism used to study M. tuberculosis cell wall physiology), affords a phenotype displaying retarded growth, and a severely altered cell wall composition which is completely deficient in arabinan (Alderwick et al. 2005). In addition, DprE1 (Rv3790) has been shown to be the molecular target of benzothiazinones, a new class of antimycobacterial agents, which can effectively kill MDR and XDR strains of TB by blocking the C2–OH epimerization of DPR to DPA (Makarov et al. 2009). Some of the current front line drugs used to treat TB include cell wall inhibitors. However, the incidence of MDR and XDR-TB cases is alarmingly high and continues to rise (Madariaga et al. 2008). Since the M. tuberculosis cell wall is a valid drug target, we must continue to investigate the physiology, biochemistry and metabolic pathways involved in its assembly to assist our efforts of developing new anti-TB therapies. However, nondividing drug-tolerant populations of “persistent” TB bacilli fail to be eradicated from the host, giving rise to a latent infection as a result of subsequent granuloma formation (Barry et al. 2009). Inhibition of the cell wall biosynthetic processes of TB bacilli responsible for latent infections is of no consequence, this being due to the fact that persistent TB bacilli are not undergoing active cell growth and division. However, these “dormant” TB bacilli still require a basal level of “energy” metabolites in order to maintain critical cellular functions (Koul et al. 2008).
pRpp is a central metabolite ubiquitous in nature providing a high-energy substrate for the biosynthesis of histidine, tryphophan, NAD, NADP and both the de novo and the salvage pathways involved in nucleoside metabolism (Hove-Jensen 1988; Figure 1). In addition to these central metabolic processes, the Corynebacterineae have evolved a unique pathway in which pRpp is siphoned into cell wall arabinan biosynthesis (Figure 1). In M. tuberculosis, Mt-PrsA is entirely responsible for the provision of pRpp and its cellular function bridges five different metabolic pathways (Figure 1). The omphalic nature of Mt-PrsA places a heavy burden on its biological function, especially during cell growth and division, when the cellular requirement for pRpp increases significantly. In this study, we describe the biochemical characterization of Mt-PrsA, which displays the highest recorded specific activity for a pRpp synthetase of 530 µmol−1 min−1 mg−1. The Km for R5P was calculated to be in the submicromolar range of 8.2 µM, suggesting a rapid enzyme turnover with a calculated specificity constant (Kcat/Km) of 7.4 × 106 M−1 s−1. This relatively high efficiency of Mt-PrsA fits well with the physiological function of the enzyme during cell wall biosynthesis, considering the increased demand for pRpp that is required for antecedent DPA biosynthesis. Mt-PrsA displays clear homotropic cooperativity for ATP, with an apparent S0.5 62.65 µM, which is extremely low considering the intracellular concentration of ATP is considered to be between 3 and 5 mM. The homology model generated for Mt-PrsA contains many structural features of other pRpp synthetases that have been solved to date. The majority of the active site residues modeled in Mt-PrsA are conserved with those from other Mt-PrsA homologs, and it appears that the appropriate configuration of amino acid side chains compares well with the B. subtilis structure. This is also the case for the regulatory site in which ADP binds to allosterically modulate the enzyme. However, an interesting observation is the presence of His109 in the flexible loop region of Mt-PrsA, a residue exclusively conserved within members of the Corynebacterineae. Considering the close proximity of His109 to both the catalytic and the regulatory sites of Mt-PrsA, and its potential to make direct contacts with both ATP and ADP ligands, the presence of this conserved His in the flexible region of mycobacterial pRpp synthetases might provide some explanation for the relatively high Kcat. pRpp synthetase superactivity is observed in human type-1 N114S mutants (Becker et al. 1995). This increase in activity is a result of the loss of a salt bridge formed between two monomers which induces small but significant changes to the catalytic and regulatory sites of the enzyme (Liu et al. 2009). Interestingly, the homology model of Mt-PrsA suggests that a salt bridge between the equivalent residues does not occur thus reducing the stability of the dimer interface. In mycobacteria, the biosynthesis of pRpp is required at a rate which is sufficient to not only supply central metabolic processes, such as de novo nucleoside biosynthesis, but also at a necessary level to meet the increased demands for pRpp during cell wall biosynthesis (Scherman et al. 1995, 1996). It seems plausible that pRpp synthetases from Corynebacterineae species have evolved a highly efficient version of the enzyme to account for the increased metabolic flux of pRpp, during various physiological states. pRpp synthetases in other organisms undergo stringent control by virtue of allosteric modulation from ADP. In this regard, Mt-PrsA also appears to be no different, displaying clear inhibition of enzyme activity between 0.5 and 2.0 mM ADP. When taken together, the data relating to ADP inhibition and the subsequent complex formation with Mt-PrsA suggest that the mechanism employed for the allosteric regulation of Mt-PrsA by ADP is through the formation of an Mt-PrsA hexamer (Arnvig et al. 1990). This net increase in Mt-PrsA hexamer oligomerization suggests that the efficacy of ATP for its Mt-PrsA catalytic site decreases, as a consequence of ADP allosteric modulation. The three human PrsA isoforms share a very high sequence identity (91–95% between all three); however, the subtle differences that do occur appear in the regions of the enzyme that are important for catalysis and allosteric regulation. Inhibition studies using two aminopyrimidopyrimidine nucleotide analogs 4-methoxy-8-(β-d-ribofuranosylamino)-pyrimido-[5,4-d]-pyrimidine and 4-amino-8-(β-d-ribofuranosylamino) -pyrimido-[5,4-d]-pyrimidine show differential levels of inhibition of human pRpp synthetases (Fry et al. 1995). Carbocyclic analogs of R5P are another class of compounds that show differential inhibition when tested against Salmonella typhimurium, B. subtilis and human pRpp synthetases (Parry et al. 1996). In this regard, the development of a similar library of compounds targeted for the specific inhibition of Mt-PrsA might warrant further investigation. Understanding the metabolic pathways that are essential for persistent TB infections, such as mycobacterial energy metabolism, in combination with pathways involved in cell wall assembly seems a plausible strategy for the development of new antimycobacterial drugs.
