

Abstract
Aging is accompanied by a progressive decline in the integrity of the immune system, a process known as immunosenescence. Rheumatoid arthritis (RA), an autoimmune disease whose incidence increases with age, is characterized by pathological features typical of immune dysfunction in the elderly, encompassing dysregulation of both innate and adaptive immune responses. Recent evidence suggests that certain features of immunosenescence, such as the decrease in T-cell generation and diversity, may contribute to the development of RA. Thus, physiological immunosenescence may render the elderly susceptible to RA, while premature immunosenescence may contribute to the development of RA in young adults. In addition, other features of immunosenescence may result from the chronic immune stimulation that occurs in RA and lead to worsening of the disease. Here we review the immunopathological features common to aging and RA, and discuss the mechanisms by which immunosenescence may contribute to the development or progression of RA.
Keywords: immunosenescence, rheumatoid arthritis, aging, autoimmunity, CD28
INTRODUCTION
Aging is characterized by a gradual deterioration of the immune system known as immunosenescence. The resulting failure to mount effective immune responses renders elderly individuals more susceptible to infections and to development of certain cancers. Paradoxically, aging is also associated with an increase in incidence of the autoimmune disease rheumatoid arthritis (RA).1–4 Individuals with RA are immunocompromised,5, 6 and accumulating evidence suggests that premature aging of the immune system contributes to the pathogenesis of RA.7–10 The progressive deterioration of the immune system with advancing age may therefore predispose to the development of RA late in life. In addition, the chronic immune stimulation characteristic of RA may itself age the immune system and lead to progression of the disease.11–13 Here, after briefly outlining of the regulation of the immune response and the pathogenesis of RA, we discuss the mechanisms by which immunosenescence spawns autoimmunity and the evidence implicating immunosenescence in the development or progression of RA.
QUALITY CONTROL IN THE IMMUNE SYSTEM
The immune system can be broadly divided into an innate component, comprising natural killer (NK) cells, macrophages, dendritic cells (DCs), and complement factors, and an adaptive component, comprising T and B cells. The innate and adaptive immune responses act in concert to defend the body against infection. Innate immunity serves as an immunological sentinel that is poised to counteract pathogens in a rapid and non-specific manner. Adaptive immunity, on the other hand, is a slow starter but, once activated, is exquisitely specific to the inciting antigen and results in the generation of immunological memory. The partition into distinct innate and adaptive responses is not black and white, as extensive crosstalk between the two systems occurs during an immune response. For instance, cytokines secreted by various immune cells modulate the activity of both innate and adaptive immune cells. Moreover, the adaptive immune response springs into action only once it has received an alert from its innate counterpart, and cells of the innate system are summoned to eliminate incapacitated pathogens and to clear cell debris in the wake of an adaptive immune response.
Induction of a primary adaptive immune response usually relies on DCs of the innate immune system taking up pathogens and presenting the pathogen antigens to naïve T cells of the adaptive immune system (Figure 1). T-cell receptors (TCRs) on the surface of T cells recognize antigen only when it is bound to major histocompatibility complex (MHC) molecules on the surface of DCs or other antigen-presenting cells.14 T cells that bear a TCR specific for the antigen presented are activated to proliferate (a process known as clonal expansion) and differentiate into effector T cells. Effector T cells include cytotoxic CD8+ T cells, which destroy infected cells, and helper CD4+ T cells, which regulate other immune cells such as B cells. B cells can function as both antigen-presenting cells and antigen-specific effector cells. B cells bearing immunoglobulin receptors specific for the antigen encountered take up the antigen and present it to antigen-specific CD4+ T cells, which in turn induce the B cells to clonally expand and differentiate into antibody-producing plasma cells (Figure 1). The antibodies produced tackle extracellular pathogens by blocking a pathogen’s access to cells (a process known as neutralization), by coating the pathogen and thereby targeting it for phagocytosis by innate immune cells (a process known as opsonization), or by activating the complement system, which promotes pathogen destruction. Whereas most activated T and B cells become effector cells, which die by apoptosis once the primary adaptive immune response is over, a few of them become long-lived memory cells, which persist in a quiescent state in the absence of their cognate antigen. If these memory cells later encounter their cognate antigen, they become activated and mount a secondary immune response that is faster and stronger than the primary immune response from which they arose.
Figure 1.
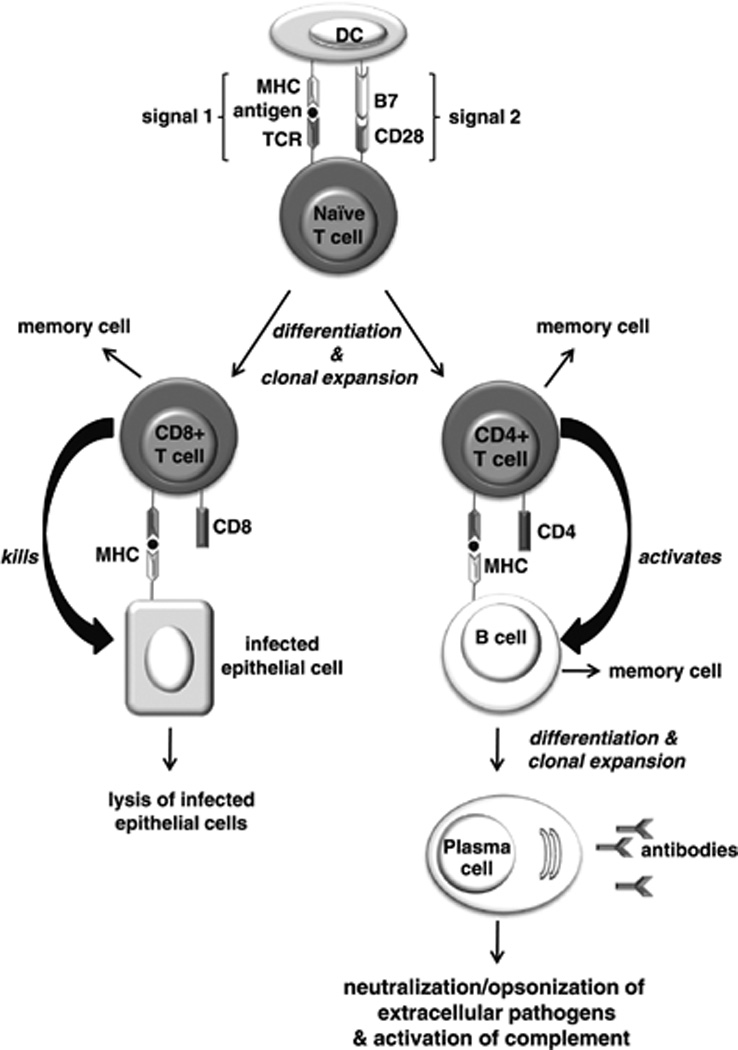
Induction of a primary adaptive immune response. Antigen-presenting cells such as dendritic cells (DCs) take up pathogens and present the pathogen antigens to naïve T cells. Those T cells that bear a T-cell receptor (TCR) specific for the antigen presented undergo clonal expansion and differentiate into CD8+ and CD4+ effector T cells. For this to occur, the T cell must receive two signals from the DC. Signal 1 is delivered by the interaction of the TCR with the antigen/ major histocompatibility complex (MHC) on the DC. Signal 2 is delivered upon binding of the B7 molecule on the DC to the CD28 receptor on the T cell. Cytotoxic CD8+ T cells recognize the antigen/MHC complex on the surface of cells, and are activated to destroy the infected cells. Naïve B cells bearing immunoglobulin receptors specific for the antigen internalize the antigen and present it to antigen-specific CD4+ T cells, which in turn induce the B cells to clonally expand and differentiate into antibody-producing plasma cells. The antibodies produced target extracellular pathogens and their toxic products via the processes of neutralization, opsonization, or complement activation. A small proportion of activated T and B cells form long-lived memory cells, which are responsible for mounting a stronger, secondary immune response upon subsequent encounters with their cognate antigen.
To mount effective adaptive immune responses without attacking the body’s own cells and tissues, the immune system must be able to distinguish self from non-self. Multiple mechanisms are in place to ensure such self-tolerance. Central tolerance mechanisms in the thymus—the site of T-cell differentiation and maturation—eliminate newly generated T cells that are strongly autoreactive.14 Only those precursor T cells whose TCRs interact weakly with self-peptide/self-MHC complexes survive and mature, a process known as positive selection. Conversely, precursor T cells whose TCRs interact strongly with self-peptide/self-MHC complexes are eliminated, and this is known as negative selection. Thus, central tolerance results in mature T cells that are both self-tolerant and self-MHC-restricted. Another level of control is imposed by peripheral tolerance, which relies largely on the interaction between T cells and DCs. Early findings gave rise to the two-signal model of activation, which laid the foundation for the concept that combined triggering of both the TCR and one or more co-stimulatory receptors is needed for the full activation of naïve T cells (Figures 1 and 2). Recent findings suggest that the two-signal model applies to the activation of memory T cells, too.15 Signal 1 is delivered by the TCR when it binds to the antigen/self-MHC complex; signal 2 is a co-stimulatory signal elicited primarily by the CD28 receptor on the T-cell surface when it binds to the B7 molecule on the DC surface. In the absence of infection, DCs do not express B7. Therefore, a mature T cell that interacts strongly with self-peptide (rather than a pathogenic antigen) in the periphery will receive signal 1 but not signal 2. This either kills the T cell or renders it refractory to activation, a state known as anergy (Figure 2). However, weak interaction between the TCR and self-peptide/MHC complexes in the periphery is required for the survival and homeostatic replication of mature T cells, as discussed below.
