

Abstract
The eukaryotic translation initiation factor 4G (eIF4G) proteins play a critical role in the recruitment of the translational machinery to mRNA. The eIF4Gs are phosphoproteins. However, the location of the phosphorylation sites, how phosphorylation of these proteins is modulated and the identity of the intracellular signaling pathways regulating eIF4G phosphorylation have not been established. In this report, two-dimensional phosphopeptide mapping demonstrates that the phosphorylation state of specific eIF4GI residues is altered by serum and mitogens. Phosphopeptides resolved by this method were mapped to the C–terminal one-third of the protein. Mass spectrometry and mutational analyses identified the serum-stimulated phosphorylation sites in this region as serines 1108, 1148 and 1192. Phosphoinositide–3–kinase (PI3K) inhibitors and rapamycin, an inhibitor of the kinase FRAP/mTOR (FKBP12–rapamycin-associated protein/mammalian target of rapamycin), prevent the serum-induced phosphorylation of these residues. Finally, the phosphorylation state of N–terminally truncated eIF4GI proteins acquires resistance to kinase inhibitor treatment. These data suggest that the kinases phosphorylating serines 1108, 1148 and 1192 are not directly downstream of PI3K and FRAP/mTOR, but that the accessibility of the C–terminus to kinases is modulated by this pathway(s).
Keywords: eukaryotic translation initiation factor 4G/FKBP12–rapamycin-associated protein/mRNA translation/phosphoinositide–3–kinase/protein synthesis
Introduction
A key regulatory step in protein synthesis is the recruitment of the translational machinery to the 5′ end of mRNA. This bridging process is accomplished via multiple protein–protein and protein–RNA interactions, coordinated primarily by two large, modular scaffolding factors: the eukaryotic translation initiation factor 4G (eIF4G) proteins (for reviews, see Hentze, 1997; Morley et al., 1997; Gingras et al., 1999b). eIF4GI was originally identified as a constituent of eIF4F, a tripartite complex also containing an mRNA 5′ cap binding protein, eIF4E, and an RNA helicase, eIF4A (reviewed in Gingras et al., 1999b). Viral proteases cleave the mammalian eIF4GI protein into three fragments (N–terminal, middle and C–terminal; Lamphear et al., 1995); biochemical and functional analyses of these fragments have clarified the role that each plays in the formation of a functional translation initiation complex. The N–terminal fragment interacts directly with eIF4E (Lamphear et al., 1995; Mader et al., 1995) and the poly(A) binding protein (PABP; Imataka et al., 1998). The middle fragment of eIF4GI possesses binding sites for eIF3, a multisubunit factor associated with the 40S ribosomal subunit, and eIF4A (Lamphear et al., 1995; Imataka and Sonenberg, 1997). The middle fragment also possesses RNA binding activity (Pestova et al., 1996). The C–terminal fragment of eIF4GI contains a second, independent binding site for eIF4A (Lamphear et al., 1995; Imataka and Sonenberg, 1997), and interacts with Mnk1 (MAP kinase signal-integrating kinase 1; Fukunaga and Hunter, 1997; or MAP kinase-interacting kinase 1; Waskiewicz et al., 1997), a MAP kinase-activated serine/threonine kinase that phosphorylates eIF4E in vitro (Waskiewicz et al., 1997) and in vivo (Pyronnet et al., 1999; Waskiewicz et al., 1999). A second eIF4G protein, eIF4GII, was recently characterized (Gradi et al., 1998a), which shares 46% identity with eIF4GI at the amino acid level. eIF4GI and eIF4GII are functional homologs, in that all of the features described above for eIF4GI are conserved in eIF4GII (Gradi et al., 1998a; Imataka et al., 1998; Pyronnet et al., 1999).
Through the aforementioned protein–protein and protein–RNA interactions, the eIF4G proteins perform several critical functions in translation initiation, including: (i) recruitment of the 40S ribosomal subunit to the 5′ end of mRNA via interactions with eIF4E and eIF3; (ii) relief of inhibitory secondary structure in the mRNA 5′ UTR, by delivering the eIF4A helicase to this region (Rozen et al., 1990); (iii) circularization of mRNA through interactions with both eIF4E and PABP (Wells et al., 1998); and (iv) delivery of the kinase Mnk1 to its substrate, eIF4E (Pyronnet et al., 1999; Waskiewicz et al., 1999). How all of these functions are coordinated and regulated by the eIF4G proteins is not understood.
The human eIF4G family also includes two other proteins: p97 and PAIP–1. p97 is homologous only to the C–terminal two-thirds of the eIF4Gs, and does not possess a region corresponding to the N–terminal one-third of the eIF4Gs. Like the eIF4Gs, it contains binding sites for eIF3, eIF4A and Mnk1. However, p97 does not interact with eIF4E or PABP (Imataka et al., 1997; Levy-Strumpf et al., 1997; Yamanaka et al., 1997), and inhibits both cap-dependent and cap-independent translation in vivo, presumably by forming non-functional initiation complexes (Imataka et al., 1997). A more distant eIF4G homolog, PAIP–1, possesses binding sites for PABP and eIF4A, and appears to act as a translational enhancer in vivo (Craig et al., 1998).
eIF4G homologs have been identified in many species (e.g. Browning et al., 1987; Goyer et al., 1993; Morley et al., 1997; Hernandez et al., 1998). While all of the eIF4G-like proteins identified so far possess a region homologous to the middle fragment of the mammalian eIF4G proteins, the N- and C–terminal regions of these proteins diverge significantly. For example, an extended eIF4G C–terminus is a feature present only in certain organisms: the mammalian (Yan et al., 1992; Lamphear et al., 1993), Drosophila (Hernandez et al., 1998) and putative zebrafish eIF4G homologs possess an elongated C–terminal region. However, a wheat eIF4G homolog (Browning et al., 1987) possesses a much smaller C–terminal region, and the Saccharomyces cerevisiae (Goyer et al., 1993) and Schizosaccharomyces pombe (Morley et al., 1997) eIF4G homologs do not possess such a region at all.
These observations suggest that the middle region of eIF4G is the ‘core’ unit required for translation. Consistent with this hypothesis, the middle fragment alone is sufficient for cap-independent translation from the encephalomyocarditis virus (EMCV) internal ribosome entry site (IRES; Pestova et al., 1996). The middle one-third of the protein plus the eIF4E binding site is the minimal fragment required for cap-dependent translation of the β–globin mRNA (Morino et al., 2000), and the middle region alone, fused to iron regulatory protein–1, can direct translation of an RNA containing an iron response element (DeGregorio et al., 1999). Thus, the termini of the mammalian eIF4GI protein are largely dispensable for translation. However, recent evidence suggests that both the N- and C–termini play important roles in enhancing and/or modulating translation in mammalian cells. For example, the eIF4G–PABP interaction, mediated by the eIF4G N–terminus (Imataka et al., 1998), is posited to inhibit the synthesis of truncated proteins from mRNAs that are not full-length (Sachs et al., 1997). The interaction between eIF4A and the eIF4GI C–terminus also appears to be important, as mutation of the C–terminal eIF4A binding site decreases the activity of eIF4GI in a reticulocyte lysate by several-fold (Morino et al., 2000). How the activity of the eIF4G termini may be regulated is not understood.
