

Abstract
Non-redundant protein datasets are of utmost importance in bioinformatics. Constructing such datasets means removing protein sequences that overreach certain similarity thresholds. Several programs such as ‘Decrease redundancy’, ‘cd-hit’, ‘Pisces’, ‘BlastClust’ and ‘SkipRedundant’ are available. The issue that we focus on here is to what extent the non-redundant datasets produced by different programs are similar to each other. A systematic comparison of the features and of the outputs of these programs, by using subsets of the UniProt database, was performed and is described here. The results show high level of overlap between non-redundant datasets obtained with the same program fed with the same initial dataset but different percentage of identity threshold, and moderate levels of similarity between results obtained with different programs fed with the same initial dataset and the same percentage of identity threshold. We must be aware that some differences may arise and the use of more than one computer application is advisable.
Keywords: protein sequence, removing redundancy, sequence alignment
Background
Protein sequence databases are extremely redundant and their redundancy must be removed in many different studies. Redundancy in a dataset occurs when several similar data are present at the same time [1]. In bioinformatics, redundancy in a collection of sequences occurs when one or more similar/homologous sequences are present in the same set of data. The inclusion of similar sequences in certain analyses will introduce undesirable biases. This occurs in particular when average values or trends have to be extracted from the data, like for example the average amino acid composition [2,3] . Another important issue is that the biological databases, such as UniProt [4], are growing at an astronomical rate, together with the redundancy of their entries. As a consequence, hardware requirements (CPU and memory) become increasingly expensive and redundancy reduction results also in lower computational costs.
Different computer programs for removing redundancy are available. They utilize different alignment methods (global or local alignment) and clustering algorithms. In the present work, several computer programs are compared systematically. Beside a general evaluation of their results, the main concern is to what extent the results of different methods are similar or different. Certain differences have been observed when using programs that utilize different methodologies. This indicates that any research that requires non-redundant sequence datasets should be taken with consciousness. If possible, it would be preferable that the research should be conducted using several non-redundant datasets obtained by using different programs.
Software
Five different computer programs were used: Decrease Redundancy [5], cd-hit [6], Pisces [7, 8] , BlastClust [9, 10] and SkipRedundant [11]. Some of them, like cd-hit and Pisces, are standalone applications, others are part of standalone packages such as BLAST (BlastClust) and EMBOSS (SkipRedundant), while one is available as a web based server (Decrease Redundancy). The computer programs used in this study along with version number (where known) and URL are listed in Table 1. Four different percentages of identity (PID) thresholds were used: 40%, 50%, 75% and 90%. Smaller and more stringent values were not used, since they cannot be used by all the computer programs.
Test environment
All standalone applications including those that are part of standalone program packages were tested on the same test platform: Intel Xenon 3.4GHz (4 CPU) with 2GB RAM, running Red Hat 4.1.2-13 (Linux version 2.6.22.2-42.fc6). All the tested standalone applications don't impose any CPU or memory requirements on the computer. However, as the applications utilize Needleman-Wunsch or Smith-Waterman sequence alignment algorithms, which both have time and space complexity O(MxN) (M is number of sequences and N is length of sequences), the computational requirements depend on the number and size of the sequences and not as much of the program itself. The program Decrease redundancy was tested using its web server, since the standalone software is not distributed.
Dataset
The sets of protein sequences, necessary for testing the various computer programs, were extracted from the UniProt database using a random number generator. Extracted sequences were used to build random sets of 100, 1,000, and 10,000 protein sequences. This procedure was repeated in order to build datasets containing only small proteins with less than 100 residues, datasets with larger proteins (100-200 residues, 200-300 residues and so on), and datasets containing only large proteins with more than 1,000 residues. In such way 33 datasets, summarized in Table 2 (see supplementary material), were constructed. All of them were used as inputs for the computer programs of Table 1 (see supplementary material), with the exception of Decrease Redundancy that, due to the web server CPU limitations, could process only datasets with 100 sequences or less.
Definition of percentage of sequence identity (PID)
The percentage of sequence identity (PID) for two aligned protein sequences is defined as number of aligned positions where the matching characters (amino acids) are identical divided by the number of aligned positions (including gaps, if any). This definition is not superfluous, since there is often some uncertainty about the operational definition of sequence identity [12].
