

Introduction
The protein kinase C (PKC) family of signal transducers are characterized by a dependence upon lipids for activity. Specifically, the classical (cPKCα, β and γ) and novel (nPKCδ, ɛ, η and θ) PKC isotypes display a physiological requirement for diacylglycerol for activity. This property of PKC has defined a now well established signalling pathway operating through receptors to phosphatidylinositol-specific phospholipase C and hence via diacylglycerol (DAG) [and inositol (1,4,5) trisphosphate Ins(1,4,5)P3/Ca2+] to PKC (Figure 1). The operation of this pathway has been described in many cell types, and numerous reviews have covered this signalling paradigm (see Nishizuka, 1986; Hug et al., 1993; Dekker and Parker, 1994; Jaken, 1996).
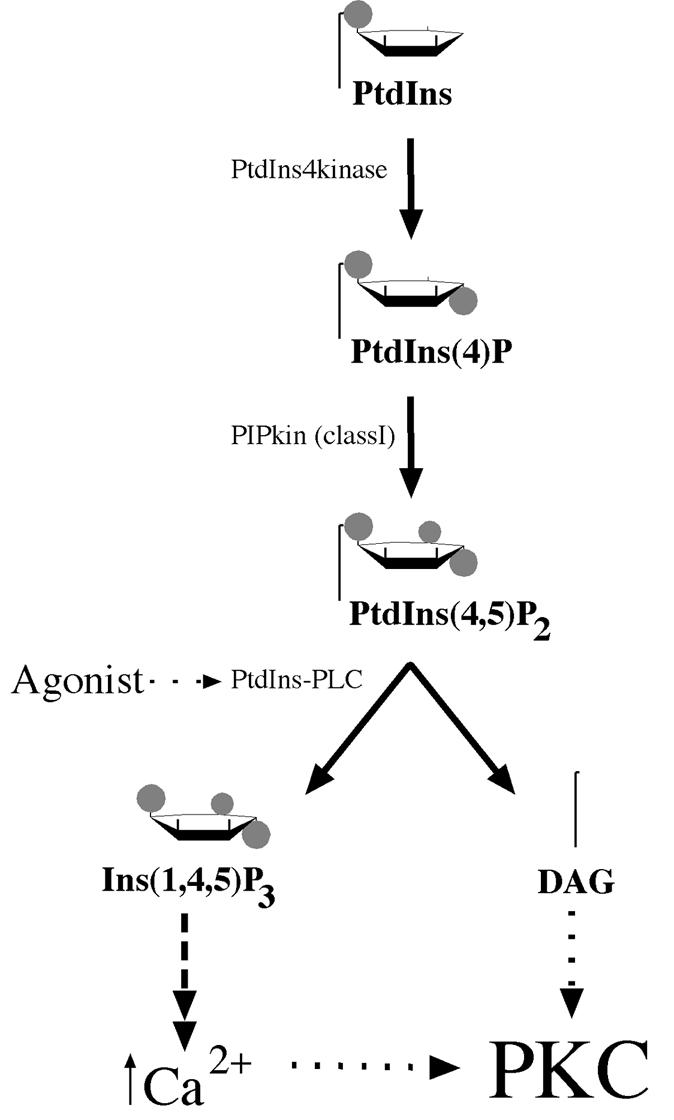
Fig. 1. The classical pathway of PKC activation. The scheme illustrates the production of the immediate precursor lipid PtdIns(4,5)P2 from its parent lipid PtdIns. Various agonists are linked to the phospholipases (PtdIns-PLC) that can cleave PtdIns(4,5)P2 to diacylglycerol (DAG) and the calcium mobilizer Ins(1,4,5)P3. Calcium can affect the cPKC class by promoting membrane recruitment, but the key allosteric activator at the membrane for both cPKC and nPKC isotypes is DAG.
More recently, attention has been drawn to the phosphorylation of PKC itself. Intriguingly, what was once considered a purely effector-driven transducer turns out to possess a complex amplitude control. The elucidation of this phosphorylation control in the cPKC isotypes has formed the basis for understanding the behaviour of the immediate family, with implications for other related AGC kinases (see Hanks and Hunter, 1995; further information is available at the Protein Kinase Resource: http://www.sdsc.edu/Kinases). The further characterization of the kinases that act upon the nPKCs provides evidence of three distinct input pathways converging upon PKC. Thus, PKC serves as an elegant example of the manner in which multiple signals are integrated in cells.
The purpose of this review is to provide: (i) a general model of phosphorylations and how they control the activity of (a) cPKC and (b) n/aPKC; (ii) a description of the current understanding of the kinases that act upon various PKCs; and (iii) a discussion of the broader implications for signalling in general.