The eIF4Gs are phosphoproteins (Duncan et al., 1987; Morley and Traugh, 1989, 1990; Donaldson et al., 1991; this study). However, the intracellular signaling pathways mediating eIF4G phosphorylation, and the location of the phosphorylation sites within the proteins, have remained uncharacterized. In this report, we map a set of phosphorylated residues in eIF4GI to the C–terminal one-third of the protein. Using mass spectrometry and mutational analyses, the serum-stimulated phosphorylation sites within this region were identified. An intracellular signaling pathway (or pathways) that may be dedicated to translational control (Brown and Schreiber, 1996) consists of phosphoinositide–3–kinase (PI3K), Akt [or protein kinase B (PKB)], and the FKBP12–rapamycin-associated protein/mammalian target of rapamycin (FRAP/mTOR; also known as RAFT1 or RAPT1; reviewed in Lawrence and Abraham, 1997; Thomas and Hall, 1997; Abraham, 1998; Gingras et al., 1999b; Raught and Gingras, 1999). This pathway(s) regulates the activity of several translation factors. We demonstrate here that PI3K and FRAP/mTOR signaling modulates the serum-stimulated phosphorylation of the C–terminal eIF4GI sites. Interestingly, the eIF4GI N–terminus appears to modulate phosphorylation of the C–terminus, because phosphorylation of truncation mutants lacking the N–terminal region are resistant to kinase inhibitor treatment. Unexpectedly, although the related eIF4GII and p97 are also phosphoproteins, the corresponding C–terminal fragments in these proteins are not phosphorylated, suggesting that this region has undergone divergent evolution.
Results
Phosphopeptide mapping of eIF4GI
To study the regulation of eIF4GI phosphorylation, two-dimensional phosphopeptide mapping of 32P–labeled, immunoprecipitated eIF4GI was conducted, essentially as described (Gingras et al., 1998). Briefly, human embryonic kidney 293 cells were metabolically labeled with [32P]orthophosphate. A subset of these cells was then stimulated with serum. The cells were lysed, and eIF4GI was immunoprecipitated using a polyclonal antiserum directed against the N–terminus of the protein (Craig et al., 1998). Immunoprecipitated material was subjected to SDS–8% PAGE, and electrotransferred to a nitrocellulose membrane. The membrane was autoradiographed, and the region harboring the protein of interest excised. Membrane pieces were subjected to tryptic digestion, and liberated phosphopeptides visualized by two-dimensional (thin-layer electrophoresis/thin-layer chromatography; TLE/TLC) mapping.
A typical autoradiogram of 32P–labeled, immunoprecipitated, gel-purified eIF4GI is shown in Figure 1A. Cerenkov counting of excised membrane pieces harboring 32P–labeled eIF4GI revealed no significant difference in the quantity of 32P incorporated into eIF4GI isolated from serum-starved (–S) versus serum-stimulated (+S) 293 cells (n = 10). However, two-dimensional tryptic phosphopeptide mapping revealed that the relative intensity of several phosphorylated peptides was significantly altered upon serum stimulation, indicating a change in the phosphorylation status of one or more amino acid residues. A highly reproducible pattern of phosphorylated peptides was observed for eIF4GI isolated from serum-starved cells (Figure 1B). Treatment of starved cells with serum (Figure 1C), insulin (data not shown) or phorbol ester (data not shown) resulted in an increase in the phosphorylation state of several tryptic peptides (serum-stimulated phosphopeptides 1–4), along with a concomitant decrease in the phosphorylation status of several others (serum-repressed phosphopeptides 7–9). No significant change in the intensity of the remaining major phosphopeptides (Figure 1C, phosphopeptides 5 and 6) was observed. Phosphopeptide mapping of eIF4GI from logarithmically growing cells yielded the same phosphopeptide pattern observed for serum-stimulated cells (data not shown). Immunoprecipitation with antiserum directed against the C–terminus of human eIF4GI yielded identical maps, confirming that all of the observed phosphopeptides are derived from eIF4GI.

Fig. 1. Phosphorylation of specific sites in eIF4GI is modulated by serum. 32P–labeled eIF4GI immunoprecipitated from 293 cells starved of serum for 36 h (–Serum or –S), or starved of serum for 36 h then stimulated with serum for 30 min (+Serum or +S), was subjected to (A) SDS–8% PAGE, then (B and C) to two-dimensional tryptic peptide mapping. The directions of chromatography (vertical) and electrophoresis (horizontal) as well as the loading origin (arrow) are indicated. Major phosphopeptides are numbered.
While the response of the phosphopeptides to serum stimulation was very consistent (i.e. peptides 1–4 increasing and peptides 7–9 decreasing in intensity), the degree of stimulation or repression varied from experiment to experiment. This variability appears to be due to a number of different reasons, including the confluence level and passage number of the cells, as well as the length of the starvation and stimulation periods. In general (as assessed by phosphoimaging and densitometry measurements), phosphopeptides 2 and 4 displayed the greatest sensitivity to serum stimulation (increasing in intensity ∼3- to 10–fold), while peptides 3 (increasing ∼2- to 5–fold) and 1 (increasing 1- to 3–fold; in some experiments no change was observed for peptide 1) were less responsive (data not shown). The intensity of peptide 7, in the serum-repressed group, also displayed acute sensitivity (decreasing by ∼2- to 10–fold) to the presence of serum, whereas peptides 8 and 9 (both decreasing by ∼2- to 5–fold) displayed a lower level of sensitivity. It is also noteworthy that while the eIF4GI phosphopeptide mapping pattern is quite reproducible, some variability is observed in the minor, unnumbered peptides as well as in peptides 4 and 6. This is most likely to be due to variations in the efficiency of tryptic digestion. Also, a streaked signal is observed directly above the loading origin in maps of endogenous eIF4GI (Figure 1B and C), suggesting that other peptides that are not clearly resolved by this method may be present. Nevertheless, the streaked signal and unnumbered phosphopeptides account for <10% of the total signal, as determined by phosphoimaging (data not shown).
Mapping of eIF4GI phosphorylation sites
To delineate the location of the eIF4GI phosphorylation sites, various influenza hemagglutinin (HA)-tagged eIF4GI protein fragments were expressed in 293T cells. (Phosphopeptide maps of eIF4GI from 293T cells are identical to those from serum-stimulated 293 cells. However, since the transfection efficiency achieved in 293T cells is much greater than that for 293 cells, they were used in all transfection protocols.) Logarithmically growing 293T cells were transiently transfected (in the presence of 10% serum), and labeled with [32P]orthophosphate. HA-tagged proteins were then immunoprecipitated with an anti-HA antibody, gel-purified and mapped. The constructs utilized in this study are shown in Figure 2F. HA-tagged eIF4GI protein fragments encompassing amino acids (aa) 614–1560 (Figure 2B) and 1045–1560 (Figure 2C) yielded phosphopeptide maps that were almost identical to the endogenous protein (Figure 2A). However, further N–terminal truncation to aa 1205 resulted in the loss of all phosphopeptides, except peptide 5 (Figure 2D). Mapping of a protein encompassing aa 1045–1372 yielded all phosphopeptides, except for peptides 5 and 6 (Figure 2E), and mapping of a fusion protein encompassing aa 1372–1560 yielded only peptide 5 (data not shown). Thus, all of the phosphopeptides mapped by this procedure were localized to the C–terminal one-third of the protein. The inability to localize peptide 6 is likely to result from differences in tryptic digestion efficiency between the larger and smaller eIF4GI fusion proteins. Interestingly, truncation of N–terminal sequences also resulted in a significant increase in the relative intensity of the serum–stimulated phosphopeptides (1–4) compared with the serum-repressed peptides (7–9; compare Figure 2A with E).