Materials and methods
Bacterial strains cloning procedures
Escherichia coli top 10 cells (Invitrogen) were used to propagate plasmids during all cloning experiments, and the overexpression of Mt-PrsA was carried out in E. coli C41 (DE3) (Lucigen). Phusion DNA polymerase was purchased from New England Biolabs, and all restriction enzymes and T4 DNA ligase were purchased from Fermentas. Oligonucleotides were purchased from Eurofins-MWG and PCR fragments were purified using QIAquick Gel Extraction Kit (Qiagen). Plasmids were purified using the Qiaprep Purification Kit (Qiagen).
Generation of C. glutamicum prsA-null mutant
For the genomic disruption of NCgl0945 (Cg-prsA), we constructed the plasmid pK18mob-Cg-prsA-int, using the primer pair pNCgl0945_intfor (GCGAGCCAATTTCTGCTCGC) and pNCgl0945_intrev (GACAACCTGGTTTGCTACCTC), to amplify C. glutamicum WT chromosomal DNA. The resulting DNA fragment was treated with AvaI/KpnI and ligated into the nonreplicative vector pK18mob (Schafer et al. 1994), which was similarly treated with AvaI/KpnI. The DNA integrity of the resulting construct pK18mob-Cg-prsA-int was confirmed by sequencing and used for the transformation of C. glutamicum by electroporation and selected for resistance to kanamycin at 25 µg/mL on BHIS medium.
Construction of Mt-PrsA expression vector
The 981 bp coding region for Mt-PrsA (annotated as Rv1017c) in the M. tuberculosis H37Rv genome was amplified by PCR using the following primer pairs (restriction sites underlined) GATCGATCGCTAGCTTGAGCCACGACTGGACCGATAATCGC (forward) and GATCGATCCTCGAGTGCGTCCCCGTCGAAAAGTCCTGTTAC (reverse). This DNA fragment was restricted with the appropriate enzymes (NheI and XhoI) and ligated into pET23b digested with identical enzymes, thus yielding pET23b-Mt-prsA, which encodes for a C-terminal His6-tagged protein. DNA sequencing and construct verification were carried out at the University of Birmingham School of Bioscience Genomics Facility.
Expression and purification of Mt-PrsA
Escherichia coli C41 (DE3) cells were transformed with pET23b-Mt-prsA and selected on the LB agar supplemented with 100 µg/mL of amplicillin. Recombinant E. coli cells harboring pET23b-Mt-prsA were used to inoculate an overnight of 5 mL LB supplemented with 100 µg/mL of amplicillin. This overnight was then used to inoculate 2 × 1 L LB medium supplemented with 100 µg/mL of amplicillin and incubated at 37°C with shaking until A600 reached 0.5. IPTG was then added at a final concentration of 1 mM and growth continued for an additional 12 h at 16°C with shaking. Cells were harvested by centrifugation at 6000 × g for 15 min and washed with 20 mL of phosphate buffered saline. Pellets were then frozen until further use. For purification, one pellet was thawed and resuspended into 50 mM KH2PO4 (pH 7.9), 300 mM NaCl, 20 mM imidazole, one complete protease inhibitor cocktail tablet (Roche) and DNAse and RNAse both at a final concentration of 50 µg/mL. The cell suspension was disrupted by sonication at a pulse rate of 30 s ON and 30 s OFF for a total of 10 cycles. The cell slurry was centrifuged for 30 min, 28,000 × g at a temperature of 4°C. The supernatant was collected and passed over a 5 mL HiTrap Ni2+-NTA agarose column (GE Healthcare), which was previously equilibrated with 50 mM KH2PO4 (pH 7.9), 300 mM NaCl and 20 mM imidazole. Elution occurred via a stepwise gradient of 50–500 mM imidazole in 50 mM KH2PO4 (pH 7.9), 300 mM NaCl and 10 mL of fractions were collected. Mt-PrsA was detected to be present and ∼ 98% pure in the 250 mM imidazole fraction, as determined by 12% SDS–PAGE analysis. The 250 mM imidazole fractions of Mt-PrsA was first dialyzed against 2 L of KH2PO4 (pH 7.9), 150 mM NaCl, 5 mM EDTA and 1 mM DTT at 4°C and then against 2 L of KH2PO4 (pH 7.9), 150 mM NaCl, 1 mM DTT and 10% glycerol. After dialysis, the protein concentration was determined using the bicinchoninic acid procedure (Pierce) and bovine serum albumin as a standard.
Preparation of [14C]-R5P
We prepared a source of [14C]-R5P using a modified process as described in Boss et al. (1984), for the subsequent use in Mt-PrsA assays. One hundred microcuries of [14C]-U-d- glucose (100 µL in 70% ethanol; ARC Radiochemicals) was dried in a 14 mL glass tube under compressed nitrogen and then resuspended in 500 µL of 50 mM KH2PO4 (pH 7.9), 150 mM NaCl, 5 mM MgCl2 and 0.5 mM MnCl2. Twenty microliters of ATP (100 mM stock) and 20 µL of NADP (100 mM stock) was added to the suspension. Twenty microliters of hexokinase/glucose-6-phosphate dehydrogenase enzyme mix (Roche) was added and incubated at 37°C for 15 min. A few grains of 6-phosphogluconic dehydrogenase (Sigma) and a further 20 µL of NADP (100 mM stock) were added and incubated for 15 min at 37°C. A few grains of phosphoribose mutase (Sigma) were added to the reaction mix and incubated for a further 15 min at 37°C followed by another addition of 20 µL of NADP (100 mM stock) and final incubation at 37°C for 15 min. The reaction mix was then diluted with H2O to a final volume of 3 mL and passed through a 10 kDa MWCO centrifugal filtration device (Millipore). The filtrate (∼3 mL) was then loaded onto a 3 mL LC-SAX anion exchange cartridge (Supelco) pre-equilibrated with H2O. The column was then washed three times with 3 mL of H2O. The column was then washed with a 100–1000 mM stepwise gradient of sodium acetate (3 mL of fractions of 100 mM increments). Each fraction (1 µL) was applied to a glass-backed PEI-cellulose TLC plate and developed in 0.85 M KH2PO4 (pH 3.4). After drying, the TLC plate was exposed overnight to a Kodak autoradiography film for visualization of radioactive phospho-sugars. [14C]-R5P eluted as a single band (when compared with the known standards) in the 200 mM sodium acetate fraction with a specific activity of 33,333 cpm/µL.