cPKC phosphorylation
Evidence that cPKCα activity is under control by phosphorylation has been available for some time, with the findings that a 12-O-tetradecanoyl-13-acetate (TPA)-induced fast migrating (dephosphorylated) form was inactive (Borner et al., 1988) and, more directly, that the purified protein could be inactivated following phosphatase treatment (Pears et al., 1992). Subsequent mutagenesis of PKCα defined a threonine residue (T497) within the activation loop of the kinase domain that was essential for activity (Cazaubon et al., 1994). This phosphorylation site is also conserved within the cAMP-dependent protein kinase (PKA; Thr197), a member of the ACG kinase superfamily. Based upon detailed structural analysis of PKA, the phosphorylated Thr197 has been shown to play a role in aligning the catalytic site of that enzyme (Knighton et al., 1991). Structural models proposed for PKCα and PKCβ suggest an equivalent role for these activation loop phosphorylation sites (Orr and Newton, 1994; Srinivasan et al., 1996). As for PKCα, an absolute requirement for phosphorylation in the activation loop T500 site in PKCβ has been established (Orr and Newton, 1994). Thus, for these cPKCs, there is evidence that activation loop phosphorylation is required for activity. Furthermore, analysis of recombinant proteins expressed in insect cells demonstrates that these sites are occupied to some degree (Keranen et al., 1995; Tsutakawa et al., 1995). Expression in mammalian cells also reveals phosphorylation of these sites in cPKC isotypes including cPKCγ (Hansra et al., 1999).
One of the original autophosphorylation sites defined for baculovirus-expressed PKCβI (=β2), T642 (Flint et al., 1990), is also occupied in purified recombinant protein and in intact cells. The phosphorylation of this site has been reported to have various effects on activity. The original mutational analysis for PKCβI indicated that its phosphorylation was essential for activity. However, the solubility of the expressed non-phosphorylated protein can be problematic; removal of phosphates from recombinant PKCα causes aggregation, paralleling the neutral detergent insolubility of the dephosphorylated forms that can accumulate in vivo (see Bornancin and Parker, 1996). Subsequently it was shown that for PKCβII, if the homologous T641 is mutated, other local sites become phosphorylated to compensate, yielding a fully functional protein (see Newton, 1997). For PKCα, the equivalent T638A mutant is not fully functional, displaying a high specific activity, but thermal instability, sensitivity to oxidation and also sensitivity to protein phosphatases (Bornancin and Parker, 1996). This phenotype suggests that even if compensating phosphorylations were to take place in PKCα, the phosphorylation of the T638 site itself plays a unique role.
The synthesis of the PKCα observations suggested that T638 occupation favours a closed conformation of the catalytic domain. Phosphorylation at the activation loop also contributes to the closed conformation. Thus, there appears to be an interaction between this C-terminal region and the core catalytic domain. The prediction of a closed conformer is supported further by partial proteolytic analysis. This demonstrated that the C-terminus is sensitive to cleavage when the T638 site is unoccupied (Bornancin and Parker, 1996). As observed for PKA, the basis of this behaviour at the molecular level may well involve an interaction of the V5 domain with the lower and upper lobes of the kinase domain core. The cooperative effect of the T497 and T638 phosphorylations would by inference reflect the appropriate folding of the core domain and/or the relative rotation of its upper/lower lobes.
A third ‘priming’ site in PKCα and PKCβ was identified by direct phosphate analysis and also through mutagenesis based upon the predicted similarity between the patterns of sites in PKC and in p70S6kinase (Keranen et al., 1995; Tsutakawa et al., 1995; Bornancin and Parker, 1997). It is located in a hydrophobic region of the C-terminal V5 subdomain of PKC, 19 amino acids following the autophosphorylation site. Mutation of this hydrophobic site in PKCα provided evidence that phosphorylation here plays a role in controlling the rate of occupation of these ‘priming’ sites (i.e. 497/638/657) in PKCα (Bornancin and Parker, 1997). Studies on PKCβII have shown that phosphorylation at the equivalent site (S660) affects Ca2+ affinity in PKCβ (Edwards and Newton, 1997). This latter property is perhaps effected through C2 domain contact with this C-terminal V5 domain, when the catalytic domain as a whole is in its closed, i.e. phosphorylated, state. While there remains some debate as to the order of all these phosphorylation events and their precise consequences, a general model based largely upon studies with PKCα is summarized in Figure 2.

Fig. 2. Model for the phosphorylation of PKC. PKC is represented by a regulatory domain, comprising a C1 domain, C2 domain and pseudosubstrate site (black circle) and a catalytic domain (C3/4) with a C-terminal, V5, extension. The unphosphorylated primary translation product is predicted to have little or no activity. On ligand binding at the membrane, PKC becomes a substrate for kinases acting upon activation loop sites (for PKCα the T497 site) and hydrophobic C–terminal sites (for PKCα the S657 site). Following subsequent autophosphorylation (for PKCα the T638 site), the kinase domain is in a closed conformation that confers stability and phosphatase resistance. Ligand dissociation allows the kinase to diffuse away from the membrane but to remain in a phosphorylated state. The latent kinase can then be recruited back to the membrane and reactivated by DAG alone.
PKC phosphorylation will be discussed here with reference to the three catalytic domain ‘priming’ phosphorylation sites noted above, i.e. the activation loop site, the autophosphorylation site and the hydrophobic, C-terminal site. Although not the central theme of this review, it should be noted that the removal of phosphate from these sites is crucial to the desensitization of cPKC (Hansra et al., 1996). This has been demonstrated clearly with phosphorylation site-specific antisera. On chronic activation, loss of immunoreactivity with these antisera precedes the characteristic down-regulation of PKC protein, i.e. dephosphorylation at these sites precedes degradation (Hansra et al., 1999). Direct [32P]orthophosphate labelling has provided similar evidence, albeit for unspecified sites in PKCα (Lee et al., 1996). The pathways leading to dephosphorylation and degradation are only now emerging and are not considered further here.
n/aPKC phosphorylation
That there is indeed some relevance to priming phosphorylations in general derives in part from the finding that equivalent ‘priming’ sites in n/aPKC isotypes are occupied in purified recombinant eukaryote expressed proteins and in intact cells (Hansra et al., 1999; Parekh et al., 1999; Ziegler et al., 1999). However, it is relevant to question whether the general scheme in Figure 2 is of utility for n/aPKC isotypes. To date, only limited studies have been carried out on the ‘priming’ phosphorylation sites in n/aPKC isotypes, but there is already information to suggest that there are both similarities and differences.