Fig. 2. Localization of eIF4GI phosphorylation sites. (A) Tryptic map of endogenous eIF4GI. (B–E) HA-tagged eIF4GI fusion protein fragments were expressed in 293T cells. Cells were metabolically labeled with 32P, and HA-tagged proteins immunoprecipitated, gel purified, and subjected to two-dimensional tryptic mapping. (F) Protein fragments tested in this study. The various protein binding regions of eIF4GI are indicated.
The regions of eIF4GII and p97 corresponding to the eIF4GI phospho-region are not phosphorylated
Alignment of the region of eIF4GI containing most of the phosphorylation sites (aa 1045–1205; hereafter referred to as the phospho-region) with the corresponding segments of two other members of the human eIF4G family, eIF4GII and p97, revealed relatively poor conservation (Figure 3A), especially compared with the surrounding regions. In fact, this region of human eIF4GI is much more closely related to the corresponding region of eIF4G proteins in other species than to the human eIF4GII and p97 proteins, sharing 93% identity with the rabbit eIF4GI protein (DDBJ/EMBL/GenBank accession No. L22090) and 47% identity with a putative zebrafish eIF4GI protein (DDBJ/EMBL/GenBank accession No. AI629389; data not shown). Thus, the newly defined phospho-region appears to have undergone divergent evolution within the human eIF4G family members.

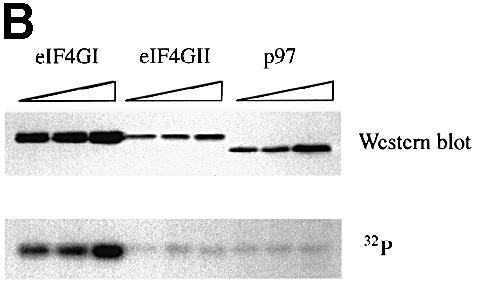
Fig. 3. The phospho-region of eIF4GI is poorly conserved within the human eIF4G family. (A) An alignment through the newly defined eIF4GI phospho-region of the members of the human eIF4G family. Locations of defined protein binding sites in eIF4G proteins are indicated, as well as percentage identity to the corresponding region in eIF4GI. Below the diagram is an alignment of the amino acid sequences of eIF4GI, eIF4GII and p97 in this region. Identical residues are boxed in black, similar residues are boxed in gray. (B) Fragments encompassing the phosphorylated region of eIF4GI (aa 1035–1206) and the corresponding regions of eIF4GII (aa 1057–1225) and p97 (aa 395–547) were expressed in 293T cells (5, 10 and 20 μg of DNA transfected) as GST fusion proteins. Cells were metabolically labeled with 32P. Proteins isolated with glutathione-coupled Sepharose beads were washed, gel-purifed, and transferred to nitrocellulose. Upper panel, Western blot using anti-GST antiserum. Lower panel, direct autoradiogram of the same nitrocellulose membrane.
Immunoprecipitation of eIF4GII and p97 from metabolically labeled 293 cells revealed that they are both phosphoproteins (data not shown). To determine whether eIF4GII and p97 are phosphorylated in the same region as eIF4GI, glutathione S–transferase (GST) fusion proteins encompassing fragments corresponding to the eIF4GI phospho-region (aa 1035–1206 for eIF4GI, aa 1057–1225 for eIF4GII, aa 395–547 for p97) were expressed in 293T cells, which were then metabolically labeled with [32P]orthophosphate. The transfected 293T cells were lysed, and GST fusion proteins isolated via incubation with glutathione-coupled Sepharose beads. Western blotting, using anti-GST antiserum (Morino et al., 2000), confirmed that all three GST fusion proteins were expressed (Figure 3B, upper panel), albeit at different levels. The same blot was then autoradiographed directly (Figure 3B, lower panel). Interestingly, only the GST–eIF4GI fusion protein incorporated significant amounts of 32P. (A weak background signal at approximately the same location as the GST–eIF4GI and GST–eIF4GII proteins is detected in all lanes; however, the intensity of this signal did not correlate with the amount of GST fusion protein expressed, and the signal did not co-migrate with GST–p97.) To ensure that all of the cells were efficiently metabolically labeled, endogenous eIF4GI was immunoprecipitated from the same lysates and found to incorporate similar amounts of 32P in all samples (data not shown). Thus, while both eIF4GII and p97 are phosphoproteins, the regions corresponding to the eIF4GI C–terminal phospho-region do not appear to be phosphorylated significantly in these members of the eIF4G family.
Identification of the serum-inducible phosphorylation sites in eIF4GI
To identify the C–terminal eIF4GI residues phosphorylated in response to serum treatment, mass spectrometric and mutational analyses were performed. Briefly, phosphopeptide mapping was conducted as above, except that eIF4GI immunoprecipitated from 2 × 108 serum-stimulated 293 cells (1/20 of which were metabolically labeled with [32P]orthophosphate) was loaded onto two TLC plates. The cellulose harboring peptides 1–4 was scraped from the plastic backing of the plates, and the phosphopeptides eluted and identified by capillary liquid chromatography–electrospray ionization tandem mass spectrometry (LC–MS/MS). The tandem mass spectrum for phosphopeptide 1 corresponds to EAALPPVSPLK, with the phosphorylation site being Ser1192 (Figure 4A). The tandem mass spectrum for phosphopeptide 3 corresponds to SFSKEVEER, with the phosphorylation site being Ser1148 (Figure 4B). The tandem mass spectrum for phosphopeptide 4 corresponds to SSLSRER, with the phosphorylation site being Ser1108 (data not shown). Mass measurements for phosphopeptide 2 predicted the peptide SSLSR (data not shown), indicating that peptide 2 is an alternative cleavage product of peptide 4. The identity of the phosphorylated residue could not be determined for this peptide.


Fig. 4. Identification of serum-stimulated phosphorylation sites by mass spectrometry. (A) Tandem mass spectrum resulting from the analysis of excised phosphopeptide 1 from the map in Figure 1C. This spectrum contained sequence information for a single phosphopeptide. Two separate ion series were recorded simultaneously; b- and y–ion series represent sequencing inward from the N- and C–termini, respectively. The computer program Sequest (Eng et al., 1994) was utilized to match this tandem mass spectrum to the sequence shown, with the serine residue being phosphorylated. (B) Tandem mass spectrum resulting from the analysis of excised phosphopeptide 3. The position of the phosphorylated serine residue is indicated.