Radiochemical assay of Mt-PrsA activity
The enzymatic activity of Mt-PrsA was determined by monitoring the transfer of PPi from ATP to the C1–OH group of enzymatically synthesized [14C]-R5P forming p[14C]Rpp and AMP. The basic reaction mix contained 50 mM KH2PO4 (pH 7.5), 150 mM NaCl, 1 mM DTT, 10% glycerol, varying amounts of [14C]-d-R5P [stored in 200 mM sodium acetate (33,333 cpm/µL)], 2 mM ATP and 30 µg of Mt-PrsA (15 µL) in a final volume of 100 µL. On occasions, MgCl2, MnCl2, CaCl2, ADP and EDTA were included in the reaction mix either separately or in combination, all at a final concentration of 2 mM. Reactions were initiated by the addition of Mt-PrsA and incubated at 37°C for 10 min. Reactions were quenched by the addition of 10 µL of 0.4 M formic acid. The mixture (1.1 µL) was applied to a glass-backed PEI-cellulose TLC plate and developed in 0.85 M KH2PO4 (pH 3.4). After drying, the plate was inspected for radioactive spots by either exposure to a Kodak film followed by autoradiography or a PhosphorImager (Molecular Dynamics). All experiments were repeated in triplicate.
Metabolism of [14C]-R5P into mycobacterial cell wall biosynthetic precursors
Membranes (containing membrane bound enzymes involved in lipid-linked cell wall biosynthetic processes) and “P60” [a percoll-derived cell-free fraction-rich carbohydrate and enzymes associated with cell wall processes as described in Besra et al. (1997)] fractions from M. smegmatis were prepared as described previously (Besra et al. 1997; Lee et al. 1997; Alderwick, Dover, et al. 2006) and resuspended in buffer A [50 mM MOPS (pH 7.9), containing 5 mM β-mercaptoethanol and 10 mM MgCl2] to a final concentration of 15 and 10 mg/mL for membrane and P60 fractions, respectively. Fifty micrograms of DP [Larodan Lipids, Malmo, Sweden; 5 mg/mL stored in CHCl3/CH3OH (2:1, v/v)] was dried in a 1.5 mL Eppendorf tube under compressed nitrogen and was resuspended by the addition of 10 µL of a solution of 50 mM MOPS (pH 7.9), 50 mM KH2PO4, 150 mM NaCl, 1 mM MnCl2, 1 mM MgCl2, 1 mM DTT and 1% IgePal CA-630 (Sigma) followed by bath sonication. The basic assay mix consisted of 0.5 mg of M. smegmatis membranes (33 µL), 2 mM ATP, 50 mM MOPS (pH 7.9), 50 mM KH2PO4, 150 mM NaCl, 1 mM MnCl2, 1 mM MgCl2, 1 mM DTT, 100,000 cpm [14C]-R5P (3 µL) in a final volume of 200 µL. NaWO4 and NaF as well as ADP were included in separate assay reactions at a final concentration of 2 mM for all inhibition studies. 0.5 mg of M. smegmatis P60 (50 µL) was added where appropriate. Reactions were initiated by the addition of 30 µg of Mt-PrsA (15 µL) and incubated at 37°C for 30 min. Assays were quenched by the addition of 1.33 mL of CHCl3/CH3OH (1:1, v/v), mixed for 30 min and centrifuged at 27,000 × g to remove the precipitated proteinaceous material. The supernatant was removed to a separate tube and combined with 670 µL of CHCl3 and 287 µL of H2O, mixed for 15 min and centrifuged for 5 min at 4000 × g to form a biphase. The lower organic phase was removed and washed twice with 765 µL of CHCl3/CH3OH/H2O (3:47:48, v/v/v) and then dried under compressed nitrogen. Samples were then resuspended in 100 µL of CHCl3/CH3OH (2:1, v/v) and an aliquot subject to TLC analysis using silica gel plates (5735 silica gel 60F254, Merck) developed in CHCl3/CH3OH/CH3COONH4/NH4OH/H2O (180:140:9:9:23, v/v/v/v/v) and visualized by autoradiography by exposure of TLCs to X-ray film (Kodak X-Omat) as described in Mikusova et al. (2005).
Metabolism of [14C]-R5P into mycobacterial cell wall biosynthetic precursors and the subsequent AraT-dependent transfer to a neoglycolipid acceptor.