Figure 2.
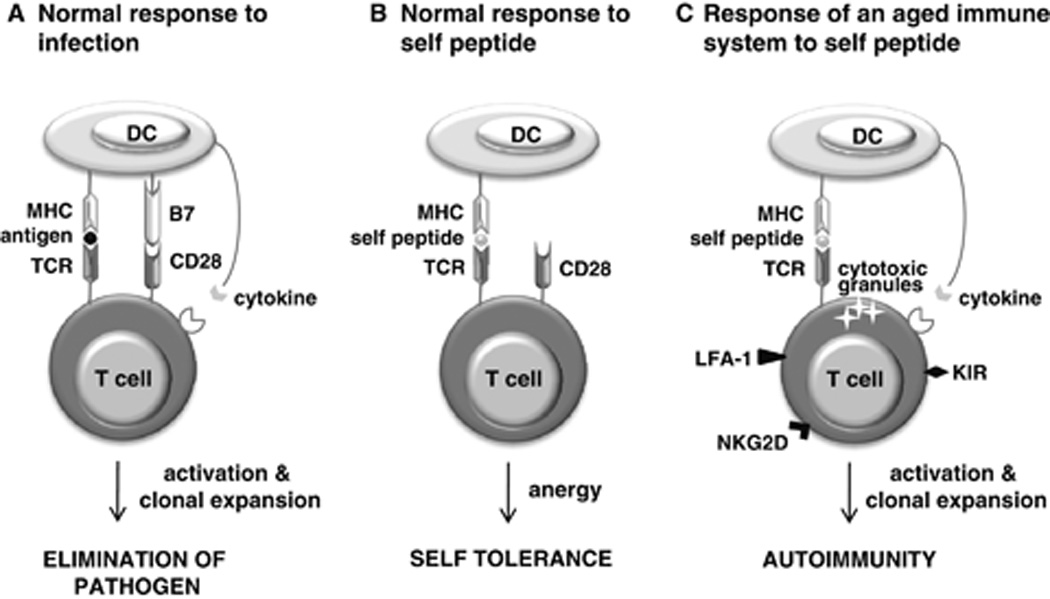
Breakdown of peripheral tolerance in aging. Normally, two signals are required for the full activation of T cells. (A) When a dendritic cell (DC) presents a pathogenic antigen to a T cell, signal 1 is delivered by the T-cell receptor (TCR) when it binds to the antigen/self-MHC complex, while signal 2 is delivered via the CD28 receptor on the T-cell surface when it binds to the B7 molecule on the DC surface. Further co-stimulatory signals are provided by secretion of cytokines from the DC. (B) In the absence of infection, DCs express no or very little B7. A T cell that interacts strongly with self-peptide (rather than with a pathogenic antigen) in the periphery will receive signal 1 but not signal 2. This either kills the T cell or renders it refractory to activation, a state known as anergy. Although originally thought to be restricted to naïve T-cell activation, CD28 co-stimulation requirement has more recently been shown to extend to memory T-cell activation. (C) In the aged immune system, the T-cell repertoire is skewed toward recognition of self-peptide, likely as a result of multiple mechanisms acting on naïve and memory T cells. The aged T-cell repertoire is also characterized by a predominance of memory cells. A subset of memory T cells have lost expression of CD28 and acquired expression of stimulatory killer immunoglobulin-like receptors (KIR), NKG2D receptors, and lymphocyte function-associated antigen 1 (LFA-1). De novo expression of these stimulatory receptors on memory CD28− T cells changes the way in which these T cells interact with their cellular environment, lowering the threshold for antigen-specific activation and even enabling activation independent of the appropriate antigen. Together with de novo expression of stimulatory receptors, development of cytotoxic granules endows CD28− T cells with certain natural killer cell functions, including cytotoxic killing. Furthermore, DCs secrete greater amounts of cytokines, and are able to induce T-cell proliferation, in response to self-peptides. These aberrations promote autoimmunity. MHC, major histocompatibility complex.
DELUDED PERPETRATORS OF RA
As outlined, intricate checkpoints exist to ensure that adaptive immune cells keep to the straight and narrow. But in certain individuals these best-laid plans go awry. The convergence of genetic predisposition and environmental triggers can result in an immune system that mistakenly recognizes some of the body’s own molecules as foreign. As a result, the immune system trains its destructive efforts on the body’s own tissues and organs, leading ultimately to the development of autoimmune disease.
Given the extensive interplay between the adaptive and innate immune responses, it comes as no surprise that both arms of the immune system play important roles in the initiation and perpetuation of RA. Several lines of evidence implicate T cells in RA pathogenesis: RA is associated with genes encoding molecules involved in T-cell activity (e.g., HLA-DR4, CTLA4, PTPN22);16 T-cell numbers are abnormally elevated in rheumatoid joints;17 and T cells drive the development of arthritis in animal models of RA.18 Evidence of B-cell involvement has come from the presence of B cells in rheumatoid joints,19 the detection of autoantibodies that are specific to RA,20 the ability of autoantibodies to exacerbate or induce arthritis in mice,21, 22 and the therapeutic efficacy of B-cell-depleting agents in RA patients.23 Innate immune cells, including DCs and macrophages, may contribute to the breach of self-tolerance in RA, by presenting arthritogenic antigens to autoreactive T cells, and to the destruction of joint cartilage, by producing proinflammatory and degradative mediators.24–26
Although characterized by inflammation, as well as cartilage and bone erosion, in the synovial joints, RA is unequivocally a systemic disorder and can affect multiple organs in addition to the joints. The mechanisms by which self-tolerance is initially breached in RA remain undefined but likely involve dysregulation of the immune system at a systemic level, followed eventually by immune targeting of the joints and hence the clinical manifestation of RA. The predilection of this systemically engendered autoimmunity for synovial joints has been attributed to the synovial microarchitecture, which facilitates immune responses, rather than to targeting of joint-specific antigens.27 RA is a heterogeneous disorder, and disease etiology likely differs between distinct subsets. To date, smoking is the only accepted environmental risk factor for RA and is thought to trigger RA by inducing protein citrullination (a posttranslational modification in which arginine residues are converted to citrulline residues) in the lungs. Citrullination of proteins results in the formation of new epitopes and thus, in certain individuals, the immune system may not recognize citrullinated proteins as self. In individuals whose genetic make-up is conducive to a strong immune response to citrullinated proteins, antibodies against citrullinated proteins develop; these are specific to RA and appear before the onset of disease.28 Protein citrullination is known to occur in synovial joints as a result of inflammation, and systemic anti-citrulline immunity elicited in the lungs may therefore go on to target citrullinated proteins in the joints, thereby precipitating the joint pathology characteristic of RA.28 Although less substantiated, a link between periodontitis and the development of RA has also been proposed. As with smoking, periodontitis has been postulated to trigger RA by inducing an immune response against citrullinated proteins, since certain periodontal pathogens can citrullinate proteins found in periodontal tissue.29
AGING T CELLS: DROP IN THE NEW AND DIFFERENT
Age-related decrease in generation of new T cells
Like RA, aging is associated with dysregulation of the immune system. One of the most widely studied phenomena in the immunobiology of aging is the decline in generation of new T cells. Since the thymus is the site of T-cell differentiation and maturation, any changes in thymic function would be expected to affect the output of T cells. Indeed, thymic involution has long been considered instrumental in the age-related decline in T-cell generation.30 Beginning around puberty, progressive atrophy of the thymus involves profound changes in the structure and cytokine milieu of the thymus, and results in a dwindling of T-cell production. Although thymic involution is a physiological process that starts early in life, the progressive decline in T-cell production eventually takes its toll on the immune system late in life (Figure 3).
Figure 3.