To confirm the mass spectrometric results, site-directed mutagenesis of serines 1108, 1148 and 1192, along with several other phosphorylation site candidates, was performed. To simplify the cloning and mapping procedures these mutants were constructed in the background of the GST fusion protein containing only the eIF4GI phospho-region (aa 1035–1206). A typical two-dimensional phosphopeptide map of the GST–eIF4GI phospho-region protein is shown in Figure 5A. As observed for the HA–1045–1372 fragment (Figure 2E), phosphorylation of the GST fusion protein is almost exclusively limited to peptides 1–4, while serum-repressed peptides 7 and 8 are almost undetectable. A phosphopeptide that migrates in the same region as peptide 9 remains visible on most maps. A novel phosphopeptide (denoted by an asterisk, Figure 5A–D) that does not co-migrate with any endogenous eIF4GI peptides is observed in maps of these proteins, and is most likely to be derived from the GST moiety.


Fig. 5. Identification of serum-inducible phosphorylation sites by mutational analysis. Mutations of serine residues identified by mass spectrometry as phosphorylation sites were introduced into the eIF4GI phospho-region (aa 1035–1206)–GST fusion proteins. Proteins were expressed in 293T cells, 32P–labeled, isolated and mapped. Tryptic maps of the GST–eIF4GI (aa 1035–1206) wild-type protein (A), and point mutants Ser1108Ala (B), Ser1148Ala (C) and Ser1192Ala (D). A novel phosphopeptide that does not co-migrate with any endogenous eIF4GI peptide is indicated with an asterisk. (E) Complete mutational analysis summary. An alignment of the eIF4GI phospho-region with the corresponding regions of eIF4GII and p97. Identical residues are boxed in black, similar residues are boxed in gray. eIF4GI residues mutated to alanine are indicated by an ‘x’ or arrow. Locations of the serum-stimulated, phosphorylated residues, as well as the phosphopeptides identified in this analysis, are also indicated.
Mutation of Ser1108 to alanine resulted in the loss of both peptides 2 and 4 (Figure 5B). Thus, tryptic cleavage after arginine 1109 appears to be relatively inefficient (most likely to be due to the presence of a phosphate group near the cleavage site), resulting in the formation of two tryptic peptides (SSLSR and SSLSRER) containing the same phosphoserine. Elimination of phosphopeptides 2 and 4 was specific for the Ser1108Ala mutant, as maps of Ser1105Ala and Ser1106Ala mutants (other serines present in the same tryptic peptide) retained peptides 1–4 (data not shown). Mutation of Ser1148 to alanine specifically abolished phosphopeptide 3 (Figure 5C), and mutation of Ser1192 to alanine eliminated phosphopeptide 1 (Figure 5D). Mutation to alanine of serines 1085, 1095, 1125, 1155, 1160 and 1170, or of threonines 1093, 1096 and 1140 had no effect on the phosphopeptide pattern (see Figure 5E for a summary of the mutational analysis). Thus, using a combination of mass spectrometric and mutational analyses, the serum-stimulated phosphorylation sites were identified as Ser1108, Ser1148 and Ser1192.
PI3K and FRAP/mTOR signaling modulates eIF4GI phosphorylation
Several translation regulatory factors have been identified as targets of the PI3K–FRAP/mTOR signaling pathway(s) (Thomas and Hall, 1997; Abraham, 1998; Gingras et al., 1999b). It was thus pertinent to test whether signaling to eIF4GI is also mediated by this pathway. Specific kinase inhibitors may be used to determine whether phosphorylation of a given protein is modulated by these kinases. Wortmannin inhibits PI3K by covalently binding to its catalytic subunit (reviewed in Ui et al., 1995), and LY294002 is a structurally unrelated, reversible PI3K inhibitor (Vlahos et al., 1994). Rapamycin is a fungal macrolide that, in a complex with the immunophilin FKBP12, binds to FRAP/mTOR to inhibit its kinase activity (Abraham, 1998).
Serum-starved 293 cells metabolically labeled with [32P]orthophosphate (Figure 6A) were pre-treated (for 20 min) with kinase inhibitors, then serum was added to the culture media to a final concentration of 10%. After 30 min in the presence of serum (Figure 6B), or serum plus inhibitor (Figure 6C and D), cells were lysed and eIF4GI was subjected to phosphopeptide mapping. LY294002 (5 μM; Figure 6C), wortmannin (100 nM; data not shown) and rapamycin (25 nM; Figure 6D) abrogated the serum-induced hyperphosphorylation of peptides 1–4, and prevented the hypophosphorylation of peptides 7–9. (In this experiment, peptide 3 responded only weakly to rapamycin addition.) Addition of solvent [dimethylsulfoxide (DMSO) for wortmannin and LY294002, ethanol for rapamycin] alone had no effect on the phosphopeptide pattern (data not shown). Thus, serum-induced modulation of eIF4GI phosphorylation appears to be mediated by PI3K and FRAP/mTOR signaling.

Fig. 6. Serum-induced phosphorylation of eIF4GI is sensitive to inhibitors of PI3K and FRAP/mTOR. Two-dimensional tryptic peptide mapping of endogenous eIF4GI isolated from 293 cells (A) starved of serum, (B) then treated with serum for 30 min, (C) pre-treated with LY294002 for 20 min, then treated with serum for 30 min, or (D) pre-treated with rapamycin for 20 min, then treated with serum for 30 min. The map in (A) is the same as that shown in Figure 1B.
The N–terminus confers kinase inhibitor sensitivity to the phospho-region
Two models may be proposed to explain the effects of kinase inhibitors on the phosphorylation state of eIF4GI: (i) the kinases phosphorylating this region are rapamycin- and LY294002-sensitive; or (ii) the kinases themselves are not sensitive to these inhibitors, but a change in the conformation of eIF4GI (due to alterations in intramolecular interactions, or in the interaction of eIF4GI with other binding partners) modulates the accessibility of the C–terminal phosphorylation sites. In this regard, the N–terminal truncation mutants used in this study were no longer sensitive to kinase inhibitor treatment. Instead, phosphorylation of the truncation mutants was constitutively in the ‘serum-stimulated’ state (data not shown). These data argue for the second model, and suggest that the N–terminal region of eIF4GI modulates the phosphorylation state of the C–terminal phospho-region.
It was of particular interest to determine whether this N–terminal regulatory region could be the eIF4E binding site, since binding of eIF4E to eIF4G is also rapamycin- and LY294002-sensitive, and because eIF4E binding induces a conformational change in the eIF4GI protein (Haghighat et al., 1996; Ohlmann et al., 1997; Hershey et al., 1999) that could theoretically alter the accessibility of the C–terminus to kinases. To address this question, HA-tagged eIF4GI fragments aa 550–1560 (consisting of the eIF4E binding site + middle + C–terminus) and aa 614–1560 (consisting only of the middle + C–terminus) were expressed in 293T cells. Cells were metabolically labeled, treated with kinase inhibitors, and both the HA-tagged eIF4GI fragments and endogenous eIF4GI were immunoprecipitated from the cell lysates and mapped (Figure 7). Both of the HA-tagged fragments were insensitive to rapamycin- (compare Figure 7B and D with A and C) and LY294002-treatment (data not shown); serines 1108, 1148 and 1192 remained phosphorylated, while phosphopeptide 7 was almost absent, even in the presence of rapamycin. Endogenous eIF4GI immunoprecipitated from the same lysates retained rapamycin- (Figure 7E and F) and LY294002-sensitivity (data not shown). (In this experiment, phosphopeptides 2, 3, 4 and 7 displayed acute sensitivity to rapamycin treatment, whereas peptide 1, which is the least responsive to serum, did not.) These data indicate that eIF4E binding per se is not responsible for conferring LY294002- and rapamycin-sensitivity upon eIF4GI.