Membranes and P60 from M. smegmatis were prepared as described in Materials and Methods. The neoglycolipid acceptor used in this study was the disaccharide α-d-Araf-(1 → 5)-α-d-Araf-O-(CH2)7CH3 (Ara2), which has been previously reported by us (Lee et al. 1997; Seidel et al. 2007). In individual assays, the 8 µL of Ara2 acceptor (stored in CHCl3/CH3OH, 2:1, v/v at 20 mM) and 50 µg of DP [Larodan Lipids; 5 mg/mL stored in CHCl3/CH3OH (2:1, v/v)] was aliquoted into 1.5 mL Eppendorf tubes and dried under nitrogen. The AraT assay was carried out as described previously (Lee et al. 1997; Seidel et al. 2007) with modifications. IgePal™ (Sigma-Aldrich) was added (0.1%, v/v) with the appropriate amount of buffer A (final volume of 100 µL). Tubes were sonicated for 15 min to resuspend lipid-linked substrates and then mixed with the remaining assay components, which included 0.5 mg of M. smegmatis membranes (33 µL), 0.5 mg of M. smegmatis P60 (50 µL), 30 µg of Mt-PrsA (15 µL), 1 mM ATP, 1 mM NADP and in some cases EMB (100 µg/mL). Assays were incubated for 1 h at 37°C and quenched by the addition of 633 µL of CHCl3/CH3OH (1:1, v/v). After mixing and centrifugation at 27,000 × g for 15 min at 4°C, the supernatant was removed and dried under nitrogen. The residue was then resuspended in 700 µL of CH3CH2OH/H2O (1:1, v/v) and loaded onto a 1 mL LC-SAX strong anion exchange cartridge (Supelco), pre-equilibrated with CH3CH2OH/H2O (1:1, v/v). The column was washed with 2 mL of CH3CH2OH and the eluate collected, dried and partitioned between the two phases arising from a mixture of n-butanol (3 mL) and water (3 mL). The resulting organic phase was recovered following centrifugation at 3500 × g and the aqueous phase again extracted twice with 3 mL of water-saturated n-butanol. The pooled extracts were back-washed twice with n-butanol-saturated water (3 mL). The n-butanol fraction was dried and resuspended in 200 µL of butanol. The extracted radiolabeled material was quantified by liquid scintillation counting using 10% of the labeled material and 5 mL of EcoScintA (National Diagnostics, Atlanta). The incorporation of [14C]-Araf into Ara2 was determined by subtracting counts present in control assays (incubations in the absence of acceptor). The remaining labeled material was subjected to TLC using CHCl3:CH2OH:H2O:NH4OH (65:25:3.6:0.5, v/v/v/v) on aluminum-backed silica gel plates (5735 silica gel 60F254, Merck) and products were visualized by autoradiography, exposing the TLCs to X-ray film (Kodak X-Omat).
Continuous enzyme-coupled spectrophotometric assay and kinetic characterization of Mt-PrsA substrates
By using a modified assay described by Braven et al. (1984), the amount of AMP produced as a product of Mt-PrsA catalysis was measured indirectly by monitoring the decrease in the absorbance of NADH at 340 nm via the following enzyme couple: 1. R5P + ATP → pRpp + AMP, catalyzed by Mt-PrsA; 2. AMP + ATP → 2 ADP, catalyzed by myokinase (MK); 3. 2 ADP + 2 phosphoenolpyruvate (PEP) → 2 pyruvate + 2 ATP, catalyzed by pyruvate kinase (PK) and 4. 2 pyruvate + 2 NADH + 2 H+ → 2 lactate + 2 NAD+, catalyzed by lactate dehydrogenase (LDH). This perpetual enzyme couple immediately replenishes the pool of ATP and results in a stoichiometry of 1 mol NADH oxidized by LDH for every 2 mol AMP produced by Mt-PrsA. The following formula was used to calculate the rate of reaction thus allowing for a detailed kinetic characterization of Mt-PrsA
| (1) |
where ν is the steady-state rate of the reaction, ΔA the change in absorbance at 340 nm and 6220 M−1 the molecular extinction coefficient of NADH measured at 340 nm. The reaction mixture comprised of 50 mM KH2PO4 (pH 7.9), 150 mM NaCl, 0.25 mM DTT, 2 mM MnCl2, 2.0 mM PEP, 1.0 mM NADH and 5 mM NaF, MK, PK and LDH were added to the mix all at a concentration of 0.2 U/mL. Variable amounts of ATP, R5P and ADP were added to the reaction mix for enzyme kinetic and inhibitor studies. Reactions were initiated by the addition of R5P. All kinetic experiments were carried out in triplicate (from three different independent protein preparations of purified recombinant Mt-PrsA), and data points (calculated mean) and standard errors (±SE) relating to the enzyme steady-state velocities (ν) (µmol min−1 mg−1) derived from Eq. (1) under various substrate and inhibitor conditions were analyzed by nonlinear regression analysis and fitted to the appropriate Eqs. (2)–(6) using the GraphPad Prism software package. Eq. (2) is the Michelis–Menten equation for hyperbolic substrate saturation kinetics and Eq. (3) applies to the nonlinear regression of substrate inhibition. Eq. (4) is the Hill equation for calculating cooperative substrate saturation kinetics, Eq. (5) applies to noncompetitive enzyme inhibition of a hyperbolic enzyme response and Eq. (6) is derived from combining Eqs. (4) and (5) to give an expression for noncompetitive enzyme inhibition of a cooperative sigmoidal response to increasing substrate concentrations.
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
where Vmax is the maximum enzyme velocity, Vapp the apparent maximal enzyme velocity, S the concentration of the substrate being varied, I the inhibitor concentration, Km the apparent Michelis–Menten constant for S, S0.5 the half-saturation concentration for S, n the apparent Hill coefficient for S, Kis and Kii the dissociation constants of the [I] for E and ES, respectively.
Analytical ultracentrifugation
Sedimentation velocity experiments were performed using a Beckman Optima XL-A analytical ultracentrifuge equipped with absorbance optics. Mt-PrsA [dialyzed into 50 mM KH2PO4 (pH 7.9), 300 mM NaCl, 1 mM DTT] was loaded into cells with two channel Epon center pieces and quartz windows. A total of 100 absorbance scans (280 nm) were recorded (40,000 rpm, 4°C) for each sample, representing the full extent of sedimentation of the sample. Data analysis was performed using the SEDFIT software fitting a single friction coefficient (Schuck 2004).