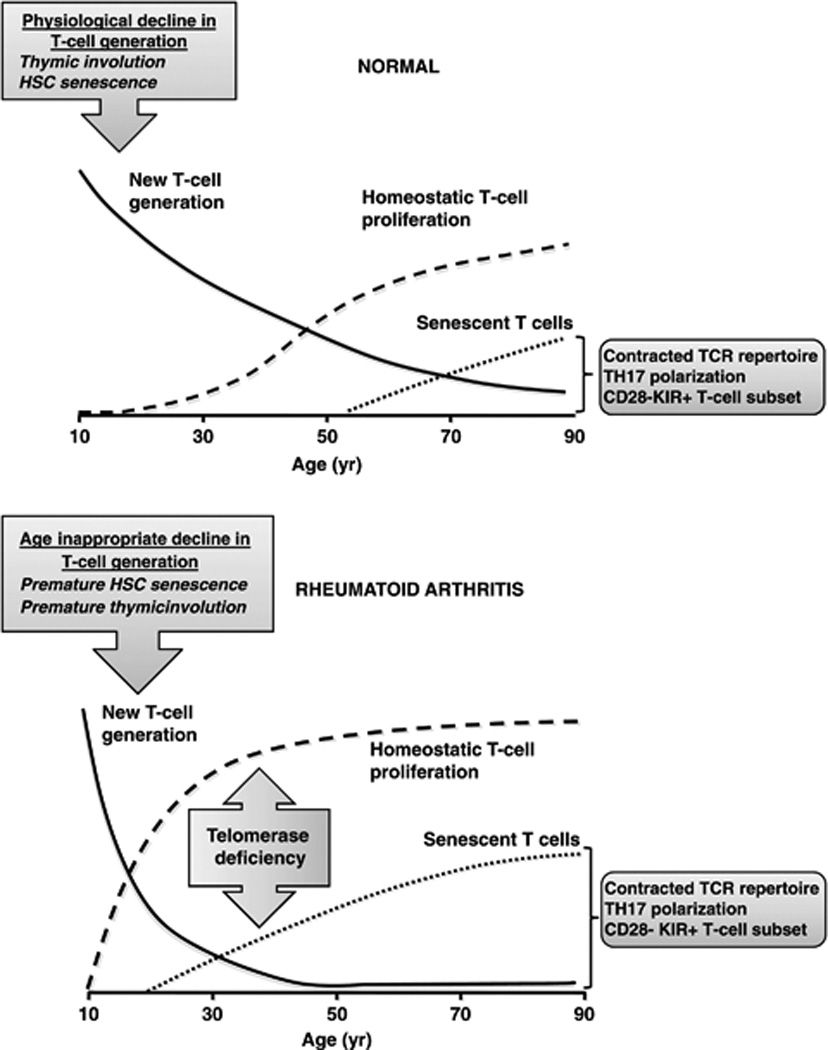
Accelerated decline in T-cell generation leads to premature emergence of senescent T cells in RA. In healthy individuals, the generation of new T cells progressively declines. This decline is compensated for by the homeostatic proliferation of mature T cells in the periphery. With age, progressively fewer new T cells enter the T-cell compartment and mature T cells must increasingly proliferate to fill the void. Eventually, the continually replicating mature T cells become exhausted and take on a senescent phenotype characterized by blunted replicative potential, contracted T-cell repertoire, TH17 polarization, loss of CD28 expression, and de novo expression of stimulatory receptors such as killer-like immunoglobulin receptors (KIR). In RA patients, T-cell generation is age-inappropriately decreased, matching that of healthy individuals 20–30 years older; consequently, homeostatic T-cell proliferation is increased, resulting in premature senescence of T cells. In (normal) aging, the progressive decline in T-cell generation hinges on involution of the thymus, but may also involve senescence of haematopoietic stem cells (HSC). In RA, the age-inappropriate decline in T-cell generation, and hence T-cell senescence, has been ascribed to premature thymic involution or premature HSC senescence. Premature T-cell senescence has also been attributed to deficiency in telomerase activity in mature T cells. The line graphs are adapted from Figure 1 in Weyand and Goronzy 2002.49
An alternative explanation for the decline in T-cell generation may lie in a drop in the supply of T-cell progenitors to the thymus. Since immature T cells in the thymus must be continually replenished by progenitor cells derived from haematopoietic stem cells (HSCs) in the bone marrow, demise of HSCs could conceivably account for the decline in T-cell generation. Much of the investigation into this possibility has exploited the ability of telomeres—structures at the end of chromosomes that protect against chromosomal instability—to serve as markers of cellular division and ultimately cellular senescence. Telomere length is referred to as a ‘mitotic clock’,31 because telomeres in dividing cells progressively erode until a limiting degree of shortening is attained and the cells permanently withdraw from the cell cycle. Demand for new immune cells, and hence for division of HSCs, is high. For this reason, HSCs express telomerase, an enzyme that attenuates telomere attrition. Nevertheless, telomere shortening does occur in HSCs as a function of age.31 Furthermore, in HSC-transfer experiments in mice, HSCs derived from aged donors repopulated the depleted lymphoid compartment of the host less effectively than did HSCs from young donors,32 suggesting that HSC replication in old age is defective.
But total numbers of HSCs do not appear to differ between the young and the elderly; rather, aging is characterized by a shift in HSC subpopulations.32 HSCs can be classified according to their fixed differentiation potential: lymphoid-biased HSCs give rise predominantly to lymphoid cells (e.g., T cells and B cells), myeloid-biased HSCs to myeloid cells (e.g., DCs and macrophages), and “balanced” HSCs to about 90% lymphoid and 10% myeloid cells. The different HSC subpopulations have distinct self-renewal capacities, such that lymphoid-biased HSCs have a shorter lifespan than do myeloid-biased HSCs.32 Indeed, studies in mice have shown that the aged HSC compartment is depleted of lymphoid-biased HSCs and enriched in myeloid-biased HSCs.33 The depletion of lymphoid-biased HSCs could translate to an insufficiency in the supply of precursor T cells and thus account for the age-related decline in T-cell generation.
Although evidence of an aging-related defect in HSC replication is increasing, thymic involution is probably the primary cause of the age-related decline in T-cell output, because naïve T-cell repopulation after bone marrow transplantation is much more severely compromised in breast cancer patients aged 50 and older than in younger patients.34
Autoimmunity rising: loss of T-cell diversity and co-stimulation control
The decline in T-cell generation, invoked as central to immunosenescence, results in a reduction in T-cell diversity.30 The T-cell system is under homeostatic control: generation of new T cells by the thymus, elimination of T cells by apoptosis, and self-replication of existing T cells in the periphery are interlinked processes that are finely counterpoised to maintain the total size of the T-cell system. Thus, a decline in generation of new T cells elicits a compensatory increase in proliferation of naïve and memory T cells in the periphery, a process that is enhanced in aging (Figure 3).30 Such homeostatic proliferation of peripheral T cells is dependent on interaction of the TCR with the self-peptide/MHC complex that was recognized by the specific TCR during positive selection of the T cell in the thymus.35, 36 Animal studies have shown that naïve T cells adoptively transferred to T-cell-depleted mice undergo homeostatic, self-peptide-driven proliferation to fill the deficient lymphoid compartment of the host.35 However, not all T cells are able to proliferate in this manner—a selectivity ascribed to differences in TCR affinity for the self-peptides inducing proliferation35—and it has thus been suggested that homeostatic proliferation involves competitive exclusion of TCRs.37 The increase in homeostatic T-cell proliferation has been proposed to synergize with the decline in T-cell generation to reduce T-cell diversity and bias the immune system toward recognition of self, i.e., autoimmunity.37
Another way in which age-dependent changes in the T-cell compartment may promote an autoimmune diathesis is through changes in the receptors expressed by T cells. With the progressive decline in T-cell generation and increase in antigenic load comes a shift towards predominance of memory T cells, many of which undergo changes in receptor expression and thereby in function. Most striking is the emergence of a subset of memory T cells that is characterized by a contracted TCR repertoire and the loss of CD28 expression. The age-related loss of CD28 expression occurs in both CD4+ and CD8+ T cells but is more common among CD8+ T cells; CD8+ and CD4+ T cells apparently undergo the same phenotypic changes with aging, but CD8+ T cells do so more rapidly.30, 38–40
Despite CD28’s role in ensuring appropriate activation of T cells,15 CD4+CD28− T cells retain their ability to produce lymphokines and appear to be autoreactive,12, 40 a phenomenon attributable to de novo receptor expression.13 In addition to losing expression of a pivotal co-stimulatory receptor, CD4+CD28− T cells acquire expression of other stimulatory receptors, such as killer immunoglobulin-like receptors (KIR), NKG2D, and lymphocyte function-associated antigen 1 (LFA-1), that are not normally expressed on T cells. De novo expression of these receptors on CD28− T cells changes the way in which these T cells interact with their cellular environment, lowering the threshold for antigen-specific activation and even enabling activation independent of the appropriate antigen.13 T cells that are both prone to react with self-antigen and are no longer restricted by a co-stimulation requirement—the central tenet of peripheral tolerance—would skew the immune system toward an autoimmune propensity. Furthermore, deficiency in co-stimulation may amplify this propensity by rendering CD28− T cells resistant to apoptosis.41, 42
The role of CD8+CD28− T cells in inflammation and autoimmunity is more complex. On the one hand, loss of CD28 renders CD8+ T cells unable to produce lymphokines and endows a certain subset of CD8+ T cells with the ability to suppress the antigen-presenting function of DCs.12 On the other hand, de novo expression of NKG2D receptor expression enables a subset of CD8+ T cells to kill cells in an antigen-independent fashion.43, 44
How these CD28− T-cell populations arise is a matter of debate. One argument is that culpability lies in the compensatory proliferation of peripheral T cells that occurs with aging, because the homeostatic cytokine interleukin (IL)-15 is able to drive the generation and proliferation of CD28− T cells.45 However, mounting evidence suggests that repeated antigenic stimulation of T cells may in fact be responsible. In vitro, repeated exposure of T cells to the same antigen suppresses CD28 expression and telomerase activity;11, 12 in vivo, CD28− T cells have shorter telomeres than their CD28+ counterparts.13, 46 Both the total antigenic load and the duration of exposure to specific antigens increase with age. In particular, chronic viral infections are thought to put increasing pressure on the immune system by imposing a need for constant immune surveillance and thus clonal expansion of immune cells. Cytomegalovirus (CMV) is a virus that establishes chronic infection and is found in as much as 50% of the adult population and 90% of elderly individuals.47 The presence of antibodies against CMV is associated with an increase in numbers of CD28− T cells—though whether or not this is simply a reflection of the increase in CMV infection with age, which is itself associated with the emergence of CD28− T cells, is unclear.47 Nonetheless, longitudinal study of CMV-seronegative patients who experienced a primary CMV infection after receiving a CMV-seropositive kidney graft showed that CMV-specific CD4+CD28− T cells develop following recovery from CMV infection, independently of patient age.48 Thus, chronic CMV infection has been postulated to be a major contributor to immunosenescence.47
T-cell aging in RA: cause or consequence?