Fig. 7. N–terminal sequences confer kinase inhibitor sensitivity to the C–terminal phospho-region. HA-tagged eIF4GI fragments were expressed in 293T cells. Cells were metabolically labeled and incubated in 10% serum (A, C and E) or 10% serum + 25 ng/ml rapamycin (B, D and F) for 1 h. Following lysis, immunoprecipitation was conducted sequentially with anti-HA and anti-eIF4GI antisera. Isolated proteins were gel-purified and mapped. The HA–eIF4GI 550–1560 fragment interacts with eIF4E, while HA–eIF4GI 614–1560 does not.
The protease sensitivity of a 98 aa N–terminal fragment of the yeast eIF4GI homolog (TIF4631) is reduced upon binding to yeast eIF4E, as it progresses from a less structured to a more structured state (Hershey et al., 1999). Thus, it remained possible that eIF4E binding to human eIF4GI could also cause a conformational change involving a much larger region of the N–terminus than is present in the aa 550–1560 fragment. To address this possibility, and to localize further the eIF4GI N–terminal region conferring rapamycin sensitivity to the C–terminal phospho-region, a much longer truncation mutant (aa 157–1560, the original eIF4GI clone; Yan et al., 1992) was also expressed in 293T cells. As with the shorter fragments, phosphorylation of this fragment displayed rapamycin resistance (data not shown). Endogenous eIF4GI immunoprecipitated from the same cell lysates retained rapamycin sensitivity (data not shown). Thus, intramolecular interactions between the extreme N–terminus (aa 1–156) and the C–terminal phospho-region, or between a second protein that interacts with the eIF4GI N–terminus (and whose binding site is not present in the truncation mutants), appear to confer rapamycin- and LY294002-sensitivity. Finally, these data also indicate that eIF4GI C–terminal phospho-region kinases are not rapamycin sensitive themselves, but that, instead, a conformational change that alters the accessibility of several key residues is effected by serum, and is inhibited by rapamycin, wortmannin and LY294002. A similar phenomenon has been reported for p70 S6 kinase 1 (Dennis et al., 1996; Mahalingam and Templeton, 1996).
Discussion
Here, we have identified a set of serum-stimulated phosphorylation sites (Ser1108, Ser1148 and Ser1192) within the highly phosphorylated C–terminal one-third of the eIF4GI protein. Phosphorylation of these sites is demonstrated to be responsive to various extracellular stimuli, and is shown to be sensitive to specific inhibitors of the PI3K–FRAP/mTOR signaling pathway(s). Interestingly, the extreme N–terminus of eIF4GI appears to modulate the phosphorylation state of the C–terminal phospho-region. Taken together, our data suggest a two-step model for the phosphorylation of eIF4GI. The eIF4GI protein in serum-starved (or kinase inhibitor-treated) cells appears to be in a repressed state, either due to intramolecular interactions between the N- and C–termini, or due to interactions with unknown eIF4G binding partners. In step one, the repressed eIF4GI molecule is derepressed by the PI3K–FRAP/mTOR pathway(s), either by direct phosphorylation of the N–terminus of the protein, or via modulation of an eIF4GI binding partner. This event renders the C–terminal serine residues accessible to other kinases. Phosphorylation of these residues results in a ‘fully active’ eIF4GI. However, how phosphorylation may affect the activity of eIF4GI in translation initiation is unknown.
This work also outlines a technique to evaluate how any extracellular stimulus or cellular stress modulates eIF4GI phosphorylation, and provides the basis for further studies to assess how phosphorylation may modulate eIF4GI activity. In addition, this study points out previously unsuspected differences between members of the human eIF4G family, in that the corresponding region of two closely related proteins, eIF4GII and p97, is not phosphorylated. This observation is intriguing, in that it suggests that the various members of the eIF4G family may be differentially regulated by intracellular signaling pathways.
Contrary to previous reports suggesting that treatment of cells with mitogenic stimuli leads to a net increase in eIF4G phosphorylation (Morley and Traugh, 1989, 1990; Donaldson et al., 1991), we did not observe a change in the quantity of 32P incorporated into eIF4GI after starvation, serum stimulation (Figure 1A) or kinase inhibitor treatment (B.Raught and A.-C.Gingras, unpublished observation). In these earlier reports, however, eIF4G (most likely to be a mixture of eIF4GI and eIF4GII) was isolated via m7GTP affinity purification as an eIF4E–eIF4G complex. Subsequent to these reports, the eIF4E-binding proteins (4E–BPs) were discovered, and found to compete with the eIF4Gs for an overlapping binding site on eIF4E (Lin et al., 1994; Pause et al., 1994; Haghighat et al., 1995; Mader et al., 1995). In quiescent or starved 293 cells, a relatively high proportion of eIF4E is bound to 4E–BPs, and a low amount of eIF4G protein is recovered after cap column purification of eIF4E (A.-C.Gingras and B.Raught, unpublished observation). Mitogenic stimulation leads to an increase in phosphorylation of the 4E–BPs, and a consequent decrease in their affinity for eIF4E. The liberated eIF4E is then free to interact with eIF4Gs, resulting in a significant increase in the amount of eIF4G recovered by cap column purification. Thus, the earlier observed increase in eIF4G phosphorylation in response to mitogen treatment probably reflected an augmentation in the quantity of eIF4G bound to eIF4E. We cannot, however, exclude the possibility that following other types of treatments, or in other cell types, the quantity of phosphate incorporated into eIF4GI changes.
It is clear that while both the human eIF4GI and eIF4GII proteins can function in translation initiation (Gradi et al., 1998a), these molecules differ in several respects. For instance, the eIF4GII protein is present at ∼1/4 of the level of eIF4GI in HeLa cells (Svitkin et al., 1999), and the cleavage kinetics of eIF4GII in virus-infected cells differs markedly from that of eIF4GI (Gradi et al., 1998b; Svitkin et al., 1999). This study also suggests that the newly defined eIF4GI phospho-region is not phosphorylated in eIF4GII. Thus, it appears that these proteins have undergone some degree of divergent evolution, and that the activity of the eIF4G proteins may be regulated quite differently. These observations may have important implications regarding the regulation of translation initiation in response to different types of intracellular signals, or in the regulation of different mRNA populations.
We previously reported that Mnk1 can phosphorylate eIF4GI in vitro (Pyronnet et al., 1999). After further study, it does not appear that this observation is relevant in vivo. Mnk1 is activated via the MAPK pathway, which is stimulated in response to serum treatment in 293 cells (von Manteuffel et al., 1996). However, truncation of the Mnk1 binding site in eIF4GI does not negatively impact upon the phosphorylation state of the serum-stimulated phosphopeptides (e.g. Figure 2E), and mapping of a recombinant eIF4GI fragment phosphorylated in vitro by Mnk1 revealed that it is phosphorylated only on serum-repressed phosphopeptides 7 and 8 (B.Raught and A.–C.Gingras, unpublished observation). Thus, it appears unlikely that this kinase plays a role in mediating eIF4GI phosphorylation.