When PKCδ is expressed in bacteria, the activation loop (T505) and C-terminal, hydrophobic (S660) sites are unphosphorylated; however, it retains catalytic activity. Proof that activation loop site phosphorylation was not required for activity was provided by studies on a T505A mutant (Stempka et al., 1997). This behaviour contrasts with that of PKCα, which is not active on expression in bacteria. Nevertheless, it has been observed that this bacterial expressed, T505/S662 unphosphorylated PKCδ protein has less than one-tenth of the activity obtained from eukaryote expressed PKCδ (Le Good et al., 1998). Interestingly, this low activity form of PKCδ is autophosphorylated at the autophosphorylation site (S643) (Le Good et al., 1998). This is consistent with its assignment as an autophosphorylation site, which has been verified directly through mutagenesis (Li et al., 1997). The reduced activity of the bacterial expressed species of PKCδ contrasts with the full activity ascribed to an equivalently phosphorylated PKCβII species (see Newton, 1997). This may reflect some of the subtle distinctions between the novel and classical isotypes. Such a distinction between PKCδ and PKCα is reflected in the finding that the PKCδ S643A mutant, unlike the equivalent S638A PKCα mutant, does not display thermal instability, but has a reduced activity (Li et al., 1997). It remains to be seen whether as described for PKCβII, local compensating phosphorylations contribute to the stability of the PKCδ S643A mutant; two proximal serine residues would be candidates. While PKCδ appears to be able to autophosphorylate at the S643 site, the S662 hydrophobic site is not occupied in bacterial PKCδ, suggesting that this is not an autophosphorylation site (see below). In general, this pattern of behaviour for PKCδ is similar to what can be extrapolated from the PKCα model, except that in the ‘basal state’ the dephosphoPKCδ appears to be folded correctly, soluble and partially (albeit ⩽10%) active.
For the atypical PKC isotypes, the C-terminal hydrophobic site, while retaining the characteristic aromatic residues, has a glutamic acid residue (FXXFEF) in place of the serine or threonine residue found in other PKC isotypes. Such a mutation in PKCα partially substitutes for phosphorylation; the T657E PKCα mutant shows an intermediate sensitivity to phosphatases, oxidants and thermal denaturation (Bornancin and Parker, 1997). It can be surmised that for the aPKC isotypes there is no phosphorylation in this conserved hydrophobic region, but that these isotypes are likely to be dephosphorylated more effectively than the c/nPKC isotypes (see further below).
The occupation of these priming sites in cPKC, nPKC and aPKC isotypes and the similarity of the kinase domains suggest that the general properties ascribed to these sites are likely to be conserved. Formal proof of this will come from further site-directed mutagenesis, reconstitution and structural analyses. As noted above, the conservation of these phosphorylation sites/motifs within the PKC family extends to other kinases of the AGC class. This includes the PKC-related kinases (PRK1/PKN, PRK2 and PKNβ/PRK4), p90rsk, p70S6kinase and PKB (also referred to as Akt) (some of these kinase domains are aligned in Figure 3). The best understood, and perhaps simplest of these is PKB. There is direct evidence that the PKBα activation loop site (T308) and the C-terminal hydrophobic site (T473) become phosphorylated and are required for activation in vivo (Alessi et al., 1996). This agonist-induced response is dependent upon phosphatidylinositol (PtdIns) 3-kinase. Mutagenesis has indicated that both the T308 and T473 sites contribute to activation in a cooperative fashion. However, disproportional effects have been observed (Stokoe et al., 1997), and it remains possible that T473 may contribute in part to stability, as observed for PKCα (Bornancin and Parker, 1997). For PKB, activation loop kinases have been identified (PDK1 and related activities) that display a requirement for PtdIns(3,4,5)P3 consistent with the PtdIns 3-kinase inhibitor sensitivity of activation loop phosphorylation in intact cells (Alessi et al., 1997; Stephens et al., 1998) [The identification of PDK1 as a PKB activation loop (T308) kinase has led to the use of the nomenclature PDK2 for the T473 kinase. However, with the possible existence of multiple PDK1-related T308 kinase activities, this nomenclature is better reserved for any mammalian PDK1 relatives and the T473 kinase named when formally identified.] The simplistic view of PKB control is of PtdIns(3,4,5)P3-dependent membrane recruitment with PDK1 and a second PKB kinase/component (see below) leading to complete phosphorylation and activation.