That T-cell aging is accelerated in RA has long been recognized. Still unclear is the extent to which such premature immunosenescence increases susceptibility to RA or is a result of RA, contributing to the pathology only once the disease is already established.
In RA patients, thymic output of T cells is age-inappropriately decreased, matching that of healthy individuals 20–30 years older (Figure 3), and the TCR repertoire is contracted in naïve and memory T cells.7–10, 37, 49 The perturbation in T-cell homeostasis is detected early in RA and is not influenced by the duration of disease,8, 50 suggesting that a premature decline in T-cell diversity may predispose to the development of RA. Indeed, HLA-DR4 alleles that are known genetic risk factors for RA have been proposed to confer susceptibility to RA in part by inducing premature immunosenescence,51, 52 and an increase in homeostatic proliferation of naïve T cells has been shown to predate the onset of RA.27 Children with the autoimmune disease juvenile idiopathic arthritis (JIA) also exhibit abnormalities in T-cell homeostasis that are evident early in the disease and do not progress over the course of the disease (though the mechanisms by which such T-cell perturbations arise may differ between JIA and RA).53, 54 Since children are relatively antigen-inexperienced, the detection of T-cell perturbations in JIA suggests that immunosenescence can potentially develop due to a primary defect that is independent of long-standing exposure to viral or other environmental antigens and lead to autoimmune disease.
Evidence for premature involution of the thymus in RA comes from the delay in reconstitution of naïve T cells following T-cell ablation and autologous haematopoietic stem cell transplantation in patients with treatment-refractory RA.55 However, the notion of HSC senescence as an alternative or additional explanation for the RA-associated reduction in T-cell generation is gaining momentum. Levels of circulating bone marrow-derived progenitor cells were shown to be diminished in RA patients; further, the remaining progenitor cells had markedly shortened telomeres and defective proliferative responses.7
Premature telomere shortening in RA is seen in lymphocytes, too.8, 51 Although accelerated telomere shortening in T cells could reflect either premature senescence of progenitor HSCs or increased homeostatic proliferation of T cells, yet another explanation has recently been advanced: that upregulation of the telomerase enzyme is faulty in RA. Naive CD4+ T cells from RA patients were shown to be defective in inducing telomerase following priming with antigen and to be more susceptible to apoptosis, a defect that was reversed when telomerase activity was artificially restored to the cells.50 However, the survival-enhancing effects of telomerase were independent of telomere length. The recent identification of telomerase as a modulator of Wnt signaling56—a pathway important in promoting cell proliferation and survival—may go some way to explaining this observation. Regardless of the means, a reduction in the survival of naïve CD4+ T cells in the periphery might place further demands on homeostatic proliferation and lead to increased remodeling of the T-cell repertoire. The deficiency in telomerase activity was found to be independent of the duration or severity of RA, suggesting that it may contribute to the development of RA.50
While a decrease in thymic output and in naïve CD4+ T-cell survival may confer susceptibility to RA, it is less clear how other features of immunosenescence—such as the emergence of memory CD28− T cells—impinge on RA. As in aging, clonally expanded CD28− T cells accumulate in RA. The focus in RA has been on CD4+ CD28− T cells, whose frequency in RA is high.9 CD4+ CD28− T cells isolated from RA patients secrete lymphokines without the need for co-stimulatory signals and appear to be autoreactive.9, 57
So, how do these CD28− T cells arise in RA, and what role do they play? They could potentially arise due to the accelerated homeostatic proliferation and, through autoreactivity, contribute to the breach of tolerance that leads to the development of RA. Although recent findings suggest that the emergence of CD4+CD28− T cells in RA does not correlate with the decline in thymic output,52 it remains possible that the increased susceptibility of naïve RA CD4+ T cells to apoptosis promotes the emergence of memory CD4+CD28− T cells through homeostatic remodeling of the T-cell compartment.50 The alternative argument is that the emergence of CD28− T cells is secondary to the development of RA. In this scenario, the generation of CD28− T cells stems from the chronic immune stimulation associated with RA, in a manner analogous to the exhaustion of the immune system in the elderly by CMV-mediated chronic stimulation of the immune system. Loss of CD27 (another T-cell co-stimulatory molecule), gain of cytotoxic potential, and evidence of antigen-driven clonal expansion are features of CD4+CD28− T cells seen both in RA and following primary CMV infection.46, 48 In fact, CD4+ CD28− T cells isolated from RA patients react to CMV, and reactivation of latent CMV by RA-associated inflammation has been proposed as yet another explanation for the generation of CD28− T cells.46, 48 CMV-induced CD28− T cells could potentially promote inflammatory disease either by continuing to respond to persistent CMV antigens or by cross-reacting with autoantigens
A further clue to the contribution of CD28− T cells in RA comes from the nature of their autoreactivity: CD28− T cells appear to react not to autoantigens known to trigger RA in animal models but to more ubiquitous autoantigens, an observation suggested to indicate that, rather than contribute to the initial breach of tolerance, these cells promote more-severe and systemic disease. 9, 13, 46 In support of this concept, accumulation of CD4+ CD28− T cells is especially marked in RA patients who have extra-articular inflammatory complications,58 and CD4+ CD28− T cells are also independently associated with atherosclerotic disease.59
In addition to promoting autoimmunity, CD28− T cells could perpetuate and amplify inflammation and tissue damage in the joints in RA. CD28− T cells exhibit increased expression of the chemokine receptor CX3CR1, which could enhance their ability to infiltrate tissues.13 Indeed, expansion of the CD28− T-cell subset is detected both in the blood and in the synovial tissue of RA patients.52 Once in the synovial joints, these cells could promote joint pathology: By acquiring KIR and NKG2D receptors and cytolytic granules (such as perforin and granzyme B), CD28− T cells take on some NK cell functions, including cytotoxic killing.
How similar, then, are the changes in the T-cell compartment in aging and in RA, and what are the implications? The age-related decline in T-cell generation and diversity appears to hinge on involution of the thymus, possibly with some involvement of HSC senescence; a decline is also seen in RA, and there is evidence that it may stem from premature HSC senescence or thymic involution—but it may also involve apoptotic hypersensitivity of naïve CD4+ T cells. Because it occurs early in the course of RA and is not associated with disease duration, the decline in T-cell generation and diversity has been proposed to contribute to the development of RA. Emergence of CD28− T cells is also common to both aging and RA, but whether this is a cause or a consequence of RA remains to be determined. Nevertheless, findings to date suggest that CD4+CD28− T cells may contribute to the pathogenesis of RA by promoting inflammation and autoimmunity. The role of CD8+CD28− T cells in RA is less clear, with different subtypes of CD8+CD28− T cells shown to have opposing effects on inflammation and tissue destruction.12, 60
THE CHANGING FACE OF NAÏVE CD4+ T CELLS
Aging is also associated with a shift in the predominance of different activated naïve CD4+ T-cell subsets. Antigen-activated naïve CD4+ T cells can be divided into distinct T-cell populations based on the cytokines they produce. For many years, such subdivision was thought to be limited to T-helper (TH) 1 CD4+ T cells and TH2 CD4+ T cells, with a high TH1 to TH2 ratio favoring T-cell-mediated inflammation and a low one favoring antibody-mediated responses. However, an expanded cast of TH cells has since been revealed, and the spotlight is currently on TH17 cells. While TH1 cells (producers of interferon γ) have long been implicated in T-cell-mediated autoimmunity, recent findings suggest that TH17 cells (producers of IL-17) are at the helm of certain autoimmune diseases. RA is one such autoimmune disease: overexpression of IL-17 in the knee joint induces RA-like features in mice; IL-17-positive cells are detected in synovial biopsies from RA patients; and neutralizing antibodies against IL-17 ameliorated disease both in mouse models of RA and in RA patients in Phase I clinical trials.61 Studies in mice showed that aging was associated with a defect in differentiation of CD4+ T cells into TH1 and TH2 cells, while differentiation into TH17 cells remained unaffected.62 Not only were TH17 cells more prevalent, but also the amount of IL-17 produced by each TH17 cell was increased in aged mice. This skewing of the CD4+ T-cell response towards a TH17 bias could potentially promote RA-associated inflammation and joint pathology in the elderly. Still unclear, however, is the extent to which these findings can be extrapolated to aging in humans, as well as the relative importance of different TH subsets in RA.