A signaling pathway that may be dedicated to translational control (Brown and Schreiber, 1996) consists of PI3K, Akt/PKB and FRAP/mTOR. These kinases signal to several factors involved in the regulation of translation, including the p70 S6 kinases (Jefferies and Thomas, 1996; Gout et al., 1998; Shima et al., 1998), eukaryotic elongation factor 2 (Redpath et al., 1996) and the 4E–BPs (Gingras et al., 1999b). eIF4GI may now be added to this list. However, even though the serum-stimulated eIF4GI phosphorylation sites identified here are rapamycin sensitive, they are not directly phosphorylated by FRAP/mTOR or p70 S6 kinases 1 and 2 in an in vitro kinase assay (A.-C.Gingras and B.Raught, unpublished data). In vivo evidence also argues against a role for rapamycin-sensitive kinases such as the p70 S6 kinases and FRAP/mTOR in the phosphorylation of the serum-stimulated eIF4GI sites: the phosphorylation state of N–terminal truncation mutants is rapamycin- and LY294002-insensitive. Thus, an N–terminal region of eIF4GI appears both to repress phosphorylation of peptides 1–4 in the serum-starved state, and confer LY294002- and rapamycin-sensitivity to the C–terminal phospho-region.
How might phosphorylation modulate eIF4GI activity? Kinase inhibitor treatment does not appear to affect the binding of several known eIF4GI binding partners, including eIF3, eIF4A (B.Raught, A.-C.Gingras, S.Morino and H.Imataka, unpublished observations) and MnkI (S.Pyronnet, personal communication). These data are consistent with the fact that the phospho-region does not overlap with the binding site of any known eIF4GI binding protein. Secondary structure predictions suggest that this region is relatively unstructured (B.Raught and A.–C.Gingras, unpublished data). The phospho-region also contains a caspase cleavage site (M.Bushell and S.J.Morley, personal communication), suggesting that it is solvent exposed. We therefore posit that this region acts as a flexible ‘hinge’ between the middle and C–terminal domains of eIF4GI. In the absence of any evidence for quantitative changes in protein–protein interactions, we suggest that phosphorylation alters intramolecular interactions to cause short- or long-range changes in eIF4GI structure. How these changes modulate the ability of eIF4GI to initiate translation remains to be determined.
Materials and methods
Cell culture/inhibitors
Cell culture and [32P]orthophosphate metabolic labeling were carried out as described (Gingras et al., 1998). Wortmannin and LY294002 were purchased from Calbiochem and diluted in DMSO at 1000×. Rapamycin was purchased from the same source, dissolved in EtOH at 25 μM, and used at a final concentration of 25 nM. 293T cells were transfected using a modified calcium phosphate procedure (Chen and Okayama, 1988). Bovine insulin was purchased from Gibco-BRL and 12–O–tetradecanoylphorbol 13–acetate (TPA) from Sigma.
Constructs
HA-tagged eIF4GI constructs were described previously (Imataka and Sonenberg, 1997). Fragments encompassing aa 1035–1206 of eIF4GI, aa 1057–1225 of eIF4GII and aa 395–547 of p97 were amplified by PCR, and cloned in-frame into EcoRI–XhoI sites of pcDNA3–GST (Imataka et al., 1998). Point mutations of eIF4GI were generated using pcDNA3–GST–eIF4G(1035–1206) as a template, and mutants were cloned into EcoRI–XhoI sites of pcDNA3–GST. All inserts were sequenced.
Antibodies
Antiserum raised against the eIF4GI N–terminus was characterized previously (Craig et al., 1998). Antiserum prepared against the eIF4GI C–terminus was derived from a New Zealand White rabbit injected with the peptide CRSRERPSQPEGLRKAASLTEDRDRGR, corresponding to aa 1154–1179 (Sheldon Biotechnologies, McGill University), conjugated to keyhole limpet hemocyanin. This antiserum does not cross-react with eIF4GII (A.Gradi and B.Raught, unpublished data).
Tryptic phosphopeptide mapping
To isolate labeled eIF4GI protein, cell culture plates were washed twice with phosphate-buffered saline, and lysed in 1 ml of lysis buffer (Gingras et al., 1998) containing a 1:500 dilution of the Sigma protease inhibitor cocktail, 10 mM NaF, 0.25 mM sodium orthovanadate and 10 nM calyculin. Cells were lysed for 15 min at 4°C, and all subsequent steps were carried out at 4°C. Lysate was scraped from the plates and centrifuged for 15 min at 14 000 g. Supernatant was pre-cleared by adding 50 μl of protein A–Sepharose, and incubated with end-over-end mixing for 60 min. eIF4GI was immunoprecipitated with 25 μl of protein A–Sepharose beads coupled to 5 μl of antiserum for 3.5 h. Beads were collected by centrifugation at 3000 g, washed twice in lysis buffer and twice in RIPA (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS). Laemmli loading buffer was added to the washed beads, and samples were boiled and subjected to SDS–8% PAGE. Separated proteins were transferred to a 0.2 μm nitrocellulose membrane (Schleicher & Schuell), which was exposed to film, and the region of the membrane harboring labeled eIF4GI was excised and Cerenkov counted. HA-tagged eIF4GI proteins were immunoprecipitated with anti-HA11 (BabCO) coupled to protein G–Sepharose beads, and subjected to SDS–12% PAGE. Tryptic digests and TLE/TLC mapping were carried out as described (Gingras et al., 1998) using Kodak (Figures 1, 2 and 6) or Merck (Figures 5 and 7) TLC plates. Phosphorylation intensity was quantified using the BAS 2000 phosphoimager (Fuji).
Mass spectrometry
Large-scale, keratin-free TLE/TLC mapping was conducted to obtain samples of sufficient mass to subject to mass spectrometry, as described (Gingras et al., 1999a). Cellulose from regions harboring radioactivity was scraped from the plate backing and prepared for analysis by LC–MS/MS, as described (Watts et al., 1994). The system used was as described (Gingras et al., 1998; Gygi et al., 1999).
Acknowledgments
Acknowledgements
The authors thank Colin Lister for exceptional technical assistance. We also thank Drs S.Pyronnet, S.J.Morley, M.Bushell, L.Beilsolell and S.K.Burley for sharing unpublished data, and Drs G.Thomas and S.Kozma for p70 S6 kinases 1 and 2. B.R. is the recipient of a Medical Research Council of Canada (MRC) post-doctoral fellowship. A.-C.G. is the recipient of an MRC doctoral award. S.P.G. is supported by National Institutes of Health grant T32HG00035 and a grant from Oxford Glycosciences. Work in R.A.'s laboratory was supported by the National Science Foundation Science and Technology Center for Molecular Biology, and by NIH grant R01 AI41109-03. Work in N.S.'s laboratory was supported by grants from the National Cancer Institute of Canada, the Medical Research Council of Canada and the Howard Hughes Medical Institute. N.S. is a distinguished MRC of Canada scientist and a Howard Hughes Medical Institute International Scholar.