Overview of different method
The principal characteristics of the computer programs used in the present work are summarized in Table 3 (see supplementary material). Most of the programs allow choosing PID threshold in range from 0 to 100 percent, but some of them, like Pisces and cd-hit, have to some extent limited ranges. The maximum accepted length of the protein is not declared in most cases with an exception of cd-hit where the maximum length is limited by the maximum integer size. The maximum number of sequences that the programs are capable of processing is also not declared by any of these programs.
All the programs accept input files in FASTA format, but only Decrease Redundancy, cd-hit and SkipRedundant provide results in the same format. Others like Pisces and BlastClust provide only a list of protein identification codes in the output file. We also verified whether the output is dependent on the input order, meaning whether the non-redundant output dataset is always the same independently of the permutations of the sequences in the input dataset. In the case of Pisces and BlastClust the output is not dependent on the order of the input. However, this is not the case with the other programs where the output is dependent on the order of the input sequences. Also, the number of non-redundant sequences outputted by all the computer programs is independent of the permutations of the input dataset.
Decrease Redundancy
The algorithm used in this program was developed by Cédric Notredame and is to date unpublished. Therefore any description of this algorithm is currently not available.
Cd-hi
Cd-hit uses the greedy incremental algorithm to cluster sequences and remove redundancy. Sequences are first sorted in a descending manner according to their length. The longest sequence is taken as the representative member of the first cluster. Then each remaining sequence is compared to the representatives of the existing clusters and if it is sufficiently similar to one of them, it is inserted into the cluster. Otherwise, a new cluster is formed with that sequence as a representative. In this way, all clusters are assembled.
Due to the large number of pairwise alignments, empirical filters are applied in order to decrease the number of alignments - the most time consuming element of the clustering algorithm. These are ‘short word’ filters that are based on the following assumption: Two similar proteins share the same number of types of dipeptides, tripeptides, and so on. Therefore the pairs of sequences that do not satisfy this condition do not have to be aligned [6, 13,14] . The alignment of two sequences that have satisfied the conditions imposed by the “short word” filters is done with the Smith-Waterman algorithms [15], which is able to delineate a biologically meaningful local alignment even if two sequences differ greatly in their length. Given the optimal alignment, the sequence identity is computed as the ratio between the number of identities and the length of the sequence which is not yet a member of any existing cluster. The identity threshold limit that must be overreached to insert a new sequence into a cluster is given by the user [16].
Pisce
This program does not provide the user with complete clusters, instead it outputs a list of cluster representatives. Other cluster members are omitted. Pisces uses the Sander and Holbohm [1] method to create non-redundant datasets. The first sequence of the input dataset is flagged as included in the non-redundant output dataset. This is a cluster representative sequence. Each subsequent sequence in the input dataset is flagged as excluded if it has a pairwise sequence identity with the first sequence lower than the user defined threshold. The excluded sequences then form a new dataset and the procedure repeats until all sequences are clustered. Pisces can use a combination of structural alignment at low sequence identity and sequence alignment at high sequence identity. Structural alignments are calculated using the CE program [17] while PSI-BLAST [9] is used for sequence alignment. If the input contains only sequences, the structural alignments are obviously bypassed. The sequence identity is defined as the number of identical pairs divided by all aligned pairs excluding gaps, if any.
BlastClust (BLAST)
The algorithm starts by pairwise comparison of all sequences using BLAST. For each pairwise comparison BLAST calculates two values ‘coverage’ and ‘score density’. The ‘coverage’ is defined as max(Cx,Cy) or min(Cx,Cy), depending on user decision, where Cx (Cy) is the ratio between the length of the high-scoring-segment-pair on sequence x (y) and the length of sequence x (y). ‘Score density’ is defined as the ratio between the number of identical residues and the length of the alignment including gaps, if any. Alternatively ‘score density’ can be defined as ratio between the BLAST score and min(HxHy) where Hx (Hy) is the length of the highscoring-segment-pair on sequence x (y).If these two values are above a certain threshold the two sequences that are compared are considered to be neighbors. In such way, a neighbor relationship list of all input sequences is determined. This list is inputted into a single-linkage clustering process. This clustering method starts with a first sequence as a cluster representative and puts any other sequence into that cluster if the sequence is a neighbor of at least one sequence already in the cluster. All remaining (not clustered) sequences are stored in the new list and the same procedure is applied again. BlastClust repeats this procedure until all sequences are clustered.