OVERREACTING MEMORY B CELLS
As in the T-cell compartment, homeostasis in the B-cell compartment appears to be actively maintained. Numbers of mature, peripheral B cells do not decrease with age.63 However, studies in mice suggest that the generation of new, mature B cells does decrease with age but that this is associated with an increase in B-cell longevity, such that total B-cell numbers remain stable.64 B-cell function, on the other hand, is markedly altered in aging. B-cell responses to primary antigenic challenge decrease with age.65, 66 This reduction in overall antibody levels is likely a corollary of the decrease in B-cell generation; the decline in T-cell helper activity also plays an important role, since the CD4+CD28− T cells that prevail in aging no longer express CD40,40 a ligand required for the production of antibodies by naïve B cells.
In contrast to total antibody levels, autoantibody levels increase with advancing age—and memory B cells formed before the onset of immunosenescence are thought to be the culprit. Stacy et al 67 propose two scenarios that might explain the late appearance of autoantibodies. One possibility is that autoantibodies are in fact present in young, healthy individuals but their levels are kept low by tolerance mechanisms; impairment of tolerance with age (as discussed above) would allow autoreactivity to develop. Alternatively, the initial B-cell response may have targeted a foreign, pathogenic antigen, and late-onset autoreactivity may then arise due to cross-recognition of a self-antigen that is newly exposed, because of tissue damage that accumulates over time, or newly formed (neo-antigens), because of inflammation-mediated modification of a self-peptide. Exposure of normally sequestered self-antigens, owing to tissue damage or cartilage destruction, is implicated in the pathogenesis of RA.68–70 In addition, generation of neo-antigens through inflammation-mediated citrullination of self-peptides and ensuing autoantibody targeting of the citrulline-modified peptides are increasingly recognized as key events in the pathogenesis of RA.20, 70, 71
DCs: WHEN THE REGULATORS DON’T REGULATE
The aberrations that typify immunosenescence do not involve changes solely in the adaptive arm of the immune system. Innate immune cells, including DCs, macrophages, neutrophils, and natural killer cells, also become dysregulated with age.72 Of primary interest in the context of this review are DCs, owing to their dual role as both presenters of antigen and inducers of self-tolerance.
The importance of DCs in maintaining self-tolerance is underscored by the finding that DC-depleted mice spontaneously develop autoimmunity.73 However, the recent discovery of functionally distinct DC subsets—plasmacytoid (pDC) and myeloid (mDC)—has hinted at greater complexity within the DC network. Evidence to date suggests that these two DC subsets exert different effects on the immune response and autoimmunity. pDCs can induce the differentiation of B cells into plasma cells and could therefore promote autoantibody production, but they appear to also play a role in the induction of tolerance.24 Consistent with a tolerance-inducing role for pDCs in inflammatory arthritis, selective pDC depletion in mice exacerbated articular pathology and enhanced the T- and B-cell autoimmune responses against the self-antigen type II collagen.74 mDCs, on the other hand, are primarily proinflammatory,24 and exogenously activated and antigen-pulsed mDCs have been shown to induce inflammatory arthritis in mice through both the priming of autoreactive T cells and the production of proinflammatory cytokines.25
As discussed, clonally expanded CD28− T cells in elderly individuals lose their requirement for CD28 co-stimulation.40 Presentation of arthritogenic self-antigens by DCs to autoreactive CD4+CD28− T cells could therefore contribute to the development of RA in the elderly. In addition, changes in the levels and activity of DCs themselves could skew the immune response in elderly individuals. Although a decrease in tolerogenic pDCs or an increase in proinflammatory mDCs could potentially favor the development of RA, such putative shifts in DC subpopulation with age have not yet been definitively demonstrated.75 In contrast, evidence of functional impairment of DCs in aging and its possible contribution to autoimmunity is substantive. DCs from elderly individuals seem to react more strongly than DCs from young individuals to apoptotic cells and to an intracellular self-antigen, human DNA.76 Moreover, the DNA-activated DCs from elderly individuals induced T cells to proliferate, whereas the less-activated DCs from young individuals were unable to do so. DC function is not restricted to antigen presentation and cytokine secretion; it also involves the clearance of cell debris and apoptotic cells through phagocytosis. Efficient clearance of apoptotic cells is important in limiting inflammation and preventing autoimmunity: if left in the tissue, apoptotic cells undergo secondary necrosis, resulting in release of proinflammatory mediators and intracellular self-antigens. DCs derived from elderly individuals were shown to have impaired phagocytic capacity, a defect that could result in the accumulation of apoptotic cells.77 Thus, aberrations in DC function with age may result not only in increased reactivity to self-antigens, but also in increased exposure to self-antigens.
CYTOKINES: STOKING THE SYNOVIAL FIRE
Both aging and RA are considered states of chronic inflammation. Serum levels of the proinflammatory cytokines tumor necrosis factor (TNF) and IL-6, key players in the pathogenesis of RA, are elevated in the elderly.78 The chronic, systemic upregulation of a cluster of proinflammatory cytokines, including TNF and IL-6, in the elderly is known as ‘inflamm-aging’78 and is thought to form part of the so-called immune-risk phenotype, a collection of immune-related parameters that are predictive of morbidity and mortality in the elderly.79
TNF and IL-6 have pleiotropic functions in the immune system; accordingly, they are involved in both the autoimmune responses and the tissue destruction that underpin RA. TNF induces (i) matrix metalloproteinase release from leukocytes, thereby driving tissue destruction; (ii) adhesion-molecule expression on endothelial cells, thus promoting infiltration of inflammatory cells into the synovium; (iii) angiogenesis and synovial-fibroblast hyperplasia, which support pannus formation; and (iv) T-cell activation, resulting in perpetuation of autoimmune responses.26 IL-6 modulates autoimmunity by promoting T-cell activation and antibody production. It also drives tissue destruction by activating macrophages and osteoclasts, which mediate cell matrix degradation and bone resorption, respectively.26, 80 The importance of TNF and IL-6 in inflammatory arthritis has been borne out by the therapeutic efficacy of biologic agents targeting these cytokines. Blockade of TNF is now the standard treatment for RA, and an antibody targeting the IL-6 receptor provides some therapeutic benefit.80, 81
The increase in TNF and IL-6 levels with advancing age would thus be expected to promote multiple processes involved in the pathogenesis of RA. Besides enhancing the recruitment and activation of immune cells, the increase in circulating TNF in the elderly may reduce both the expression of CD28 and the activity of telomerase in T cells.82 Thus, inflammaaging could potentially augment both the synovitis and the systemic autoimmunity associated with RA.
But what is the source of these elevated cytokines in the elderly? Although macrophages, innate effector cells central to the synovitis in RA, are generally thought to be the primary producers of these cytokines,26, 78 studies to date indicate that aging is in fact associated with a decrease in macrophage production of proinflammatory cytokines.83, 84 In contrast, senescence of fibroblast and epithelial cells is associated with a change in the secretory profile of these cells, involving an increase in secretion of multiple growth factors, adhesion molecules, and proinflammatory cytokines, including IL-6.85 In addition, aging is associated with low-grade inflammation in adipose tissue, and TNF and IL-6 expression was shown to be greater in adipocytes from aged mice than in adipocytes from young mice.86
UPSHOT
Increasing life expectancy means that research into the immunobiology of aging is of increasing importance. Aging has a profound effect on the immune system, and the elderly are more susceptible to infections and cancer, as well as to certain autoimmune diseases, such as RA. RA is characterized by pathological features typical of immune dysfunction in the elderly, including loss of T-cell diversity and dysregulation of T-cell reactivity, increase in levels of autoantibodies and proinflammatory cytokines, and perturbation of DC activation (Figure 4). On the one hand, the increased autoimmunity associated with aging of the immune system could underpin the systemic breach of tolerance that leads to RA, a notion in keeping with the documented increase in incidence of RA in the elderly; a defect that results in premature aging of the immune system might also predispose to the development of RA in young adults. On the other hand, certain features of immunosenescence might be the result, rather than the cause, of RA. Even so, immunosenescence arising secondary to the development of RA might perpetuate and amplify the disease, by augmenting systemic autoreactivity or synovial joint inflammation. Not all aspects of immunosenescence, however, favor the development of RA,60, 84 and differences in clinical features between early-onset and late-onset RA have been documented.87 A clear picture of the relationship between immunosenescence and RA is still being sought.