Fig. 3. AGC kinase domain alignment. The kinase domains of PKCα/δ/ζ, the PKC-related kinase PRK1, PKBα and p70S6kinase α (p70α) are aligned from their first conserved kinase sub-domain (I) through sub-domain X, to the region covering the C-terminal extension. The kinase sub-domains are indicated by the shaded areas, with regions I–X displayed with an increasingly darker hue. The three phosphorylation sites (or acidic residues) discussed in the text are indicated by the red bars. The activation loop is indicated by the dark purple block, and the hydrophobic C-terminal phosphorylation site is also boxed in dark purple.
The kinase domain of p70S6kinase shows a similar dependence upon the same class of phosphorylation sites for activation, in addition to other sites on an extended C–terminal domain (reviewed in Pullen and Thomas, 1997). In this case, the hydrophobic site (T389) is particularly sensitive to the immunosuppressant rapamycin; the activation loop site shows much less sensitivity (Ferrari et al., 1993). While other sites contribute to p70S6kinase control, it is clear that the key agonist-induced activating phosphorylations are those also conserved in PKC.
The kinase cascade to PKC activation loop phosphorylation
Various elements of conservation within this class of kinases have led to the finding that PDK1 or perhaps a close relative is responsible for PKC activation loop phosphorylation. PDK1 will phosphorylate both nPKCδ and aPKCζ in vitro (Chou et al., 1998; Le Good et al., 1998). Co-expression of PDK1 with PKCδ/ζ in mammalian cells can also induce PKC phosphorylation at activation loop sites. In intact cells, the effect of PDK1 is blocked by the PtdIns 3-kinase inhibitor LY294002, and this effect appears to be directed through PDK1 and not PKC (Le Good et al., 1998). Consistent with this, PtdIns(3,4,5)P3 cooperates with the PKC activator TPA in stimulating PDK1 phosphorylation of PKCδ in vitro. The evidence suggests that PDK1 and PKC need to be co-recruited to membranes through interaction with their respective allosteric activators in order for phosphorylation to be efficient.
A broad role for PDK1, or a relative, in PKC phosphorylation is supported by the finding that all PKC isotype subclasses can form complexes with PDK1 (Le Good et al., 1998). Whether PDK1 is itself responsible for all PKC activation loop phosphorylations in vivo remains to be determined, although recent observations indicate a role for PDK1 in cPKC phosphorylation in vivo (Dutil et al., 1998). Perhaps the most compelling, albeit circumstantial, evidence for a physiological role for PDK1 comes from studies on the PRK proteins. These latter kinases have been shown to be regulated by small G–proteins of the Rho class, through interactions at the N–terminal HR1 domains (Shibata et al., 1996; Flynn et al., 1998). It has been reported that Rho binding leads to increased activity (up to 4-fold) in vitro and hence it has been proposed that this effect is a consequence of the binding to and sequestration of the HR1 domain, where pseudosubstrate sites are present (Shibata et al., 1996). It has been found that the PRKs also interact with PDK1 via their PIF (PDK1-interacting fragment) region (Balendran et al., 1999) and they are phosphorylated in their activation loops by this kinase (Flynn et al., 2000). However, this effect is closely linked to the control through Rho. The interaction between PRK1/2 and PDK1 shows dependence upon active Rho. Thus, Rho binding to PRK1/2 leads to a conformational change that is permissive for PDK1 binding. It is proposed that the subsequent phosphorylation of PRK1/2 in the activation loop is the key activating event for PRK itself (Flynn et al., 2000). More direct evidence for this relationship derives from studies in vivo, where it is found that RhoB recruits PRK1/2 to an endosomal compartment (Mellor et al., 1998). We have found that PDK1 is also recruited to this compartment by RhoB, but only when PRK1 is co-expressed. Thus a heterotrimeric complex can be formed between RhoB, PRK1 and PDK1 in vivo (summarized in Figure 4). In addition, it has been shown that the PRK recruited to the endosome compartment by RhoB is in a hyperphosphorylated state (Mellor et al., 1998), consistent with a role for the associated PDK1 in carrying out one part of this phosphorylation.

Fig. 4. Rho-dependent PRK activation via PDK1. In the unliganded state, PRK is shown to be in an inactive conformation, with the pseudosubstrate site(s) in the HR1 domain interacting with the catalytic domain. On Rho binding to the HR1 motif in a membrane compartment, the kinase domain is released and can express catalytic activity, albeit at a low level. The exposed kinase domain can interact with PDK1 through its PIF motif (see text) and, on its association with PtdIns(3,4,5)P3, PDK1 will phosphorylate and activate PRK. The domains illustrated are summarized in the figure.
There is an obvious symmetry in the behaviour of PKC and PRK with respect to phosphorylation by PDK1, optimum phosphorylation of both classes being dependent upon their particular, membrane-associated, allosteric activators. This pattern of behaviour would apply equally to PKB with its PH domain-dependent phosphoinositide binding. It is assumed that PDK1 requires its own activator PtdIns(3,4,5)P3 for effective catalytic activity; this would be consistent with the PtdIns 3-kinase dependence of PKC activation loop phosphorylation in intact cells (Chou et al., 1998; Le Good et al., 1998). For PKC, there is direct in vivo evidence for the need for activation of the target kinase (i.e. the PKC), since calphostin C, which selectively blocks the allosteric activation of PKC by DAG, inhibits serum-induced activation loop phosphorylation, as does the PtdIns 3-kinase inhibitor LY294002, which would affect PDK1 recruitment/activation (Parekh et al., 1999).