SkipRedundant (EMBOSS)
With this method, all pairwise sequence alignments are calculated using the EMBOSS implementation of the Needleman-Wunsch global alignment algorithm [18]. The program can use two procedures for removing redundant sequences:
If a pair of proteins achieve a percentage of sequence identity greater than a threshold (specified by the user) the shortest sequence is discarded;
If a pair of proteins have a percentage of sequence identity that lies outside a range (specified by the user) the shortest sequence is discarded. After the sequences have been removed the list contains only non-redundant entries.
Results and discussion
In the following sections the results will be presented and discussed in three parts. First, the attention will be focused on some general features of the various computer programs and in particular it will be shown that all of them allow one to correctly remove sequence redundancy. Then, it will be examined what happens when the redundancy reduction becomes more and more severe by using the same computer program and it will be shown that all the programs present similar trends at this regard. Eventually, the attention will be focused on the comparison of the results obtained by using different methods and it will be shown that some differences may be observed on the outputs of different computer programs.
General considerations
The programs listed in Table 1 (see supplementary material) have been tested using the datasets extracted from UniProt database (Table 2, see supplementary material). Each program used four different PID thresholds, 40%, 50%, 75% and 90%. Smaller and more stringent values of PID were disregarded since they cannot be used by some of the programs of Table 1 (see supplementary material). As expected, if the PID threshold increases, the non-redundant datasets become larger, independently of the program. Depending on the selected PID threshold, 40%, 50%, 75% and 90%, the output dataset is in average reduced to 90%, 92%, 88% and 96% of the input dataset respectively. Moreover, the number of sequences in the non-redundant datasets, obtained by different programs fed with the same input datasets and PID thresholds are very similar. For example in the case of the D_100_500 test set with 40% PID the number of sequences in the non-redundant dataset is 97, 97, 96 and 97 for Decrease Redundancy, cd-hit, Pisces and BlastClust respectively. Table 4 (see supplementary material) shows the percentage of sequences found in the output relative to the input (Ptot) at various PID threshold values. All the programs have similar Ptot values for the same PID with the exception of SkipRedundant which showed lower Ptot for PID 90%. Not surprisingly, smaller Ptot values are observed for smaller and more stringent PID thresholds.
Moreover, we did not observe any correlation between the length of the protein sequences in the input datasets and the resulting non-redundant outputs. All the programs show a closely similar behavior at this regard.
Furthermore, we examined the level of sequence similarity within each non-redundant dataset. We used both the Smith-Waterman (local alignment) and Needleman-Wunsch (global alignment) method and we observed some differences in the output of different computer programs. The average identity is in the range of 23% to 42% and 15% to 21% for Smith-Waterman and Needleman-Wunsch method respectively. An example of the distribution of pairwise identity in non-redundant datasets (calculated using max. PID = 40%) is given in Figure 1.
Figure 1.
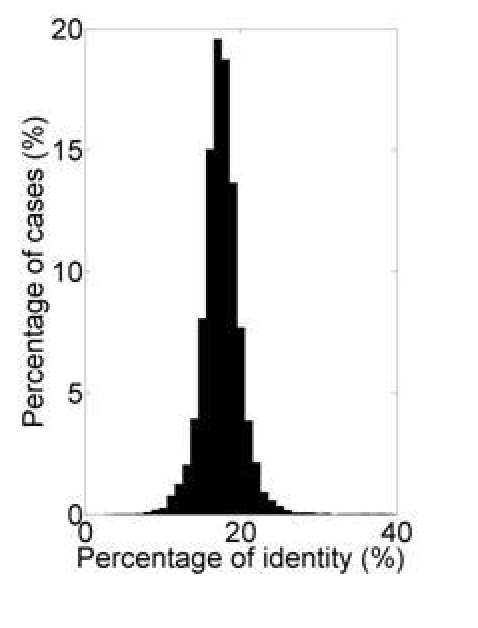
Pairwise percentages of identity calculated on the non-redundant set using the Needleman-Wunsch algorithm. Non-redundant sets were obtained using cd-hit program with max PID = 40%. Similar results were obtained for Decrease redundancy, Pisces and BlastClust.