ITF spectroscopy
ITF experiments were carried out using a PTI QuantaMaster™ 40 spectrofluorimeter and data recorded using the FeliX32 software. The excitation wavelength was set at 292 nm, and the fluorescence emission (Femission) spectra were recorded between 300 and 400 nm for each ligand aliquot added to a 200 µL solution containing 70 µM Mt-PrsA. 50 mM KH2PO4 (pH 7.9), 150 mM NaCl, 0.25 mM DTT, 2 mM MnCl2 was used as a buffer for the measurement of R5P, ATP and AMPPNP ligand binding (or a combination) to Mt-PrsA. A plot of the change in fluorescence emission (ΔFemission) intensity at λ335nm vs. [L] was fitted to Eq. (7) for R5P, Eq. (8) for ATP and ADP for independent additions, Eq. (9) for R5P in combination with AMPPNP and Eq. (10) for ATP and ADP in combination using GraphPad Prism software
| (7) |
| (8) |
| (9) |
| (10) |
where Fmax indicates the maximum change in fluorescence emission, Kd the concentration of ligand required to achieve half maximum fluorescence, L the concentration of ligand and n the Hill coefficient. Both a and b relate to the relative parameters determined for two separate ligands included in a binding assay.
Generation of a Mt-PrsA homology model
A homology model of the molecular structure of Mt-PrsA was derived based on the crystal structure of B. subtilis Prs (Eriksen et al. 2000). Models were obtained from several web-based modeling servers, including SWISS-MODEL (Schwede et al. 2003), Geno3D (Combet et al. 2002) and 3D-JIGSAW (Bates et al. 2001). The homology models were analyzed in comparison with the Bs-Prs structures in complex with SO4− or mADP (PDB entry 1DKR and 1DKU; Eriksen et al. 2000).
Conflict of interest
None declared.
Abbreviations
AG, arabinogalactan; AMPPNP, ATP analog 5'-adenylyl-β, γ-imidodiphosphate; Ara2, α-d-Araf-(1 → 5)-α-d-Araf-O- (CH2)7CH3; AraT, arabinofuranosyltransferase; [14C]-R5P, [14C]-U-d-R5P; DP, decaprenol-1-monophosphate; DPA, decaprenol-1-monophosphorarabinose; DPR, decaprenol-1-monophosphoribose; DPPR, decaprenol-1-monophosphoribose-5-phosphate; DPX, decaprenol-1-monophosphoryl-2-keto-β-erythro-pentofuranose; EMB, ethambutol; ITF, intrinsic tryptophan fluorescence; LAM, lipoarabinomannan; LDH, lactate dehydrogenase; MDR, multidrug resistant; MK, myokinase; Mt-PrsA, Mycobacterium tuberculosis phosphoribosyl-1-pyrophosphate synthetase; PEP, phosphoenolpyruvate; PK, pyruvate kinase; pRpp, phosphoribosyl-1-pyrophosphate; R5P, ribose-5-phosphate; TB, tuberculosis; WT, wild type; XDR, extensively drug resistant.
Acknowledgements
G.S.B. acknowledges support in the form of a Personal Research Chair from Mr. James Bardrick, Royal Society Wolfson Research Merit Award, as a former Lister Institute-Jenner Research Fellow, the Medical Research Council and The Wellcome Trust (081569/Z/06/Z).
References
- Alderwick LJ, Birch HL, Mishra AK, Eggeling L, Besra GS. Structure, function and biosynthesis of the Mycobacterium tuberculosis cell wall: Arabinogalactan and lipoarabinomannan assembly with a view to discovering new drug targets. Biochem Soc Trans. 2007;35:1325–1328. doi: 10.1042/BST0351325. doi:10.1042/BST0351325. [DOI] [PubMed] [Google Scholar]
- Alderwick LJ, Dover LG, Seidel M, Gande R, Sahm H, Eggeling L, Besra GS. Arabinan-deficient mutants of Corynebacterium glutamicum and the consequent flux in decaprenylmonophosphoryl-d-arabinose metabolism. Glycobiology. 2006;16:1073–1081. doi: 10.1093/glycob/cwl030. doi:10.1093/glycob/cwl030. [DOI] [PubMed] [Google Scholar]
- Alderwick LJ, Radmacher E, Seidel M, Gande R, Hitchen PG, Morris HR, Dell A, Sahm H, Eggeling L, Besra GS, et al. Deletion of Cg-emb in Corynebacterianeae leads to a novel truncated cell wall arabinogalactan, whereas inactivation of Cg-ubiA results in an arabinan-deficient mutant with a cell wall galactan core. J Biol Chem. 2005;280:32362–32371. doi: 10.1074/jbc.M506339200. doi:10.1074/jbc.M506339200. [DOI] [PubMed] [Google Scholar]
- Alderwick LJ, Seidel M, Sahm H, Besra GS, Eggeling L. Identification of a novel arabinofuranosyltransferase (AftA) involved in cell wall arabinan biosynthesis in Mycobacterium tuberculosis. J Biol Chem. 2006;281:15653–15661. doi: 10.1074/jbc.M600045200. doi:10.1074/jbc.M600045200. [DOI] [PubMed] [Google Scholar]
- Arnvig K, Hove-Jensen B, Switzer RL. Purification and properties of phosphoribosyl-diphosphate synthetase from Bacillus subtilis. Eur J Biochem. 1990;192:195–200. doi: 10.1111/j.1432-1033.1990.tb19214.x. doi:10.1111/j.1432-1033.1990.tb19214.x. [DOI] [PubMed] [Google Scholar]
- Bald D, Koul A. Respiratory ATP synthesis: The new generation of mycobacterial drug targets? FEMS Microbiol Lett. 2010;308:1–7. doi: 10.1111/j.1574-6968.2010.01959.x. doi:10.1111/j.1574-6968.2010.01959.x. [DOI] [PubMed] [Google Scholar]