Figure 4.
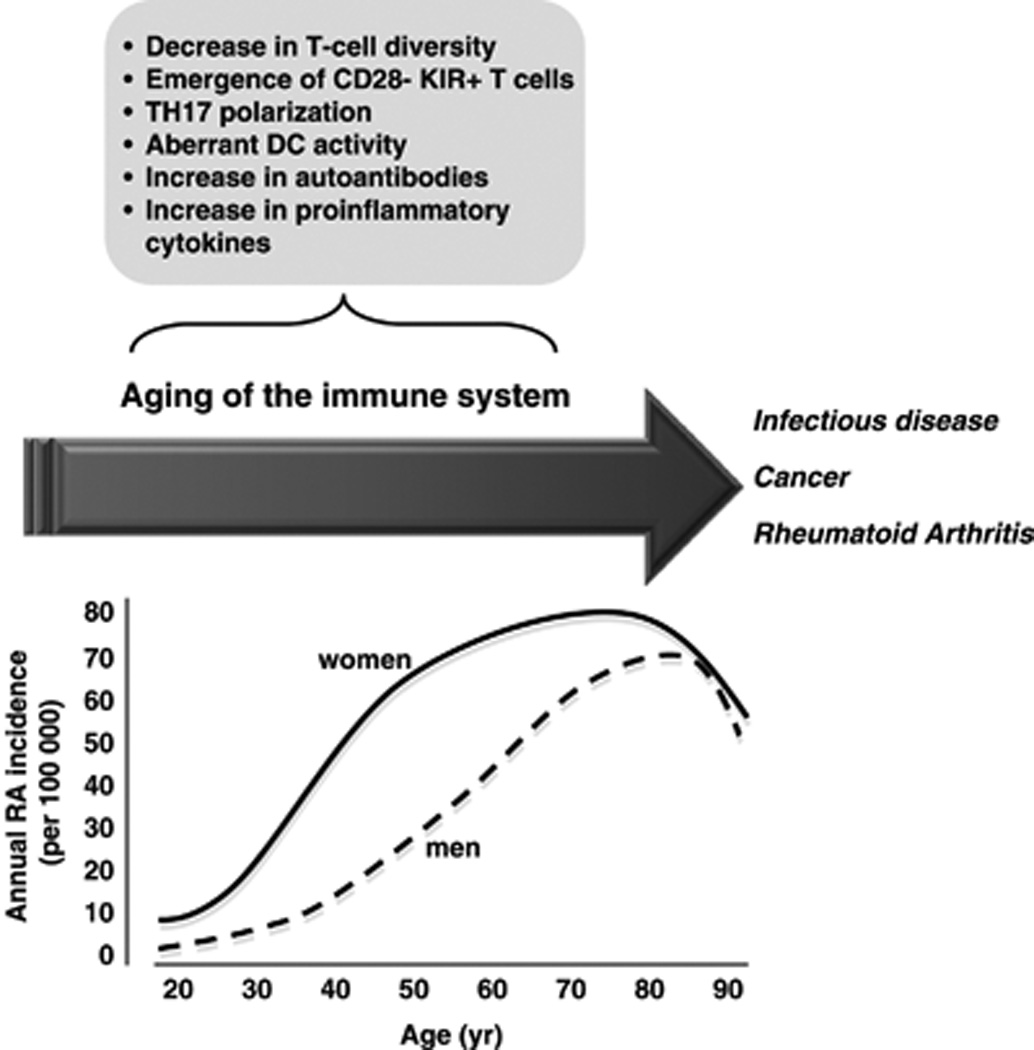
Aging of the immune system increases susceptibility to disease. The arrow indicates the progressive decline in integrity of the immune system with age. Such aging of the immune system, or immunosenescence, involves a decrease in T-cell generation and diversity, and an increase in levels of proinflammatory cytokines and autoantibodies. Loss of peripheral T-cell tolerance, owing to dysregulation of dendritic cells (DCs) and T cells, may also be involved. The decline in immune function is paralleled by an increase in incidence of several diseases, including infectious disease, cancer, and rheumatoid arthritis (RA). The line graph of RA incidence is a schematic approximation of the general trend in incidence with age. That RA incidence is generally higher in women, increases progressively with age, and decreases in the oldest age groups is a finding consistent across multiple studies; the late decrease is observed earlier among women than among men.1–4
Like RA, aging can be considered a heterogeneous, polygenic disorder. Further insight into the links between immunosenescence and RA might enable the identification of elderly individuals who are at increased risk of developing RA. Subpopulations of elderly individuals with specific immunosenescent parameters could potentially be screened for predictors of RA; although reliable predictors of the development of RA remain to be attained, anti-CCP antibodies88 appear promising and other prognostic biomarkers are being developed. In addition, unraveling the mechanisms underlying immunosenescence should shed light on the pathogenesis, and inform strategies for the treatment, of RA in young adults. Several strategies aimed at reversing T-cell senescence are close to or in clinical trials,89 and could potentially present a novel approach to the treatment of RA.
ACKNOWLEDGMENTS
This work was funded by NIH R21-AI-069160 and R01-AR-054822 to W.H.R. W.H.R. also receives funding from the Department of Veterans Affairs.
Sponsor’s Role: None
Footnotes
Conflict of Interest: The editor in chief has reviewed the conflict of interest checklist provided by the authors and has determined that the authors have no financial or any other kind of personal conflicts with this paper.
Author Contributions: Both authors contributed to the writing of this manuscript.
REFERENCES
- 1.Doran MF, Pond GR, Crowson CS, et al. Trends in incidence and mortality in rheumatoid arthritis in Rochester, Minnesota, over a forty-year period. Arthritis Rheum. 2002;46:625–631. doi: 10.1002/art.509. [DOI] [PubMed] [Google Scholar]
- 2.Pedersen JK, Svendsen AJ, Horslev-Petersen K. Incidence of Rheumatoid Arthritis in the Southern part of Denmark from 1995 to 2001. Open Rheumatol J. 2007;1:18–23. doi: 10.2174/1874312900701010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riise T, Jacobsen BK, Gran JT. Incidence and prevalence of rheumatoid arthritis in the county of Troms, northern Norway. J Rheumatol. 2000;27:1386–1389. [PubMed] [Google Scholar]
- 4.Symmons DP, Barrett EM, Bankhead CR, et al. The incidence of rheumatoid arthritis in the United Kingdom: Results from the Norfolk Arthritis Register. Br J Rheumatol. 1994;33:735–739. doi: 10.1093/rheumatology/33.8.735. [DOI] [PubMed] [Google Scholar]
- 5.Baecklund E, Iliadou A, Askling J, et al. Association of chronic inflammation, not its treatment, with increased lymphoma risk in rheumatoid arthritis. Arthritis Rheum. 2006;54:692–701. doi: 10.1002/art.21675. [DOI] [PubMed] [Google Scholar]
- 6.Doran MF, Crowson CS, Pond GR, et al. Frequency of infection in patients with rheumatoid arthritis compared with controls: A population-based study. Arthritis Rheum. 2002;46:2287–2293. doi: 10.1002/art.10524. [DOI] [PubMed] [Google Scholar]
- 7.Colmegna I, Diaz-Borjon A, Fujii H, et al. Defective proliferative capacity and accelerated telomeric loss of hematopoietic progenitor cells in rheumatoid arthritis. Arthritis Rheum. 2008;58:990–1000. doi: 10.1002/art.23287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koetz K, Bryl E, Spickschen K, et al. T cell homeostasis in patients with rheumatoid arthritis. Proc Natl Acad Sci U S A. 2000;97:9203–9208. doi: 10.1073/pnas.97.16.9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmidt D, Goronzy JJ, Weyand CM. CD4+ CD7− CD28− T cells are expanded in rheumatoid arthritis and are characterized by autoreactivity. J Clin Invest. 1996;97:2027–2037. doi: 10.1172/JCI118638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagner UG, Koetz K, Weyand CM, et al. Perturbation of the T cell repertoire in rheumatoid arthritis. Proc Natl Acad Sci U S A. 1998;95:14447–14452. doi: 10.1073/pnas.95.24.14447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valenzuela HF, Effros RB. Divergent telomerase and CD28 expression patterns in human CD4 and CD8 T cells following repeated encounters with the same antigenic stimulus. Clin Immunol. 2002;105:117–125. doi: 10.1006/clim.2002.5271. [DOI] [PubMed] [Google Scholar]
- 12.Vallejo AN. CD28 extinction in human T cells: altered functions and the program of T-cell senescence. Immunol Rev. 2005;205:158–169. doi: 10.1111/j.0105-2896.2005.00256.x. [DOI] [PubMed] [Google Scholar]
- 13.Weng NP, Akbar AN, Goronzy J. CD28(−) T cells: Their role in the age-associated decline of immune function. Trends Immunol. 2009;30:306–312. doi: 10.1016/j.it.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldrath AW, Bevan MJ. Selecting and maintaining a diverse T-cell repertoire. Nature. 1999;402:255–262. doi: 10.1038/46218. [DOI] [PubMed] [Google Scholar]
- 15.Boesteanu AC, Katsikis PD. Memory T cells need CD28 costimulation to remember. Semin Immunol. 2009;21:69–77. doi: 10.1016/j.smim.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van der Helm-van Mil AH, Wesoly JZ, Huizinga TW. Understanding the genetic contribution to rheumatoid arthritis. Curr Opin Rheumatol. 2005;17:299–304. doi: 10.1097/01.bor.0000160780.13012.be. [DOI] [PubMed] [Google Scholar]
- 17.Thomas R, McIlraith M, Davis LS, et al. Rheumatoid synovium is enriched in CD45RBdim mature memory T cells that are potent helpers for B cell differentiation. Arthritis Rheum. 1992;35:1455–1465. doi: 10.1002/art.1780351209. [DOI] [PubMed] [Google Scholar]
- 18.Panayi GS. Even though T-cell-directed trials have been of limited success, is there reason for optimism? Nat Clin Pract Rheumatol. 2006;2:58–59. doi: 10.1038/ncprheum0094. [DOI] [PubMed] [Google Scholar]
- 19.Schroder AE, Greiner A, Seyfert C, et al. Differentiation of B cells in the nonlymphoid tissue of the synovial membrane of patients with rheumatoid arthritis. Proc Natl Acad Sci U S A. 1996;93:221–225. doi: 10.1073/pnas.93.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rantapaa-Dahlqvist S, de Jong BA, Berglin E, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48:2741–2749. doi: 10.1002/art.11223. [DOI] [PubMed] [Google Scholar]
- 21.Khachigian LM. Collagen antibody-induced arthritis. Nat Protoc. 2006;1:2512–2516. doi: 10.1038/nprot.2006.393. [DOI] [PubMed] [Google Scholar]