References
- Abraham R.T. (1998) Mammalian target of rapamycin—immunosuppressive drugs uncover a novel pathway of cytokine receptor signaling. Curr. Opin. Immunol., 10, 330–336. [DOI] [PubMed] [Google Scholar]
- Brown E.J. and Schreiber, S.L. (1996) A signaling pathway to translational control. Cell, 86, 517–520. [DOI] [PubMed] [Google Scholar]
- Browning K.S., Maia, D.M., Lax, S.R. and Ravel, J.M. (1987) Identification of a new protein synthesis initiation factor from wheat germ. J. Biol. Chem., 262, 538–541. [PubMed] [Google Scholar]
- Chen C.A. and Okayama, H. (1988) Calcium phosphate-mediated gene transfer: a highly efficient transfection system for stably transforming cells with plasmid DNA. Biotechniques, 6, 632–638. [PubMed] [Google Scholar]
- Craig A.W.B., Haghighat, A., Yu, A.T.K. and Sonenberg, N. (1998) Interaction of polyadenylate-binding protein with the eIF4G homologue PAIP enhances translation. Nature, 392, 520–523. [DOI] [PubMed] [Google Scholar]
- DeGregorio E., Preiss, T. and Hentze, M.W. (1999) Translation driven by an eIF4G core domain in vivo. EMBO J., 18, 4865–4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis P.B., Pullen, N., Kozma, S.C. and Thomas, G. (1996) The principal rapamycin-sensitive p70s6k phosphorylation sites, T-229 and T-389, are differentially regulated by rapamycin-insensitive kinase kinases. Mol. Cell. Biol., 16, 6242–6251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson R.W., Hagedorn, C.H. and Cohen, S. (1991) Epidermal growth factor or okadaic acid stimulates phosphorylation of eukaryotic initiation factor 4F. J. Biol. Chem., 266, 3162–3166. [PubMed] [Google Scholar]
- Duncan R., Milburn, S.C. and Hershey, J.W. (1987) Regulated phosphorylation and low abundance of HeLa cell initiation factor eIF–4F suggest a role in translational control. Heat shock effects on eIF–4F. J. Biol. Chem., 262, 380–388. [PubMed] [Google Scholar]
- Eng J., McCormack, A.L. and Yates, J.R. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom., 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Fukunaga R. and Hunter, T. (1997) MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J., 16, 1921–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras A.-C., Kennedy, S.G., O'Leary, M.A., Sonenberg, N. and Hay, N. (1998) 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt (PKB) signaling pathway. Genes Dev., 12, 502–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras A.-C., Gygi, S.P., Raught, B., Polakiewicz, R.D., Abraham, R.T., Hoekstra, M.F., Aebersold, R. and Sonenberg, N. (1999a) Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev., 13, 1422–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras A.-C., Raught, B. and Sonenberg, N. (1999b) eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem., 68, 913–963. [DOI] [PubMed] [Google Scholar]
- Gout I., Minami, T., Hara, K., Tsujishita, Y., Filonenko, V., Waterfield, M.D. and Yonezawa, K. (1998) Molecular cloning and characterization of a novel p70 S6 kinase, p70 S6 kinase β containing a proline-rich region. J. Biol. Chem., 273, 30061–30064. [DOI] [PubMed] [Google Scholar]
- Goyer C., Altmann, M., Lee, H.S., Blanc, A., Deshmukh, M., Woolford, J.L.,Jr, Trachsel, H. and Sonenberg, N. (1993) TIF4631 and TIF4632: two yeast genes encoding the high-molecular-weight subunits of the cap-binding protein complex (eukaryotic initiation factor 4F) contain an RNA recognition motif-like sequence and carry out an essential function. Mol. Cell. Biol., 13, 4860–4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradi A., Imataka, H., Svitkin, Y.V., Rom, E., Raught, B., Morino, S. and Sonenberg, N. (1998a) A novel functional human eukaryotic translation initiation factor 4G. Mol. Cell. Biol., 18, 334–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradi A., Svitkin, Y.V., Imataka, H. and Sonenberg, N. (1998b) Proteolysis of human eukaryotic translation initiation factor eIF4GII, but not eIF4GI, coincides with the shutoff of host protein synthesis after poliovirus infection. Proc. Natl Acad. Sci. USA, 95, 11089–11094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gygi S.P., Rochon, Y., Franza, B.R. and Aebersold, R. (1999) Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol., 19, 1720–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighat A., Mader, S., Pause, A. and Sonenberg, N. (1995) Repression of cap-dependent translation by 4E-binding protein 1: competition with p220 for binding to eukaryotic initiation factor-4E. EMBO J., 14, 5701–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighat A., Svitkin, Y., Novoa, I., Kuechler, E., Skern, T. and Sonenberg, N. (1996) The eIF4G–eIF4E complex is the target for direct cleavage by the rhinovirus 2A proteinase. J. Virol., 70, 8444–8450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze M.W. (1997) eIF4G: a multipurpose ribosome adapter? Science, 275, 500–501. [DOI] [PubMed] [Google Scholar]
- Hernandez G., del Mar Castellano, M., Agudo, M. and Sierra, J.M. (1998) Isolation and characterization of the cDNA and the gene for eukaryotic translation initiation factor 4G from Drosophila melanogaster. Eur. J. Biochem., 253, 27–35. [DOI] [PubMed] [Google Scholar]
- Hershey P.E.C., McWhirter, S.M., Gross, J.D., Wagner, G., Alber, T. and Sachs, A.B. (1999) The cap-binding protein eIF4E promotes folding of a functional domain of yeast translation initiation factor eIF4GI. J. Biol. Chem., 274, 21297–21304. [DOI] [PubMed] [Google Scholar]
- Imataka H. and Sonenberg, N. (1997) Human eukaryotic translation initiation factor 4G (eIF4G) possesses two separate and independent binding sites for eIF4A. Mol. Cell. Biol., 17, 6940–6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imataka H., Olsen, H.S. and Sonenberg, N. (1997) A new translational regulator with homology to eukaryotic translation initiation factor 4G. EMBO J., 16, 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imataka H., Gradi, A. and Sonenberg, N. (1998) A newly identified N–terminal amino acid sequence of human eIF4G binds poly(A) binding protein and functions in poly(A)-dependent translation. EMBO J., 17, 7480–7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferies H.B. and Thomas,G. (1996) Ribosomal protein S6 phosphorylation and signal transduction. In Hershey,J.W.B., Mathews,M. and Sonenberg,N. (eds), Translational Control. Cold Spring Harbor Laboratory Press, Plainview, NY, pp. 389–409. [Google Scholar]
- Lamphear B.J., Yan, R., Yang, F., Waters, D., Liebig, H.D., Klump, H., Kuechler, E., Skern, T. and Rhoads, R.E. (1993) Mapping the cleavage site in protein synthesis initiation factor eIF-4 γ of the 2A proteases from human Coxsackievirus and rhinovirus. J. Biol. Chem., 268, 19200–19203. [PubMed] [Google Scholar]
- Lamphear B.J., Kirchweger, R., Skern, T. and Rhoads, R.E. (1995) Mapping of functional domains in eukaryotic protein synthesis initiation factor 4G (eIF4G) with picornaviral proteases. Implications for cap-dependent and cap-independent translational initiation. J. Biol. Chem., 270, 21975–21983. [DOI] [PubMed] [Google Scholar]