- Banerjee A, Dubnau E, Quemard A, Balasubramanian V, Um KS, Wilson T, Collins D, de Lisle G, Jacobs WR., Jr inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science. 1994;263:227–230. doi: 10.1126/science.8284673. [DOI] [PubMed] [Google Scholar]
- Barry CE, 3rd, Boshoff HI, Dartois V, Dick T, Ehrt S, Flynn J, Schnappinger D, Wilkinson RJ, Young D. The spectrum of latent tuberculosis: Rethinking the biology and intervention strategies. Nat Rev. 2009;7:845–855. doi: 10.1038/nrmicro2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates PA, Kelley LA, MacCallum RM, Sternberg MJ. Enhancement of protein modeling by human intervention in applying the automatic programs 3D-JIGSAW and 3D-PSSM. Proteins. 2001;45(Suppl. 5):39–46. doi: 10.1002/prot.1168. doi:10.1002/prot.1168. [DOI] [PubMed] [Google Scholar]
- Becker MA, Smith PR, Taylor W, Mustafi R, Switzer RL. The genetic and functional basis of purine nucleotide feedback-resistant phosphoribosylpyrophosphate synthetase superactivity. J Clin Invest. 1995;96:2133–2141. doi: 10.1172/JCI118267. doi:10.1172/JCI118267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger AE, Besra GS, Ford ME, Mikusova K, Belisle JT, Brennan PJ, Inamine JM. The embAB genes of Mycobacterium avium encode an arabinosyl transferase involved in cell wall arabinan biosynthesis that is the target for the antimycobacterial drug ethambutol. Proc Natl Acad Sci USA. 1996;93:11919–11924. doi: 10.1073/pnas.93.21.11919. doi:10.1073/pnas.93.21.11919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besra GS, Khoo KH, McNeil MR, Dell A, Morris HR, Brennan PJ. A new interpretation of the structure of the mycolyl-arabinogalactan complex of Mycobacterium tuberculosis as revealed through characterization of oligoglycosylalditol fragments by fast-atom bombardment mass spectrometry and 1H nuclear magnetic resonance spectroscopy. Biochemistry. 1995;34:4257–4266. doi: 10.1021/bi00013a015. doi:10.1021/bi00013a015. [DOI] [PubMed] [Google Scholar]
- Besra GS, Morehouse CB, Rittner CM, Waechter CJ, Brennan PJ. Biosynthesis of mycobacterial lipoarabinomannan. J Biol Chem. 1997;272:18460–18466. doi: 10.1074/jbc.272.29.18460. doi:10.1074/jbc.272.29.18460. [DOI] [PubMed] [Google Scholar]
- Birch HL, Alderwick LJ, Bhatt A other authors. Biosynthesis of mycobacterial arabinogalactan: Identification of a novel α(1→3) arabinofuranosyltransferase. Mol Microbiol. 2008;69:1191–1206. doi: 10.1111/j.1365-2958.2008.06354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boss GR, Idriss SD, Willis RC, Seegmiller JE. Synthesis of (14C)-ribose-5-phosphate and (14C)-phosphoribosylpyrophosphate and their use in new enzyme assays. Adv Exp Med Biol. 1984;165(Pt B):11–13. doi: 10.1007/978-1-4757-0390-0_3. [DOI] [PubMed] [Google Scholar]
- Braven J, Hardwell TR, Seddon R, Whittaker M. A spectrophotometric assay of phosphoribosyl pyrophosphate synthetase. Ann Clin Biochem. 1984;21(Pt 5):366–371. doi: 10.1177/000456328402100505. [DOI] [PubMed] [Google Scholar]
- Briken V, Porcelli SA, Besra GS, Kremer L. Mycobacterial lipoarabinomannan and related lipoglycans: From biogenesis to modulation of the immune response. Mol Microbiol. 2004;53:391–403. doi: 10.1111/j.1365-2958.2004.04183.x. doi:10.1111/j.1365-2958.2004.04183.x. [DOI] [PubMed] [Google Scholar]
- Carter AT, Beiche F, Hove-Jensen B, Narbad A, Barker PJ, Schweizer LM, Schweizer M. PRS1 is a key member of the gene family encoding phosphoribosylpyrophosphate synthetase in Saccharomyces cerevisiae. Mol Gen Genet. 1997;254:148–156. doi: 10.1007/s004380050402. doi:10.1007/s004380050402. [DOI] [PubMed] [Google Scholar]
- Cole ST, Barrell BG. Analysis of the genome of Mycobacterium tuberculosis H37Rv. Novartis Found Symp. 1998;217:160–172. discussion 172–177 doi:10.1002/0470846526.ch12. [PubMed] [Google Scholar]
- Combet C, Jambon M, Deleage G, Geourjon C. Geno3D: Automatic comparative molecular modelling of protein. Bioinformatics. 2002;18:213–214. doi: 10.1093/bioinformatics/18.1.213. doi:10.1093/bioinformatics/18.1.213. [DOI] [PubMed] [Google Scholar]
- Daffé M, Brennan PJ, McNeil M. Predominant structural features of the cell wall arabinogalactan of Mycobacterium tuberculosis as revealed through characterization of oligoglycosyl alditol fragments by gas chromatography/mass spectrometry and by 1H and 13C NMR analyses. J Biol Chem. 1990;265:6734–6743. [PubMed] [Google Scholar]
- Dye C. Global epidemiology of tuberculosis. Lancet. 2006;367:938–940. doi: 10.1016/S0140-6736(06)68384-0. doi:10.1016/S0140-6736(06)68384-0. [DOI] [PubMed] [Google Scholar]
- Eriksen TA, Kadziola A, Bentsen AK, Harlow KW, Larsen S. Structural basis for the function of Bacillus subtilis phosphoribosyl-pyrophosphate synthetase. Nat Struct Biol. 2000;7:303–308. doi: 10.1038/74069. doi:10.1038/74069. [DOI] [PubMed] [Google Scholar]