The broad conclusion from these studies is that there is a cascade of kinases involving PtdIns 3-kinase, PDK1 and various members of the PKC superfamily. The specificity of function is driven, at least in part, by effector/second messenger interaction with the target kinase. For PKC, the consequence of phosphorylation is an increase in latent catalytic activity, without bypassing the requirement for allosteric activators.
Transphosphorylation at the PKC C-terminal domain
A membrane-associated activity that phosphorylates the hydrophobic C-terminal site in the V5 domain of PKCα has been detected, whose presence/activity in the membrane fraction is compromised if cells are pre-treated with rapamycin or the PtdIns 3-kinase inhibitor LY294002 (Ziegler et al., 1999). Consistent with a role for this activity in PKC phosphorylation in vivo, these inhibitors are also found to block the serum-stimulated phosphorylation of the equivalent hydrophobic sites in PKCδ and PKCɛ in intact cells. Purification of the membrane-associated kinase that phosphorylates the C-terminal hydrophobic motif in PKC has led to the identification of an atypical PKCζ/ι as a component of a complex. Consistent with a role for aPKC is the finding that expression of an activated form of PKCζ induces the phosphorylation of co-expressed PKCδ in serum-starved cells. This phosphorylation is not inhibited by rapamycin, demonstrating that the overexpressed, active PKCζ overcomes the normal serum-dependent control hierarchy. Nevertheless, in the presence of activated PKCζ, the extent of PKCδ C–terminal phosphorylation can be increased still further on serum stimulation; this is also rapamycin-insensitive, indicating that the serum-dependent step is probably PKCδ directed (e.g. through agonist-induced DAG production). This is reminiscent of the situation with PDK1 where PKC-directed allosteric interaction supports phosphorylation. The sensitivity to rapamycin and LY294002 is indicative of a role for mTOR in controlling this phosphorylation. In fact, it has been shown that a rapamycin-resistant mTOR relieves the rapamycin sensitivity of the PKCδ (and ɛ) phosphorylation pathway at the hydrophobic site (Parekh et al., 1999). Whether the elements between mTOR and PKC phosphorylation involve kinases, phosphatases or both has yet to be elucidated. However, based upon the behaviour of an N-terminal truncated p70S6kinase mutant (Mahalingam and Templeton, 1996), the simplest model would be that mTOR controls a phosphatase that acts upon this PKC site. The overexpression of the activated aPKCζ mutant must be sufficient to promote the phosphorylation of this site, despite the relief of phosphatase inhibition through mTOR inhibition by rapamycin (see Figure 5, below).

Fig. 5. A general scheme of PKC controls. The figure illustrates the upstream inputs to membrane recruitment and phosphorylation of PKC that are required to generate the ‘mature’ phosphorylated form. For reasons of clarity, this is a simplified view of the process that excludes the action of PKC-interacting proteins that are likely to play roles in localization and membrane targeting. Some of the inhibitors that can block specific steps in the input pathways are included (in red), as well as the kinases and phosphatases (undefined) predicted to play key roles in effecting the modifications. External influences are known to control many of these steps, including activation of PtdIns-PLC, PtdIns 3-kinase and mTOR. Inputs to the aPKC complex (?) are as yet undefined.
It has been shown that the serum-induced phosphorylation of p70S6kinase in the rapamycin-sensitive T389 site is sensitive to amino acid starvation. Similar studies on PKCδ indicate that the acute serum-induced phosphorylation in the equivalent hydrophobic, C-terminal site is also inhibited under conditions of amino acid deprivation (Parekh et al., 1999). Hence there is present in mammalian cells an amino acid-sensing pathway that is likely to operate through mTOR to control the phosphorylation of multiple cellular kinases.
PKC phosphorylation and its implications
The synthesis of all the disparate experimental data on PKC and its relatives provides a testable model of the controls acting upon PKC (Figure 5). It is certainly the case that elements within this model remain unknown, and further that this is likely to be an oversimplification. Nevertheless, it does provide a basis upon which to reach a precise molecular description of the controls.
There are some interesting implications deriving from the pattern of controls acting upon c/nPKC isotypes. With respect to the ‘priming’ phosphorylations themselves, there is little doubt that at least in combination these act as an amplitude control, i.e. when phosphorylated in these three priming sites, PKC has a higher specific activity than when unphosphorylated (the latter may have low or undetectable activity). Thus, DAG (+Ca2+) will acutely switch these transducers on (c/nPKC isotypes), but the volume control is a function of phosphorylation. While this distinction exists between allosteric effector and phosphorylation, the evidence for PKC is that phosphorylation occurs efficiently in vivo when PKC is in an active, i.e. effector-bound, conformation. This parallels the behaviour of PKB where its interaction with phosphoinositides through its regulatory PH domain is permissive for its phosphorylation by PDK1 (Alessi et al., 1997, 1998; Stephens et al., 1998). Extrapolating to p70S6kinase, it is likely that interactions with (N-terminus) and/or phosphorylation of (C-terminus) this kinase is also required for optimum phosphorylation by PDK1; in vitro, effective phosphorylation by PDK1 is observed for the truncated protein (Alessi et al., 1998; Pullen et al., 1998). Certainly for PRK1/2, its interaction with its selective regulator Rho⋅GTP is required for PDK1 to bind and phosphorylate (Flynn et al., 2000). It is a consequence of these requirements that PDK1 specificity is built into the system through the dependence of its targets on their co–factors/associated proteins, i.e. phosphorylation by PDK1 minimally involves the coincident detection of PtdIns(3,4,5)P3 plus a specific effector-bound kinase in the same compartment.