- 22.Uysal H, Bockermann R, Nandakumar KS, et al. Structure and pathogenicity of antibodies specific for citrullinated collagen type II in experimental arthritis. J Exp Med. 2009;206:449–462. doi: 10.1084/jem.20081862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edwards JC, Szczepanski L, Szechinski J, et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 24.Lebre MC, Tak PP. Dendritic cells in rheumatoid arthritis: Which subset should be used as a tool to induce tolerance? Hum Immunol. 2009;70:321–324. doi: 10.1016/j.humimm.2009.02.006. Epub 2009 Feb 21. [DOI] [PubMed] [Google Scholar]
- 25.Leung BP, Conacher M, Hunter D, et al. A novel dendritic cell-induced model of erosive inflammatory arthritis: Distinct roles for dendritic cells in T cell activation and induction of local inflammation. J Immunol. 2002;169:7071–7077. doi: 10.4049/jimmunol.169.12.7071. [DOI] [PubMed] [Google Scholar]
- 26.McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7:429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- 27.Goronzy JJ, Weyand CM. Rheumatoid arthritis. Immunol Rev. 2005;204:55–73. doi: 10.1111/j.0105-2896.2005.00245.x. [DOI] [PubMed] [Google Scholar]
- 28.Klareskog L, Padyukov L, Ronnelid J, et al. Genes, environment and immunity in the development of rheumatoid arthritis. Curr Opin Immunol. 2006;18:650–655. doi: 10.1016/j.coi.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 29.de Pablo P, Chapple IL, Buckley CD, et al. Periodontitis in systemic rheumatic diseases. Nat Rev Rheumatol. 2009;5:218–224. doi: 10.1038/nrrheum.2009.28. [DOI] [PubMed] [Google Scholar]
- 30.Goronzy JJ, Lee WW, Weyand CM. Aging and T-cell diversity. Exp Gerontol. 2007;42:400–406. doi: 10.1016/j.exger.2006.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaziri H, Dragowska W, Allsopp RC, et al. Evidence for a mitotic clock in human hematopoietic stem cells: loss of telomeric DNA with age. Proc Natl Acad Sci U S A. 1994;91:9857–9860. doi: 10.1073/pnas.91.21.9857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muller-Sieburg C, Sieburg HB. Stem cell aging: survival of the laziest? Cell Cycle. 2008;7:3798–3804. doi: 10.4161/cc.7.24.7214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cho RH, Sieburg HB, Muller-Sieburg CE. A new mechanism for the aging of hematopoietic stem cells: Aging changes the clonal composition of the stem cell compartment but not individual stem cells. Blood. 2008;111:5553–5561. doi: 10.1182/blood-2007-11-123547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hakim FT, Memon SA, Cepeda R, et al. Age-dependent incidence, time course, and consequences of thymic renewal in adults. J Clin Invest. 2005;115:930–939. doi: 10.1172/JCI22492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ernst B, Lee DS, Chang JM, et al. The peptide ligands mediating positive selection in the thymus control T cell survival and homeostatic proliferation in the periphery. Immunity. 1999;11:173–181. doi: 10.1016/s1074-7613(00)80092-8. [DOI] [PubMed] [Google Scholar]
- 36.Goldrath AW, Bevan MJ. Low-affinity ligands for the TCR drive proliferation of mature CD8+ T cells in lymphopenic hosts. Immunity. 1999;11:183–190. doi: 10.1016/s1074-7613(00)80093-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goronzy JJ, Weyand CM. Thymic function and peripheral T-cell homeostasis in rheumatoid arthritis. Trends Immunol. 2001;22:251–255. doi: 10.1016/s1471-4906(00)01841-x. [DOI] [PubMed] [Google Scholar]
- 38.Czesnikiewicz-Guzik M, Lee WW, Cui D, et al. T cell subset-specific susceptibility to aging. Clin Immunol. 2008;127:107–118. doi: 10.1016/j.clim.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Posnett DN, Sinha R, Kabak S, et al. Clonal populations of T cells in normal elderly humans: The T cell equivalent to "benign monoclonal gammapathy". J Exp Med. 1994;179:609–618. doi: 10.1084/jem.179.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weyand CM, Brandes JC, Schmidt D, et al. Functional properties of CD4+ CD28− T cells in the aging immune system. Mech Ageing Dev. 1998;102:131–147. doi: 10.1016/s0047-6374(97)00161-9. [DOI] [PubMed] [Google Scholar]
- 41.Vallejo AN, Schirmer M, Weyand CM, et al. Clonality and longevity of CD4+CD28null T cells are associated with defects in apoptotic pathways. J Immunol. 2000;165:6301–6307. doi: 10.4049/jimmunol.165.11.6301. [DOI] [PubMed] [Google Scholar]
- 42.Spaulding C, Guo W, Effros RB. Resistance to apoptosis in human CD8+ T cells that reach replicative senescence after multiple rounds of antigen-specific proliferation. Exp Gerontol. 1999;34:633–644. doi: 10.1016/s0531-5565(99)00033-9. [DOI] [PubMed] [Google Scholar]
- 43.Bauer S, Groh V, Wu J, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285:727–729. doi: 10.1126/science.285.5428.727. [DOI] [PubMed] [Google Scholar]
- 44.Verneris MR, Karami M, Baker J, et al. Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ T cells. Blood. 2004;103:3065–3072. doi: 10.1182/blood-2003-06-2125. [DOI] [PubMed] [Google Scholar]
- 45.Chiu WK, Fann M, Weng NP. Generation and growth of CD28nullCD8+ memory T cells mediated by IL-15 and its induced cytokines. J Immunol. 2006;177:7802–7810. doi: 10.4049/jimmunol.177.11.7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thewissen M, Somers V, Hellings N, et al. CD4+CD28null T cells in autoimmune disease: Pathogenic features and decreased susceptibility to immunoregulation. J Immunol. 2007;179:6514–6523. doi: 10.4049/jimmunol.179.10.6514. [DOI] [PubMed] [Google Scholar]
- 47.Vasto S, Colonna-Romano G, Larbi A, et al. Role of persistent CMV infection in configuring T cell immunity in the elderly. Immun Ageing. 2007;4:2. doi: 10.1186/1742-4933-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Leeuwen EM, Remmerswaal EB, Vossen MT, et al. Emergence of a CD4+CD28− granzyme B+, cytomegalovirus-specific T cell subset after recovery of primary cytomegalovirus infection. J Immunol. 2004;173:1834–1841. doi: 10.4049/jimmunol.173.3.1834. [DOI] [PubMed] [Google Scholar]
- 49.Weyand CM, Goronzy JJ. Premature immunosenescence in rheumatoid arthritis. J Rheumatol. 2002;29:1141–1146. [PubMed] [Google Scholar]
- 50.Fujii H, Shao L, Colmegna I, et al. Telomerase insufficiency in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2009;106:4360–4365. doi: 10.1073/pnas.0811332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schonland SO, Lopez C, Widmann T, et al. Premature telomeric loss in rheumatoid arthritis is genetically determined and involves both myeloid and lymphoid cell lineages. Proc Natl Acad Sci U S A. 2003;100:13471–13476. doi: 10.1073/pnas.2233561100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thewissen M, Somers V, Venken K, et al. Analyses of immunosenescent markers in patients with autoimmune disease. Clin Immunol. 2007;123:209–218. doi: 10.1016/j.clim.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 53.Lorenzi AR, Morgan TA, Anderson A, et al. Thymic function in juvenile idiopathic arthritis. Ann Rheum Dis. 2009;68:983–990. doi: 10.1136/ard.2008.088112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prelog M, Schwarzenbrunner N, Sailer-Hock M, et al. Premature aging of the immune system in children with juvenile idiopathic arthritis. Arthritis Rheum. 2008;58:2153–2162. doi: 10.1002/art.23599. [DOI] [PubMed] [Google Scholar]