- Fry DW, Becker MA, Switzer RL. Inhibition of human 5-phosphoribosyl-1-pyrophosphate synthetase by 4-amino-8-(beta- D-ribofuranosylamino)-pyrimido[5,4-d]pyrimidine-5'-monophosphate: Evidence for interaction at the ADP allosteric site. Mol Pharmacol. 1995;47:810–815. [PubMed] [Google Scholar]
- Gande R, Gibson KJ, Brown AK other authors. Acyl-CoA carboxylases (accD2 and accD3), together with a unique polyketide synthase (Cg-pks), are key to mycolic acid biosynthesis in Corynebacterianeae such as Corynebacterium glutamicum and Mycobacterium tuberculosis. J Biol Chem. 2004;279:44847–44857. doi: 10.1074/jbc.M408648200. doi:10.1074/jbc.M408648200. [DOI] [PubMed] [Google Scholar]
- Gibson KJ, Eggeling L, Maughan WN, Krumbach K, Gurcha SS, Nigou J, Puzo G, Sahm H, Besra GS. Disruption of Cg-Ppm1, a polyprenyl monophosphomannose synthase, and the generation of lipoglycan-less mutants in Corynebacterium glutamicum. J Biol Chem. 2003;278:40842–40850. doi: 10.1074/jbc.M307988200. doi:10.1074/jbc.M307988200. [DOI] [PubMed] [Google Scholar]
- Gibson KJ, Schubert KR, Switzer RL. Binding of the substrates and the allosteric inhibitor adenosine 5'-diphosphate to phosphoribosylpyrophosphate synthetase from Salmonella typhimurium. J Biol Chem. 1982;257:2391–2396. [PubMed] [Google Scholar]
- Hove-Jensen B. Cloning and characterization of the prs gene encoding phosphoribosylpyrophosphate synthetase of Escherichia coli. Mol Gen Genet. 1985;201:269–276. doi: 10.1007/BF00425670. doi:10.1007/BF00425670. [DOI] [PubMed] [Google Scholar]
- Hove-Jensen B. Mutation in the phosphoribosylpyrophosphate synthetase gene (prs) that results in simultaneous requirements for purine and pyrimidine nucleosides, nicotinamide nucleotide, histidine, and tryptophan in Escherichia coli. J Bacteriol. 1988;170:1148–1152. doi: 10.1128/jb.170.3.1148-1152.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hove-Jensen B. Phosphoribosylpyrophosphate (PRPP)-less mutants of Escherichia coli. Mol Microbiol. 1989;3:1487–1492. doi: 10.1111/j.1365-2958.1989.tb00134.x. doi:10.1111/j.1365-2958.1989.tb00134.x. [DOI] [PubMed] [Google Scholar]
- Huang H, Scherman MS, D'Haeze W, Vereecke D, Holsters M, Crick DC, McNeil MR. Identification and active expression of the Mycobacterium tuberculosis gene encoding 5-phospho-α-D-ribose-1-diphosphate: Decaprenyl-phosphate 5-phosphoribosyltransferase, the first enzyme committed to decaprenylphosphoryl-D-arabinose synthesis. J Biol Chem. 2005;280:24539–24543. doi: 10.1074/jbc.M504068200. doi:10.1074/jbc.M504068200. [DOI] [PubMed] [Google Scholar]
- Koul A, Dendouga N, Vergauwen K other authors. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat Chem Biol. 2007;3:323–324. doi: 10.1038/nchembio884. doi:10.1038/nchembio884. [DOI] [PubMed] [Google Scholar]
- Koul A, Vranckx L, Dendouga N other authors. Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J Biol Chem. 2008;283:25273–25280. doi: 10.1074/jbc.M803899200. doi:10.1074/jbc.M803899200. [DOI] [PubMed] [Google Scholar]
- Krath BN, Hove-Jensen B. Organellar and cytosolic localization of four phosphoribosyl diphosphate synthase isozymes in spinach. Plant Physiol. 1999;119:497–506. doi: 10.1104/pp.119.2.497. doi:10.1104/pp.119.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RE, Brennan PJ, Besra GS. Mycobacterial arabinan biosynthesis: The use of synthetic arabinoside acceptors in the development of an arabinosyl transfer assay. Glycobiology. 1997;7:1121–1128. doi: 10.1093/glycob/7.8.1121. doi:10.1093/glycob/7.8.1121. [DOI] [PubMed] [Google Scholar]
- Li S, Lu Y, Peng B, Ding J. Crystal structure of human phosphoribosylpyrophosphate synthetase 1 reveals a novel allosteric site. Biochem J. 2007;401:39–47. doi: 10.1042/BJ20061066. doi:10.1042/BJ20061066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Peng X, Zhao F, Zhang G, Tao Y, Luo Z, Li Y, Teng M, Li X, Wei S. N114S mutation causes loss of ATP-induced aggregation of human phosphoribosylpyrophosphate synthetase 1. Biochem Biophys Res Commun. 2009;379:1120–1125. doi: 10.1016/j.bbrc.2009.01.034. doi:10.1016/j.bbrc.2009.01.034. [DOI] [PubMed] [Google Scholar]
- Madariaga MG, Lalloo UG, Swindells S. Extensively drug-resistant tuberculosis. Am J Med. 2008;121:835–844. doi: 10.1016/j.amjmed.2008.04.015. doi:10.1016/j.amjmed.2008.04.015. [DOI] [PubMed] [Google Scholar]
- Makarov V, Manina G, Mikusova K other authors. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science. 2009;324:801–804. doi: 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil M, Daffe M, Brennan PJ. Location of the mycolyl ester substituents in the cell walls of mycobacteria. J Biol Chem. 1991;266:13217–13223. [PubMed] [Google Scholar]