The similarities between PKC phosphorylation control and that of its AGC kinase family relatives are clear. Yet the importance of these events for PKC have been slow to be appreciated, because of a fundamental distinction between PKC and the rest. This difference is a temporal one that is a consequence of turnover. Unlike PKB, when PKC (n/cPKC) releases its activator (DAG), its ‘priming’ phosphorylations are not rapidly lost. On the contrary, it seems as though the inactive, closed conformer of PKC is in fact relatively resistant to phosphatases. For PKC, it is the allosterically activated form that appears to be targeted for dephosphorylation (and degradation) (Hansra et al., 1996, 1999; Lee et al., 1996). A very important consequence of this behaviour of PKC is that the accumulation of phosphorylated PKC isotypes serves to integrate information over time. For nPKCδ and nPKCɛ to become phosphorylated at their priming sites, it is clear that at least four criteria have to be fulfilled: (i) DAG must be provided; (ii) sufficient PtdIns(3,4,5)P3 must be present to recruit/activate PDK1; (iii) PKCζ and its partner(s) must be located/activated appropriately; and (iv) mTOR must be active. Because the phosphorylations are relatively stable, the amplitude control exerted by these modifications remains in place for tens of minutes to hours.
Why might such a system evolve? A possible explanation is that these permissive amplitude controls sense events for which the cell would wish to buffer consequences. Thus, for example, a cell transiently exposed to an amino acid-depleted environment might shut down new protein synthesis rapidly, but would not be expected to commit itself immediately to apoptosis. Perhaps the essential survival role played by PKCα phosphorylation (Whelan and Parker, 1998) reflects such a protective strategy.
It has been implicit in the above commentary that the effects of PKC phosphorylation are concerned primarily with its intrinsic catalytic activity and ability to phosphorylate and modify the actions of its own downstream targets. This may not be the entire case. The evidence on PRK2–PDK1 interaction is that in this complexed state, PDK1 acquires the ability to act not only on the activation loop site of PKB, but also on the hydrophobic C-terminal site (Balendran et al., 1999). The action of such a complex is probably reflected in the behaviour of the PKCζ complex that will promote the phosphorylation of the hydrophobic C-terminal site in PKCδ, as discussed above. It follows then that since all PKC isotypes tested can form complexes with PDK1, any one such complex may be responsible for the targeting of PKB or related AGC kinases. c/nPKC phosphorylation within the FXXFSY motif may thus have an additional role in promoting the PDK1 scaffolding properties of these kinases. How such a role is integrated with intrinsic catalytic function is a key question for the future.
Table I. Priming phosphorylation sites in the PKC superfamily.
| PKC isotypes | Activation loop | Effect of no phosphate/Ala mutation | C-terminal autophosphorylation | Effect of no phosphate/Ala mutation | C-terminal hydrophobic | Effect of no phosphate/Ala mutation |
|---|---|---|---|---|---|---|
| Classical | ||||||
| α | T497 | inactive | T638 | inactivation-sensitive | S657 | inactivation-sensitive |
| TFCGT | TPPDQ | FSYVN | ||||
| β1(II) | T500 | inactive | T641 | inactivea | S660 | lower |
| TFCGT | TPPDQ | FSFVN | relative Ca2+sensitivity | |||
| β2(I) | T500 | T642 | inactive (insoluble?) | S661 | ||
| TFCGT | TPTDK | FSYTN | ||||
| γ | T514 | T655 | T674 | |||
| TFCGT | TPPDR | FTYVN | ||||
| Novel | ||||||
| δ | T505 | low activity | S643 | low activity | S662 | low activity |
| TFCGT | SFSDK | FSFVN | ||||
| ɛ | T566 | T710 | low activity | S729 | low activity | |
| TFCGT | TLVDE | FSYFG | ||||
| η | T513 | T655 | S674 | |||
| TFCGT | TPIDE | FSYVS | ||||
| θ | T538 | S676 | S695 | |||
| TFCGT | SFADR | FSFIN | ||||
| Atypical | ||||||
| ζ | T410 | low activity | T560 | E579 | ||
| TFCGT | TPDDE | FEFIN | ||||
| ι | T403 | T574 | E555 | |||
| TFCGT | TPDDD | FEYIN |
The amino acid number of the sites listed varies by one or two residues between different species. The available information on the effect of a lack of phosphate, or an alanine mutation at the priming phosphorylation site, on the catalytic activity is included. The effects are discussed further in the text.
Residues flanking the T641 site in PKCβ2(I) can still be autophosphorylated, and compensate for the lack of phosphate at this site. When the flanking autophosphorylation sites are also mutated to alanine residues, the lack of phosphate at T641 results in an inactive protein.