Our study showed inconclusive data for the program SkipRedundant. For percentages of identity of 50% and lower, independently of the input dataset, the program reported results containing only few cluster representatives which were not further analyzed. Therefore the results obtained by this program with percentages of identity of 40% and 50% are not taken into further consideration in this study.
These results clearly indicate that all the programs, even within the limitations shown by some of them, are able to produce dataset of sequences that are really non-redundant, as far as the redundancy is related to the level of sequence similarity. Apparently, therefore, any of these programs can be used and there is no reason to prefer one or the other.
Overlaps at different threshold
Previously, we have shown that the size of the non-redundant datasets increases with the percentage of identity threshold used by the programs. For example there are 91 sequences outputted by Pisces fed with the dataset D_100_800 if the PID threshold is 40% while 92 sequences are outputted if the threshold is 50%. It is also interesting to examine the overlap between these two datasets of 91 and 92 sequences, produced by the same program fed with the same input and with different PID thresholds. The overlap is measured as the percentage of the proteins of the smaller dataset that are observed also in the larger dataset. The results show that the overlap is in most cases 100%. Although in few cases this value is slightly smaller than 100%, it is never smaller than 96%.
Alternatively, this can be described as the invariance of the output content on the progressive strengthening of the non-redundancy threshold: if the PID threshold is made more stringent, for example by lowering it from 50% to 40%, the sequences that are retained in the 40% non-redundant output were already present in the 50% output. This can be explained by the fact that the number of sequences in the output is dependent of the percentage of identity threshold defined by the user but the selection of the cluster representatives is independent of that same threshold.
Overlap between different computer programs
A crucial issue is the comparison between the outputs of different programs fed with the same input dataset and the same PID threshold value. The overlap between two non-redundant datasets is measured as the percentage of proteins of the smaller dataset that are found in the larger dataset. Table 5 shows the overlap values between different programs and for the same threshold values. The average overlap is around 90%, ranging from 88% to 100%.The discrepancy between the results shown in Table 5 can be explained by several facts; first, different programs use different alignment algorithms ‐ this particularly refers to SkipRedundant which uses global alignment while the others use local alignment; second, even if two or more programs use the same alignment algorithm, the fine tuning of the algorithm can be different; third, the selection of cluster representatives is done in different ways, depending on the clustering algorithm.
An example of overlap of four non-redundant datasets, each obtained with a different program, based on the same input dataset and the same PID threshold, is given in Figure 2. This figure supports the findings that the non-redundant sets obtained by different programs fed with the same input dataset and maximum PID have a considerable degree of similarity in size and content.
Figure 2.
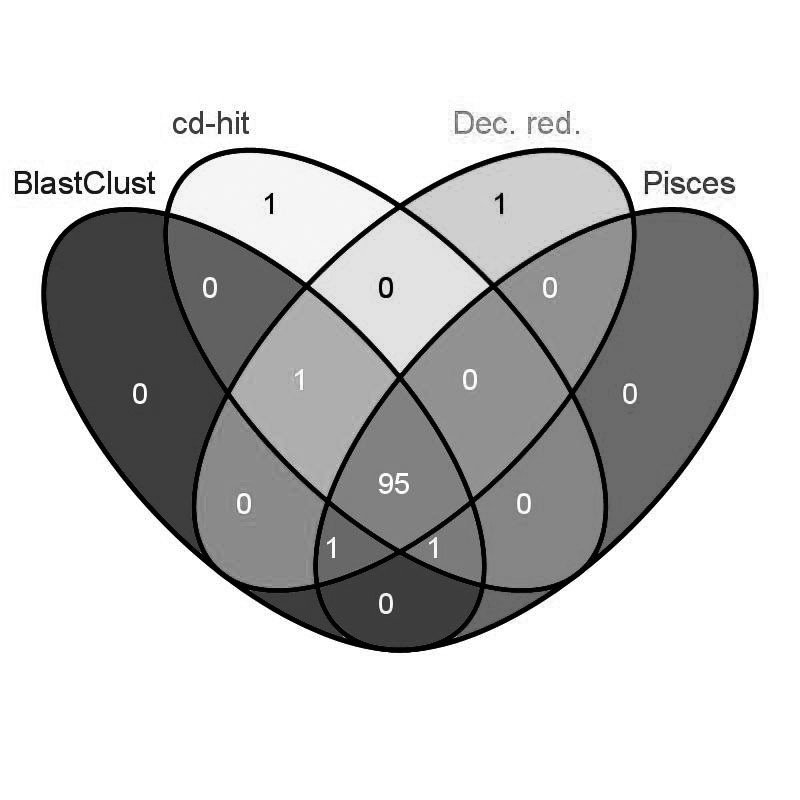
Venn diagram. Overlap of four non-redundant datasets, each obtained with a different program, based on the same input dataset (D_100_100) and the same PID (40%). 95 sequences are common to the non-redundant outputs of BlastClust, cd-hit, Decrese redundancy, and Pisces.