- 55.Kapoor S, Wilson AG, Sharrack B, et al. Haemopoietic stem cell transplantation--an evolving treatment for severe autoimmune and inflammatory diseases in rheumatology, neurology and gastroenterology. Hematology. 2007;12:179–191. doi: 10.1080/10245330701255106. [DOI] [PubMed] [Google Scholar]
- 56.Park JI, Venteicher AS, Hong JY, et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature. 2009;460:66–72. doi: 10.1038/nature08137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Park W, Weyand CM, Schmidt D, et al. Co-stimulatory pathways controlling activation and peripheral tolerance of human CD4+CD28− T cells. Eur J Immunol. 1997;27:1082–1090. doi: 10.1002/eji.1830270507. [DOI] [PubMed] [Google Scholar]
- 58.Martens PB, Goronzy JJ, Schaid D, et al. Expansion of unusual CD4+ T cells in severe rheumatoid arthritis. Arthritis Rheum. 1997;40:1106–1114. doi: 10.1002/art.1780400615. [DOI] [PubMed] [Google Scholar]
- 59.Nadareishvili ZG, Li H, Wright V, et al. Elevated pro-inflammatory CD4+CD28− lymphocytes and stroke recurrence and death. Neurology. 2004;63:1446–1451. doi: 10.1212/01.wnl.0000142260.61443.7c. [DOI] [PubMed] [Google Scholar]
- 60.Davila E, Kang YM, Park YW, et al. Cell-based immunotherapy with suppressor CD8+ T cells in rheumatoid arthritis. J Immunol. 2005;174:7292–7301. doi: 10.4049/jimmunol.174.11.7292. [DOI] [PubMed] [Google Scholar]
- 61.van den Berg WB, Miossec P. IL-17 as a future therapeutic target for rheumatoid arthritis. Nat Rev Rheumatol. 2009;5:549–553. doi: 10.1038/nrrheum.2009.179. [DOI] [PubMed] [Google Scholar]
- 62.Eaton SM, Burns EM, Kusser K, et al. Age-related defects in CD4 T cell cognate helper function lead to reductions in humoral responses. J Exp Med. 2004;200:1613–1622. doi: 10.1084/jem.20041395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Davey FR, Huntington S. Age-related variation in lymphocyte subpopulations. Gerontology. 1977;23:381–389. doi: 10.1159/000212212. [DOI] [PubMed] [Google Scholar]
- 64.Kline GH, Hayden TA, Klinman NR. B cell maintenance in aged mice reflects both increased B cell longevity and decreased B cell generation. J Immunol. 1999;162:3342–3349. [PubMed] [Google Scholar]
- 65.Goidl EA, Innes JB, Weksler ME. Immunological studies of aging. II. Loss of IgG and high avidity plaque-forming cells and increased suppressor cell activity in aging mice. J Exp Med. 1976;144:1037–1048. doi: 10.1084/jem.144.4.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zharhary D, Klinman NR. Antigen responsiveness of the mature and generative B cell populations of aged mice. J Exp Med. 1983;157:1300–1308. doi: 10.1084/jem.157.4.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stacy S, Krolick KA, Infante AJ, et al. Immunological memory and late onset autoimmunity. Mech Ageing Dev. 2002;123:975–985. doi: 10.1016/s0047-6374(02)00035-0. [DOI] [PubMed] [Google Scholar]
- 68.Hueber W, Kidd BA, Tomooka BH, et al. Antigen microarray profiling of autoantibodies in rheumatoid arthritis. Arthritis Rheum. 2005;52:2645–2655. doi: 10.1002/art.21269. [DOI] [PubMed] [Google Scholar]
- 69.Kinloch A, Tatzer V, Wait R, et al. Identification of citrullinated alpha-enolase as a candidate autoantigen in rheumatoid arthritis. Arthritis Res Ther. 2005;7:R1421–R1429. doi: 10.1186/ar1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Klareskog L, Ronnelid J, Lundberg K, et al. Immunity to citrullinated proteins in rheumatoid arthritis. Annu Rev Immunol. 2008;26:651–675. doi: 10.1146/annurev.immunol.26.021607.090244. [DOI] [PubMed] [Google Scholar]
- 71.Lundberg K, Nijenhuis S, Vossenaar ER, et al. Citrullinated proteins have increased immunogenicity and arthritogenicity and their presence in arthritic joints correlates with disease severity. Arthritis Res Ther. 2005;7:R458–R467. doi: 10.1186/ar1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Plackett TP, Boehmer ED, Faunce DE, et al. Aging and innate immune cells. J Leukoc Biol. 2004;76:291–299. doi: 10.1189/jlb.1103592. [DOI] [PubMed] [Google Scholar]
- 73.Ohnmacht C, Pullner A, King SB, et al. Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J Exp Med. 2009;206:549–559. doi: 10.1084/jem.20082394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jongbloed SL, Benson RA, Nickdel MB, et al. Plasmacytoid dendritic cells regulate breach of self-tolerance in autoimmune arthritis. J Immunol. 2009;182:963–968. doi: 10.4049/jimmunol.182.2.963. [DOI] [PubMed] [Google Scholar]
- 75.Shurin MR, Shurin GV, Chatta GS. Aging and the dendritic cell system: Implications for cancer. Crit Rev Oncol Hematol. 2007;64:90–105. doi: 10.1016/j.critrevonc.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Agrawal A, Tay J, Ton S, et al. Increased reactivity of dendritic cells from aged subjects to self-antigen, the human DNA. J Immunol. 2009;182:1138–1145. doi: 10.4049/jimmunol.182.2.1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Agrawal A, Agrawal S, Cao JN, et al. Altered innate immune functioning of dendritic cells in elderly humans: A role of phosphoinositide 3-kinase-signaling pathway. J Immunol. 2007;178:6912–6922. doi: 10.4049/jimmunol.178.11.6912. [DOI] [PubMed] [Google Scholar]
- 78.Franceschi C, Bonafe M, Valensin S, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- 79.Boren E, Gershwin ME. Inflamm-aging: autoimmunity, and the immune-risk phenotype. Autoimmun Rev. 2004;3:401–406. doi: 10.1016/j.autrev.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 80.Smolen JS, Aletaha D, Koeller M, et al. New therapies for treatment of rheumatoid arthritis. Lancet. 2007;370:1861–1874. doi: 10.1016/S0140-6736(07)60784-3. [DOI] [PubMed] [Google Scholar]
- 81.Scheinecker C, Smolen J, Yasothan U, et al. Nat Rev Drug Discov. 2009;8:273–274. doi: 10.1038/nrd2863. [DOI] [PubMed] [Google Scholar]
- 82.Parish ST, Wu JE, Effros RB. Modulation of T lymphocyte replicative senescence via TNF-{alpha} inhibition: Role of caspase-3. J Immunol. 2009;182:4237–4243. doi: 10.4049/jimmunol.0803449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Beharka AA, Meydani M, Wu D, et al. Interleukin-6 production does not increase with age. J Gerontol A Biol Sci Med Sci. 2001;56:B81–B88. doi: 10.1093/gerona/56.2.b81. [DOI] [PubMed] [Google Scholar]
- 84.Renshaw M, Rockwell J, Engleman C, et al. Cutting edge: Impaired Toll-like receptor expression and function in aging. J Immunol. 2002;169:4697–4701. doi: 10.4049/jimmunol.169.9.4697. [DOI] [PubMed] [Google Scholar]
- 85.Coppe JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wu D, Ren Z, Pae M, et al. Aging up-regulates expression of inflammatory mediators in mouse adipose tissue. J Immunol. 2007;179:4829–4839. doi: 10.4049/jimmunol.179.7.4829. [DOI] [PubMed] [Google Scholar]
- 87.Ramos-Casals M, Garcia-Carrasco M, Brito MP, et al. Autoimmunity and geriatrics: clinical significance of autoimmune manifestations in the elderly. Lupus. 2003;12:341–355. doi: 10.1191/0961203303lu383ed. [DOI] [PubMed] [Google Scholar]
- 88.Nielen MM, van Schaardenburg D, Reesink HW, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: A study of serial measurements in blood donors. Arthritis Rheum. 2004;50:380–386. doi: 10.1002/art.20018. [DOI] [PubMed] [Google Scholar]
- 89.Goldberg GL, Zakrzewski JL, Perales MA, et al. Clinical strategies to enhance T cell reconstitution. Semin Immunol. 2007;19:289–296. doi: 10.1016/j.smim.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]