- Mikusova K, Huang H, Yagi T other authors. Decaprenylphosphoryl arabinofuranose, the donor of the D-arabinofuranosyl residues of mycobacterial arabinan, is formed via a two-step epimerization of decaprenylphosphoryl ribose. J Bacteriol. 2005;187:8020–8025. doi: 10.1128/JB.187.23.8020-8025.2005. doi:10.1128/JB.187.23.8020-8025.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortalo-Magne A, Dupont MA, Lemassu A, Andersen AB, Gounon P, Daffe M. Molecular composition of the outermost capsular material of the tubercle bacillus. Microbiology. 1995;141(Pt 7):1609–1620. doi: 10.1099/13500872-141-7-1609. [DOI] [PubMed] [Google Scholar]
- Parry RJ, Burns MR, Skae PN, Hoyt JC, Pal B. Carbocyclic analogues of D-ribose-5-phosphate: Synthesis and behavior with 5-phosphoribosyl α-1-pyrophosphate synthetase. Bioorg Med Chem. 1996;4(7):1077–1068. doi: 10.1016/0968-0896(96)00090-9. doi:10.1016/0968-0896(96)00090-9. [DOI] [PubMed] [Google Scholar]
- Rao SP, Alonso S, Rand L, Dick T, Pethe K. The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2008;105:11945–11950. doi: 10.1073/pnas.0711697105. doi:10.1073/pnas.0711697105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. doi:10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- Schafer A, Tauch A, Jager W, Kalinowski J, Thierbach G, Puhler A. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: Selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene. 1994;145:69–73. doi: 10.1016/0378-1119(94)90324-7. doi:10.1016/0378-1119(94)90324-7. [DOI] [PubMed] [Google Scholar]
- Scherman MS, Kalbe-Bournonville L, Bush D, Xin Y, Deng L, McNeil M. Polyprenylphosphate-pentoses in mycobacteria are synthesized from 5-phosphoribose pyrophosphate. J Biol Chem. 1996;271:29652–29658. doi: 10.1074/jbc.271.47.29652. doi:10.1074/jbc.271.47.29652. [DOI] [PubMed] [Google Scholar]
- Scherman M, Weston A, Duncan K, Whittington A, Upton R, Deng L, Comber R, Friedrich JD, McNeil M. Biosynthetic origin of mycobacterial cell wall arabinosyl residues. J Bacteriol. 1995;177:7125–7130. doi: 10.1128/jb.177.24.7125-7130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert KR, Switzer RL, Shelton E. Studies of the quaternary structure and the chemical properties of phosphoribosylpyrophosphate synthetase from Salmonella typhimurium. J Biol Chem. 1975;250:7492–7500. [PubMed] [Google Scholar]
- Schuck P. A model for sedimentation in inhomogeneous media. I. Dynamic density gradients from sedimenting co-solutes. Biophys Chem. 2004;108:187–200. doi: 10.1016/j.bpc.2003.10.016. doi:10.1016/j.bpc.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. doi:10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel M, Alderwick LJ, Birch HL, Sahm H, Eggeling L, Besra GS. Identification of a novel arabinofuranosyltransferase AftB involved in a terminal step of cell wall arabinan biosynthesis in Corynebacterianeae, such as Corynebacterium glutamicum and Mycobacterium tuberculosis. J Biol Chem. 2007;282:14729–14740. doi: 10.1074/jbc.M700271200. doi:10.1074/jbc.M700271200. [DOI] [PubMed] [Google Scholar]
- Stankiewicz PJ, Gresser MJ. Inhibition of phosphatase and sulfatase by transition-state analogues. Biochemistry. 1988;27:206–212. doi: 10.1021/bi00401a031. doi:10.1021/bi00401a031. [DOI] [PubMed] [Google Scholar]
- Switzer RL, Sogin DC. Regulation and mechanism of phosphoribosylpyrophosphate synthetase. V. Inhibition by end products and regulation by adenosine diphosphate. J Biol Chem. 1973;248:1063–1073. [PubMed] [Google Scholar]
- Tozzi MG, Camici M, Mascia L, Sgarrella F, Ipata PL. Pentose phosphates in nucleoside interconversion and catabolism. FEBS J. 2006;273:1089–1101. doi: 10.1111/j.1742-4658.2006.05155.x. doi:10.1111/j.1742-4658.2006.05155.x. [DOI] [PubMed] [Google Scholar]
- Turnock DC, Ferguson MA. Sugar nucleotide pools of Trypanosoma brucei, Trypanosoma cruzi, and Leishmania major. Eukaryot Cell. 2007;6:1450–1463. doi: 10.1128/EC.00175-07. doi:10.1128/EC.00175-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne LG, Sohaskey CD. Nonreplicating persistence of Mycobacterium tuberculosis. Annu Rev Microbiol. 2001;55:139–163. doi: 10.1146/annurev.micro.55.1.139. doi:10.1146/annurev.micro.55.1.139. [DOI] [PubMed] [Google Scholar]
- Willemoes M, Hove-Jensen B, Larsen S. Steady state kinetic model for the binding of substrates and allosteric effectors to Escherichia coli phosphoribosyl-diphosphate synthase. J Biol Chem. 2000;275:35408–35412. doi: 10.1074/jbc.M006346200. doi:10.1074/jbc.M006346200. [DOI] [PubMed] [Google Scholar]
- Wojtkiewicz B, Szmidzinski R, Jezierska A, Cocito C. Identification of a salvage pathway for D-arabinose in Mycobacterium smegmatis. Eur J Biochem. 1988;172:197–203. doi: 10.1111/j.1432-1033.1988.tb13873.x. doi:10.1111/j.1432-1033.1988.tb13873.x. [DOI] [PubMed] [Google Scholar]
- Wolucka BA, McNeil MR, de Hoffmann E, Chojnacki T, Brennan PJ. Recognition of the lipid intermediate for arabinogalactan/arabinomannan biosynthesis and its relation to the mode of action of ethambutol on mycobacteria. J Biol Chem. 1994;269:23328–23335. [PubMed] [Google Scholar]