Conclusion
A large scale comparison of various computer programs that are commonly used to remove redundancy from protein sequence databases has been performed. Different computational approaches clearly produce slightly different results. If this is not completely unexpected, since several differences are observed in all these algorithms, it is nevertheless intriguing. The preference for a program is largely dependent on technical issues like the availability of a simple and user friendly stand alone executable, which is particularly attractive for large scale studies, rather than the availability of a web based server, which eliminate the problems to install local copies of the software. Most of the bioinformaticians assume that redundancy reduction is a routine and simple step, which can be performed with any of the available programs. However, we have documented that the lists of non-redundant protein sequences outputted by different programs are different, even when the redundant input set of sequences is the same and the threshold of percentage of sequence identity is the same. Discrepancies up to about 10% are quite common.
This does not indicate that these computer programs have difficulties in removing the redundancy. In fact, all of their outputs are more than acceptable in terms of residual similarity between the entries that are grouped in the outputs. However, this clearly suggests that a good practice in bioinformatics should be the use of more than one computer application for removing redundancy from protein sequence ensembles. In this way, all the computations would be done in parallel by using two or more nonredundant sets of sequences and the comparison of the final results of the computations would reinforce their intrinsic value, a sort of precaution to avoid possible biases and mistakes. It is eventually important to point out that it is impossible to rank the different programs according to the quality of their outputs. To do it, it would be necessary to have a benchmark dataset, which can be produced only by using the same programs that we compared. Any other benchmark, constructed with human intervention, would be biased by subjectivity.
Supplementary material
Acknowledgments
Funding from BIN-II and BIN-III (GEN-AU Austrian research program) is gratefully acknowledged. Grant from the Ministry of Science, Republic of Croatia, number 036-0362214-1987 is gratefully acknowledged
Footnotes
Citation:Kresimir & Oliviero, Bioinformation 5(6): 234-239 (2010)
References
- 1.Hobohm U, et al. Protein Sci. 1992;1:409. doi: 10.1002/pro.5560010313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carugo O. Protein Sci. 2008;17:2189. doi: 10.1110/ps.037762.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cornish-Bowden A. Biochem J. 1983;213:271. doi: 10.1042/bj2130271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Apweiler A, et al. Nucleic Acids Res. 2004;32:D115. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gasteiger E, et al. Nucleic Acids Res. 2003;31:3784. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li W, et al. Bioinformatics. 2006;22:1658. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 7.Wang G, et al. Nucleic Acids Res. 2005;33 doi: 10.1093/nar/gki492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang G, et al. Bioinformatics. 2003;19:1589. doi: 10.1093/bioinformatics/btg224. [DOI] [PubMed] [Google Scholar]
- 9.Altschul SF, et al. Journal of Molecular Biology. 1990;215:403. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 10.Wheeler D, et al. 2007 [Google Scholar]
- 11.Rice P, et al. Trends in Genetics. 2000;16:276. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 12.Cameron M, et al. Structure. 2004;12:737. [Google Scholar]
- 13.Li W, et al. Bioinformatics. 2001;17:282. doi: 10.1093/bioinformatics/17.3.282. [DOI] [PubMed] [Google Scholar]
- 14.Li W, et al. Bioinformatics. 2002;18:77. doi: 10.1093/bioinformatics/18.1.77. [DOI] [PubMed] [Google Scholar]
- 15.Smith TF, et al. Journal of Molecular Biology. 1981;147:195. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 16.Holm L, et al. Bioinformatics. 1998;14:423. doi: 10.1093/bioinformatics/14.5.423. [DOI] [PubMed] [Google Scholar]
- 17.Shindyalov I, et al. Protein Engineering. 1998;11:739. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- 18.Needelman S, et al. Journal of Molecular Biology. 1970;48:443. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.