- Lawrence J.C. Jr and Abraham, R.T. (1997) PHAS/4E-BPs as regulators of mRNA translation and cell proliferation. Trends Biochem. Sci., 22, 345–349. [DOI] [PubMed] [Google Scholar]
- Levy-Strumpf N., Deiss, L.P., Berissi, H. and Kimchi, A. (1997) DAP-5, a novel homolog of eukaryotic translation initiation factor 4G isolated as a putative modulator of γ interferon-induced programmed cell death. Mol. Cell. Biol., 17, 1615–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T.A., Kong, X., Haystead, T.A., Pause, A., Belsham, G., Sonenberg, N. and Lawrence, J.C.,Jr (1994) PHAS-I as a link between mitogen-activated protein kinase and translation initiation. Science, 266, 653–656. [DOI] [PubMed] [Google Scholar]
- Mader S., Lee, H., Pause, A. and Sonenberg, N. (1995) The translation initiation factor eIF-4E binds to a common motif shared by the translation factor eIF-4 γ and the translational repressors 4E-binding proteins. Mol. Cell. Biol., 15, 4990–4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalingam M. and Templeton, D.J. (1996) Constitutive activation of S6 kinase by deletion of amino-terminal autoinhibitory and rapamycin sensitivity domains. Mol. Cell. Biol., 16, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morino S., Imataka,H., Svitkin,Y.V., Pestova,T.V. and Sonenberg,N. (2000) Eukaryotic translation initiation factor (eIF4E) binding site and the middle one-third of eIF4GI constitute the core domain for cap-dependent translation and the C–terminal one-third functions as a modulatory region. Mol. Cell. Biol., 20, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley S.J. and Traugh, J.A. (1989) Phorbol esters stimulate phosphorylation of eukaryotic initiation factors 3, 4B and 4F. J. Biol. Chem., 264, 2401–2404. [PubMed] [Google Scholar]
- Morley S.J. and Traugh, J.A. (1990) Differential stimulation of phosphorylation of initiation factors eIF-4F, eIF-4B, eIF-3 and ribosomal protein S6 by insulin and phorbol esters. J. Biol. Chem., 265, 10611–10616. [PubMed] [Google Scholar]
- Morley S.J., Curtis, P.S. and Pain, V.M. (1997) eIF4G: translation's mystery factor begins to yield its secrets. RNA, 3, 1085–1104. [PMC free article] [PubMed] [Google Scholar]
- Ohlmann T., Pain, V.M., Wood, W., Rau, M. and Morley, S.J. (1997) The proteolytic cleavage of eukaryotic initiation factor (eIF) 4G is prevented by eIF4E binding protein (PHAS-I; 4E-BP1) in the reticulocyte lysate. EMBO J., 16, 844–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pause A., Belsham, G.J., Gingras, A.-C., Donzé, O., Lin, T.A., Lawrence, J.C., Jr and Sonenberg, N. (1994) Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature, 371, 762–767. [DOI] [PubMed] [Google Scholar]
- Pestova T.V., Hellen, C.U. and Shatsky, I.N. (1996) Canonical eukaryotic initiation factors determine initiation of translation by internal ribosomal entry. Mol. Cell. Biol., 16, 6859–6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyronnet S., Imataka, H., Gingras, A.-C., Fukunaga, R., Hunter, T. and Sonenberg, N. (1999) Human eukaryotic translation initiation factor 4G (eIF4G) recruits MNK1 to phosphorylate eIF4E. EMBO J., 18, 270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raught B. and Gingras, A.-C. (1999) eIF4E activity is regulated at multiple levels. Int. J. Biochem. Cell Biol., 31, 43–57. [DOI] [PubMed] [Google Scholar]
- Redpath N.T., Foulstone, E.J. and Proud, C.G. (1996) Regulation of translation elongation factor-2 by insulin via a rapamycin-sensitive signalling pathway. EMBO J., 15, 2291–2297. [PMC free article] [PubMed] [Google Scholar]
- Rozen F., Edery, I., Meerovitch, K., Dever, T.E., Merrick, W.C. and Sonenberg, N. (1990) Bidirectional RNA helicase activity of eucaryotic translation initiation factors 4A and 4F. Mol. Cell. Biol., 10, 1134–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs A.B., Sarnow, P. and Hentze, M.W. (1997) Starting at the beginning, middle and end: translation initiation in eukaryotes. Cell, 89, 831–838. [DOI] [PubMed] [Google Scholar]
- Shima H., Pende, M., Chen, Y., Fumagalli, S., Thomas, G. and Kozma, S.C. (1998) Disruption of the p70S6k/p85S6k gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J., 17, 6649–6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkin Y.V., Gradi, A., Imataka, H., Morino, S. and Sonenberg, N. (1999) Eukaryotic initiation factor 4GII (eIF4GII), but not eIF4GI, cleavage correlates with inhibition of host cell protein synthesis after human rhinovirus infection. J. Virol., 73, 3467–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G. and Hall, M.N. (1997) TOR signalling and control of cell growth. Curr. Opin. Cell Biol., 9, 782–787. [DOI] [PubMed] [Google Scholar]
- Ui M., Okada, T., Hazeki, K. and Hazeki, O. (1995) Wortmannin as a unique probe for an intracellular signalling protein, phosphoinositide 3-kinase. Trends Biochem. Sci., 20, 303–307. [DOI] [PubMed] [Google Scholar]
- Vlahos C.J., Matter, W.F., Hui, K.Y. and Brown, R.F. (1994) A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem., 269, 5241–5248. [PubMed] [Google Scholar]
- von Manteuffel S.R., Gingras, A.-C., Ming, X.F., Sonenberg, N. and Thomas, G. (1996) 4E-BP1 phosphorylation is mediated by the FRAP–p70s6k pathway and is independent of mitogen-activated protein kinase. Proc. Natl Acad. Sci. USA, 93, 4076–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waskiewicz A.J., Flynn, A., Proud, C.G. and Cooper, J.A. (1997) Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J., 16, 1909–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waskiewicz A.J., Johnson, J.C., Penn, B., Mahalingam, M., Kimball, S.R. and Cooper, J.A. (1999) Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol. Cell. Biol., 19, 1871–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts J.D., Affolter, M., Krebs, D.L., Wange, R.L., Samelson, L.E. and Aebersold, R. (1994) Identification by electrospray ionization mass spectrometry of the sites of tyrosine phosphorylation induced in activated Jurkat T cells on the protein tyrosine kinase ZAP-70. J. Biol. Chem., 269, 29520–29529. [PubMed] [Google Scholar]
- Wells S.E., Hillner, P.E., Vale, R.D. and Sachs, A.B. (1998) Circularization of mRNA by eukaryotic translation initiation factors. Mol. Cell, 2, 135–140. [DOI] [PubMed] [Google Scholar]
- Yamanaka S., Poksay, K.S., Arnold, K.S. and Innerarity, T.L. (1997) A novel translational repressor mRNA is edited extensively in livers containing tumors caused by the transgene expression of the apoB mRNA-editing enzyme. Genes Dev., 11, 321–333. [DOI] [PubMed] [Google Scholar]
- Yan R., Rychlik, W., Etchison, D. and Rhoads, R.E. (1992) Amino acid sequence of the human protein synthesis initiation factor eIF-4 γ. J. Biol. Chem., 267, 23226–23231. [PubMed] [Google Scholar]