References
- Alessi D.R., Andjelkovic, M., Caudwell, B., Cron, P., Morrice, N., Cohen, P. and Hemmings, B.A. (1996) Mechanism of activation of protein-kinase-B by insulin and IGF-1. EMBO J., 15, 6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Alessi D.R., James, S.R., Downes, C.P., Holmes, A.B., Gaffney, P.R.J., Reese, C.B. and Cohen, P. (1997) Characterization of a 3-phosphoinositide-dependent protein-kinase which phosphorylates and activates protein-kinase B-α. Curr. Biol., 7, 261–269. [DOI] [PubMed] [Google Scholar]
- Alessi D.R., Kozlowski, M.T., Weng, Q.P., Morrice, N. and Avruch, J. (1998) 3-Phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro.Curr. Biol., 8, 69–81. [DOI] [PubMed] [Google Scholar]
- Balendran A., Casamayor, A., Deak, M., Paterson, A., Gaffney, P., Currie, R., Downes, C.P. and Alessi, D.R. (1999) PDK1 acquires PDK2 activity in the presence of a synthetic peptide derived from the carboxyl terminus of PRK2. Curr. Biol., 9, 393–404. [DOI] [PubMed] [Google Scholar]
- Bornancin F. and Parker, P.J. (1996) Phosphorylation of threonine-638 critically controls the dephosphorylation and inactivation of protein-kinase C-α. Curr. Biol., 6, 1114–1123. [DOI] [PubMed] [Google Scholar]
- Bornancin F. and Parker, P.J. (1997) Phosphorylation of protein kinase C-α on serine 657 controls the accumulation of active enzyme and contributes to its phosphatase-resistant state. J. Biol. Chem., 272, 3544–3549. [DOI] [PubMed] [Google Scholar]
- Borner C., Eppenberger, U., Wyss, R. and Fabbro, D. (1988) Continuous synthesis of two protein kinase C-related proteins after down-regulation by phorbol esters. Proc. Natl Acad. Sci. USA, 85, 2110–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazaubon S., Bornancin, F. and Parker, P.J. (1994) Threonine-497 is a critical site for permissive activation of protein kinase Cα. Biochem. J., 301, 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou M.M., Hou, W., Johnson, J., Graham, L.K., Lee, M.H., Chen, C.S., Newton, A.C., Schaffhausen, B.S. and Toker, A. (1998) Regulation of protein kinase C ζ by PI 3-kinase and PDK-1. Curr. Biol., 8, 1069–1077. [DOI] [PubMed] [Google Scholar]
- Dekker L.V. and Parker, P.J. (1994) Protein kinase C—a question of specificity. Trends Biochem. Sci., 19, 73–77. [DOI] [PubMed] [Google Scholar]
- Dutil E.M., Toker, A. and Newton, A.C. (1998) Regulation of conventional protein kinase C isozymes by phosphoinositide-dependent kinase 1 (PDK-1). Curr. Biol., 8, 1366–1375. [DOI] [PubMed] [Google Scholar]
- Edwards A.S. and Newton, A.C. (1997) Phosphorylation at conserved carboxyl-terminal hydrophobic motif regulates the catalytic and regulatory domains of protein kinase C. J. Biol. Chem., 272, 18382–18390. [DOI] [PubMed] [Google Scholar]
- Ferrari S., Pearson, R.B., Siegmann, M., Kozma, S.C. and Thomas, G. (1993) The immunosuppressant rapamycin induces inactivation of p70s6k through dephosphorylation of a novel set of sites. J. Biol. Chem., 268, 16091–16094. [PubMed] [Google Scholar]
- Flint A.J., Paladini, R.D. and Koshland, D.E.,Jr (1990) Autophosphorylation of protein kinase C at three separated regions of its primary sequence. Science, 249, 408–411. [DOI] [PubMed] [Google Scholar]
- Flynn P., Mellor, H., Palmer, R., Panayotou, G. and Parker, P.J. (1998) Multiple interactions of PRK1 with RhoA. Functional assignment of the HR1 repeat motif. J. Biol. Chem., 273, 2698–2705. [DOI] [PubMed] [Google Scholar]
- Flynn P., Mellor,H., Casamassima,A. and Parker,P.J. (2000) Rho-GTPase control of PRK activation by PDK1. J. Biol. Chem., in press. [DOI] [PubMed] [Google Scholar]
- Hanks S.K. and Hunter, T. (1995) Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J., 9, 576–596. [PubMed] [Google Scholar]
- Hansra G., Bornancin, F., Whelan, R., Hemmings, B.A. and Parker, P.J. (1996) 12-O-Tetradecanoylphorbol-13-acetate-induced dephosphorylation of protein kinase Cα correlates with the presence of a membrane associated protein phosphatase 2A heterotrimer. J. Biol. Chem., 271, 32785–32788. [DOI] [PubMed] [Google Scholar]
- Hansra G., Garcia-Paramio, P., Prevostel, C., Whelan, R.D., Bornancin, F. and Parker, P.J. (1999) Multisite dephosphorylation and desensitisation of conventional protein kinase C isotypes. Biochem. J., 342, 337–344. [PMC free article] [PubMed] [Google Scholar]
- Hug H. and Sarre, T.F. (1993) Protein kinase C isoenzymes: divergence in signal transduction?Biochem. J., 291, 329–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaken S. (1996) Protein kinase C isozymes and substrates. Curr. Opin. Cell Biol., 8, 168–173. [DOI] [PubMed] [Google Scholar]
- Keranen L.M., Dutil, E.M. and Newton, A.C. (1995) Protein kinase C is regulated in vivo by three functionally distinct phosphorylations. Curr. Biol., 5, 1394–1403. [DOI] [PubMed] [Google Scholar]
- Knighton D., Zheng, J., Teneyck, L., Ashford, V., Xuong, N., Taylor, S. and Sowadski, J. (1991) Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science, 253, 407–414. [DOI] [PubMed] [Google Scholar]
- Le Good J.A., Ziegler, W.H., Parekh, D.B., Alessi, D.R., Cohen, P. and Parker, P.J. (1998) Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science, 281, 2042–2045. [DOI] [PubMed] [Google Scholar]
- Lee H.W., Smith, L., Pettit, G.R. and Smith, J.B. (1996) Dephosphorylation of activated protein kinase C contributes to down-regulation by bryostatin. Am. J. Physiol., 40, C304–C311. [DOI] [PubMed] [Google Scholar]
- Li W.Q., Zhang, J.C., Bottaro, D.P., Li, W. and Pierce, J.H. (1997) Identification of serine 643 of protein kinase C-δ as an important autophosphorylation site for its enzymatic activity. J. Biol. Chem., 272, 24550–24555. [DOI] [PubMed] [Google Scholar]
- Mahalingam M. and Templeton, D.J. (1996) Constitutive activation of S6 kinase by deletion of amino-terminal autoinhibitory and rapamycin sensitivity domains. Mol. Cell. Biol., 16, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor H., Flynn, P., Nobes, C.D., Hall, A. and Parker, P.J. (1998) PRK1 is targeted to endosomes by the small GTPase, RhoB. J. Biol. Chem., 273, 4811–4814. [DOI] [PubMed] [Google Scholar]
- Newton A.C. (1997) Regulation of protein kinase C. Curr. Opin. Cell Biol., 9, 161–167. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y. (1986) Studies and perspectives of protein kinase C. Science, 233, 305–312. [DOI] [PubMed] [Google Scholar]
- Orr J.W. and Newton, A.C. (1994) Requirement for negative charge on ‘activation loop’ of protein kinase C. J. Biol. Chem., 269, 27715–27718. [PubMed] [Google Scholar]
- Parekh D., Ziegler, W., Yonezawa, K., Hara, K. and Parker, P.J. (1999) mTOR controls one of two kinase pathways acting upon nPKCδ and nPKCɛ. J. Biol. Chem., 274, 34758–34764. [DOI] [PubMed] [Google Scholar]
- Pears C., Stabel, S., Cazaubon, S. and Parker, P.J. (1992) Studies on the phosphorylation of protein kinase C-α. Biochem. J., 283, 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pullen N. and Thomas, G. (1997) The modular phosphorylation and activation of p70S6K. FEBS Lett., 410, 78–82. [DOI] [PubMed] [Google Scholar]
- Pullen N., Dennis, P.B., Andjelkovic, M., Dufner, A., Kozma, S.C., Hemmings, B.A. and Thomas, G. (1998) Phosphorylation and activation of p70s6k by PDK1. Science, 279, 707–710. [DOI] [PubMed] [Google Scholar]
- Shibata H., Mukai, H., Inagaki, Y., Homma, Y., Kimura, K., Kaibuchi, K., Narumiya, S. and Ono, Y. (1996) Characterization of the interaction between RhoA and the amino-terminal region of PKN. FEBS Lett., 385, 221–224. [DOI] [PubMed] [Google Scholar]
- Srinivasan N., Bax, B., Blundell, T.L. and Parker, P.J. (1996) Structural aspects of the functional modules in human protein kinase C α deduced from comparative analyses. Proteins, 26, 217–235. [DOI] [PubMed] [Google Scholar]
- Stempka L., Girod, A., Muller, H.J., Rincke, G., Marks, F., Gschwendt, M. and Bossemeyer, D. (1997) Phosphorylation of protein kinase C δ (PKCδ) at threonine 505 is not a prerequisite for enzymatic activity. J. Biol. Chem., 272, 6805–6811. [DOI] [PubMed] [Google Scholar]
- Stephens L., et al. (1998)Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science, 279, 710–714. [DOI] [PubMed] [Google Scholar]
- Stokoe D., Stephens, L.R., Copeland, T., Gaffney, P.R., Reese, C.B., Painter, G.F., Holmes, A.B., McCormick, F. and Hawkins, P.T. (1997) Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase. Science, 277, 567–570. [DOI] [PubMed] [Google Scholar]
- Tsutakawa S.E., Medzihradszky, K.F., Flint, A.J., Burlingame, A.L. and Koshland, D.E.,Jr (1995) Determination of in vivo phosphorylation sites in protein kinase C. J. Biol. Chem., 270, 26807–26812. [DOI] [PubMed] [Google Scholar]
- Whelan D.H. and Parker, P.J. (1998) Loss of protein kinase C function induces an apoptotic response. Oncogene, 15, 1939–1944. [DOI] [PubMed] [Google Scholar]
- Ziegler W.H., Parekh, D.B., Le Good, J.A., Whelan, R.D.H., Kelly, J.J., Frech, M.M., Hemmings, B.A. and Parker, P.J. (1999) Rapamycin-sensitive phosphorylation of PKC on a carboxy-terminal site by an atypical PKC complex. Curr. Biol., 9, 522–529. [DOI] [PubMed] [Google Scholar]